Introduction
Pseudoachondroplasia (PSACH; MIM no. 177170) is an
autosomal dominant osteochondrodysplasia characterized by
short-limb short stature, brachydactyly and early-onset
osteoarthropathy (1). Affected
individuals appear normal at birth. A waddling gait is often the
presenting feature at the onset of walking (1). Typically, at approximately two years
of age, the rate of growth falls below the standard growth curve,
causing a moderately severe form of disproportionate short-limb
short stature (1). Vertebral
anomalies, present in childhood, usually resolve with age, however
osteoarthritis is progressive and severe.
Previous genetic studies have demonstrated that
PSACH is caused by heterozygous mutations in the cartilage
oligomeric matrix protein (COMP; MIM no. 600310) gene (2,3).
COMP is located on chromosome 19p13.1 (4). COMP rat and bovine sequencing
indicate that it is a member of the thrombospondin gene family
(4). It contains 19 exons,
encoding the epidermal growth factor-like (type II) repeats,
calmodulin-like (type III) repeats and the C-terminal domain. Exons
4–19 correspond in sequence and intron location to the
thrombospondin genes, whereas exons 1–3 are unique to COMP
(2).
It is notable that mutations in COMP produce
clinical phenotypes ranging from the severe end of the spectrum,
PSACH, to a mild condition, multiple epiphyseal dysplasia (MED).
Briggs et al (5) suggested
that missense mutations in the type III repeats (amino acid
residues 268–528) are the major cause of PSACH and MED, exhibit
significant phenotypic correlations and derived preliminary
genotype to phenotype correlations. However, the correlations
remain unclear, and more data is required to further deduce
genotype-phenotype correlations. Although numerous mutations in
COMP in patients with PSACH have been reported, data on Chinese
patients with PSACH remain limited (6–10).
The current study presents the molecular and clinical
characteristics of six Chinese patients with PSACH.
Materials and methods
Patients
The current study was approved by the Ethics
Committee of the Sixth People's Hospital Affiliated to Shanghai
Jiao Tong University (Shanghai, China). Prior to initiation of the
study, written informed consent for genetic tests and publication
of the case details was obtained from all adult participants and
the parents or the legal guardians on behalf of children involved
in the study.
Mutation analysis
Informed consent was obtained from the families and
from 250 healthy ethnically matched volunteers prior to blood
sampling and DNA analysis. These 250 Chinese volunteers (125 males
and 125 females) were recruited from the Department of
Osteoporosis, Sixth People's Hospital Affiliated to Shanghai Jiao
Tong University. This is the same control cohort used in our
previous studies (11,12). The DNA was extracted from
peripheral white blood cells by proteinase K (Kurabo Industries,
Ltd., Tokyo, Japan) digestion followed by purification with
phenol/chloroform and isopropyl alcohol precipitation (Ling Feng
Chemical Reagent Co., Ltd., Shanghai, China). The DNA sequence for
the COMP gene was obtained from the available online database
(GenBank accession no. NC_000019). The COMP gene primers (Table I) were designed using Primer3
software (version 0.4.0; http://primer3.wi.mit.edu/). Template DNA extracted
from peripheral white blood cells, and all exons and their
exon-intron boundaries in the COMP gene were amplified via
polymerase chain reaction (PCR). The reaction mixture for COMP
10F/R was as follows: 20 µl Mixture including 1X GC buffer I
(Takara Bio, Inc., Otsu, Japan), 2.5 mM Mg2+, 0.2 mM
dNTP, 0.2 µM of each primer, 1 unit HotStarTaq polymerase
(Qiagen, Inc., Valencia, CA, USA) and 1 µl template DNA. The
reaction mixture for COMP 1F/R, 15F/R and 16F/R was as follows: A
20 µl mixture including 1X HotStarTaq buffer, 2.0 mM
Mg2+, 0.2 mM dNTP, 0.2 µM of each primer, 1 unit
HotStarTaq polymerase and 1 µl template DNA. The reaction
mixture for the remaining fragments was as follows: A 20µl
mixture including 1X GC buffer I (Takara Bio, Inc.), 2.5 mM
Mg2+, 0.2 mM dNTP, 0.2 µM each primer, 1 unit
HotStarTaq polymerase and 1 µl template DNA. The thermal
cycling conditions for COMP 10F/R were as follows: 95°C for 15 min;
11 cycles of 94°C for 15 sec, 62–0.5°C per cycle for 40 sec, 72°C
for 1 min; 24 cycles of 94°C for 15 sec, 56°C for 30 sec, 72°C for
1 min; and 72°C for 2 min. The thermal cycling conditions for COMP
1F/R, 15F/R and 16F/R were as follows: 95°C for 15 min; 11 cycles
of 94°C for 15 sec, 62–0.5°C per cycle for 40 sec, 72°C for 1 min;
24 cycles of 94°C for 15 sec, 56°C for 30 sec, 72°C for 1 min; and
72°C for 2 min. The thermal cycling conditions for the remaining
fragments were as follows: 95°C for 15 min; 35 cycles of 96°C for
10 sec and 68°C for 4 min. Direct sequencing was performed using
the BigDye Terminator Cycle Sequencing Ready Reaction kit, version
3.1 (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), and the sequencing was analyzed with an ABI Prism 3130
automated sequencer (Applier Biosystems; Thermo Fisher Scientific,
Inc.). To assess the damaging effects of missense mutations in
silico, the online database, Protein Variation Effect Analyzer
(PROVEAN; http://provean.jcvi.org/index.php) was used (13).
 | Table IPolymerase chain reaction primer
sequences of the COMP gene. |
Table I
Polymerase chain reaction primer
sequences of the COMP gene.
| Gene | Forward primer | Reverse primer |
|---|
| COMP-1 |
5′-GACGCTGGTGACTCTGTTTCC-3′ |
5′-GGGCCTATTTATCCCCAAGG-3′ |
| COMP-2 |
5′-GCAAGAGGATGCGATACAGCCACACC-3′ |
5′-AGCCTGGATCCCGGGCCTCTC-3′ |
| COMP-3 |
5′-GGAGGTCCGGGTTCGCTGCAA-3′ |
5′-CCATTCCCGTTCGTCCGTCCA-3′ |
| COMP-4 |
5′-ACGGGGCAGAAAAGCGGGACA-3′ |
5′-CGGGGATGGTCCTTGGGTTGG-3′ |
| COMP-5 |
5′-TGCCCGCCCTCATCCTTCCTC-3′ |
5′-TCACCACCCCACGCAGACACC-3′ |
| COMP-6 |
5′-CCCTCCTCCCCCAGTGCAACG-3′ |
5′-ACGCCTGGTCGCCCACGAAG-3′ |
| COMP-7 |
5′-GGTAAGGCCCGCTGGGGAGGA-3′ |
5′-GGGCGGGCACAGAAGGTGTGA-3′ |
| COMP-8 |
5′-GGAGCGCCAGTGCCGTAAGGTG-3′ |
5′-TCCCTAGGCCCCGCTCACAGC-3′ |
| COMP-9 |
5′-GATGGGGCGTGGCTTGGATGA-3′ |
5′-CTGGCGCCCCACCATGGTCTT-3′ |
| COMP-10 |
5′-TGAAGTTGGGACTCTGTTCCAG-3′ |
5′-TGGATAGGTGGGATCCAGAGA-3′ |
| COMP-11 |
5′-AGCCTGCGGTGGGGGTTTCCT-3′ |
5′-CCACGCCGTCCCCTGAGAGGT-3′ |
| COMP-12 |
5′-CCACGATGGCCAGGGTGATGC-3′ |
5′-CCCGCTCCGTGGCAGGATAGC-3′ |
| COMP-13 |
5′-GGGCGGGCCCTGACTTTAGCC-3′ |
5′-CCCTCTGTCCCCGCCCTTCCT-3′ |
| COMP-14 |
5′-GGAGGGGCGTGGCCAATGGT-3′ |
5′-GCCCAGAGGAGGGCTGGGACA-3′ |
| COMP-15 | 5′
CTGTGACCTATCCCCAAGCTG 3′ |
5′-GGCTCATTCTAACTGCCCTGT-3′ |
| COMP-16 |
5′-CCAGTCAGGGGTACCCATCTT-3′ |
5′-AGCACTGAGGCCTTGGTTTG-3′ |
Results
Patients
The present study included six Chinese families with
PSACH. Detailed clinical information of these families is described
in the following sections, and the clinical characteristics are
summarized in Table II.
| Table IISummary of clinical manifestations of
six Chinese patients with pseudoachondroplasia. |
Table II
Summary of clinical manifestations of
six Chinese patients with pseudoachondroplasia.
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 |
|---|
| Gender | Male | Male | Female | Male | Female | Male |
| Age (years) | 20 | 5 | 8 | 2 | 4 | 6 |
| Height (cm) | 149 | 81 | 110 | 82 | 85 | 84 |
| Spine changes | Mild scoliosis, | Mild scoliosis,
platyspondyly and beaking of vertebrae | Mild scoliosis,
platyspondyly and beaking of vertebrae | Mild scoliosis,
platyspondyly and beaking of vertebrae | Mild scoliosis,
platyspondyly and beaking of vertebrae | Mild scoliosis,
platyspondyly and beaking of vertebrae |
| Brachydactyly | + | + | + | + | + | + |
| Irregular
epiphyses | − | + | + | + | + | + |
| Irregular
metaphyses | − | + | + | + | + | + |
| Restricted
extension of the joints | − | Shoulders and
elbows | − | − | Elbows | Shoulders and
elbows |
| Deformities of the
lower limbs | − | Genu varum | Genu varum | Genu varum | Genu varum | Genu varum |
| Osteoarthritis | Left knee | − | − | − | − | − |
Family 1
The proband was a 20-year-old male of Han ethnicity.
He was the second of two siblings in a healthy non-consanguineous
couple. The pregnancy and delivery of this individual were
considered normal, and no abnormalities were identified at birth.
The patient began to walk at the age of 16 months, and slowness in
linear growth was noticed at 5 years of age. Simultaneously, a limp
and brachydactyly were noted. Physical examination at the age of 20
years demonstrated disproportional short stature with short limbs:
Height, 149 cm (−3 standard deviations); arm span, 137 cm.
Generalized brachydactyly of the hands was observed. No restricted
extension of the joints was identified, and intelligence was
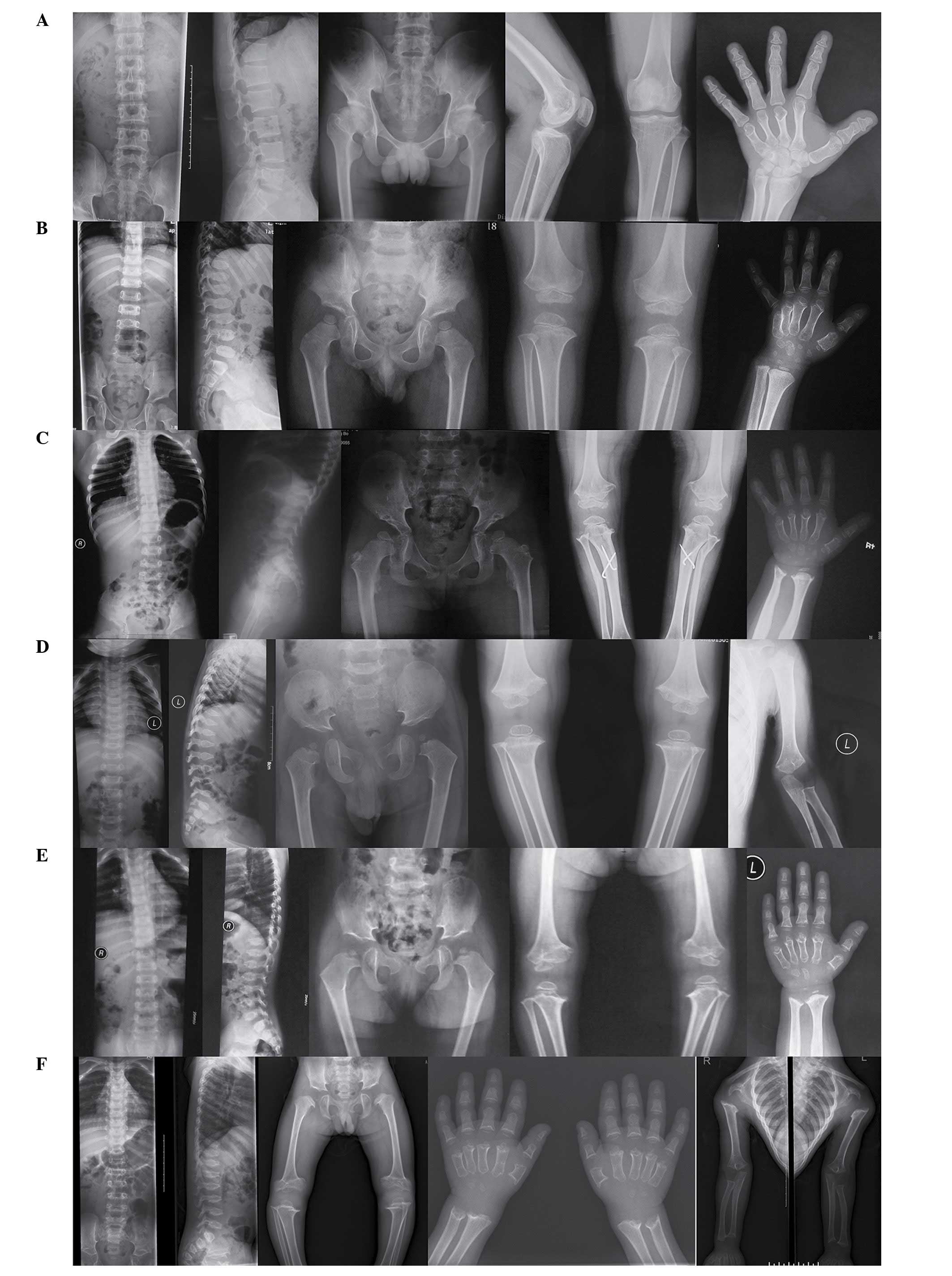
considered normal. The radiographic images presented in Fig. 1A exhibited clinical features of
PSACH disease. The patient exhibited vertebral bodies of a normal
height (anteroposterior and lateral views), flat bilateral femoral
capitals of the pelvis (anteroposterior view), mild osteoarthritis
of the left knee (anteroposterior and lateral views) and a hand
with brachydactyly (posterior-anterior view). His parents, brother
and relatives were healthy with no similar features observed.
Family 2
The proband was a 5-year-old Han boy. He was the
only child of a healthy non-consanguineous couple. The pregnancy
and delivery of this individual were considered normal, however no
birth parameters were available. Genu varum and slowness in linear
growth were noted at the age of 20 months, and the individual began
to walk at the age of 2. Physical examination at age of 5 observed
disproportional short stature with short limbs: Height, 81 cm (−3
standard deviations); arm span, 78 cm; upper segment/lower segment
ratio of 1.57. The hands exhibited generalized brachydactyly and
the shoulders and elbows presented with restricted extension. clear
genu varum was noted and intelligence was considered normal. The
radiographic images presented in Fig.
1B exhibited clinical features of PSACH disease. The patient
presented with mild scoliosis, platyspondyly and anterior beaking
of the vertebral bodies (anteroposterior and lateral views), small
capital femoral epiphyses of the pelvis (anteroposterior view),
irregular epiphyses in the knees (anteroposterior view) and a hand
with brachydactyly (posterior-anterior view). All of his parents
and relatives were healthy with no similar features.
Family 3
The 8-year-old proband was born to
non-consanguineous and healthy parents. She was born at full term
with a birth height of 51 cm. A waddling gait was recognized upon
learning to walk, and during physical examination at 8 years of
age, her height was measured as 110 cm (−3 standard deviations).
Genu varum was clear and moderate generalized brachydactyly of the
hands was observed. The extension of the shoulders and elbows was
restricted and the individual additionally presented with pectus
carinatum. Her intelligence was normal. The radiographic images
presented in Fig. 1C exhibited
clinical features of PSACH disease. The patient had similar
radiographic characteristics to that of patient 2. Laboratory tests
including mucopolysaccharide analysis of urine and thyroid function
tests were normal, and all of her parents and relatives were
healthy with no similar features.
Family 4
The proband was a 2-year-old (35-month-old) Han boy.
He was the first of two siblings in a healthy non-consanguineous
couple. Results of a prenatal fetal ultrasound were normal, as was
the delivery of this child. His birth weight was 3.2 kg and his
birth height was 52 cm. He manifested with a delayed odontogenesis
at 6 months. The anterior fontanelle closed at 17 months. He was
unable to walk until 19 months of age. Physical examination
observed disproportional short stature with short limbs: Height, 82
cm (−3 standard deviations); arm span, 78 cm; upper segment/lower
segment ratio, 1.73. He additionally presented with clear pectus
carinatum and genu varum. His enunciation was neither clear nor
continuous, however he responded well. The radiographic images
presented in Fig. 1D exhibited
clinical features of PSACH disease. The patient had similar
radiographic characteristics to that of patient 2, however
irregular metaphyses of the left elbow (anteroposterior view) was
additionally observed. His parents and relatives were healthy with
no similar features observed.
Family 5
The proband was a 4-year-old Han girl. She was the
second of three siblings in a healthy nonconsanguineous couple. The
pregnancy and delivery of this individual were considered normal.
She was born with a birth weight of 5 kg and a birth height of 50
cm. Developmental progress was normal until 1.5 years of age, when
the patient learnt to walk and presented with mild bowing of the
legs. Slowness in linear growth was noted at 2.5 years, with the
individual reported to be of short stature in comparison with her
peers. Physical examination at the age of 4 years demonstrated
disproportional short stature with short limbs: Height, 85 cm (−3
standard deviations); arm span, 72 cm; upper segment/lower segment
ratio, 1.43. There was moderate generalized brachydactyly of the
hands and restricted extension of the elbows and genu varum were
clear. Her intelligence was normal. The radiographic images
presented in Fig. 1E exhibited
clinical features of PSACH disease. The patient had similar
radiographic characteristics to that of patient 2. Her parents and
relatives were healthy with no similar features.
Family 6
The proband was a 6-year-old Han boy. He was the
only child of a healthy non-consanguineous couple. The pregnancy
and delivery of this individual were considered normal, however no
birth parameters were available Genu varum and slowness in linear
growth were noted at the age of 2. Physical examination
demonstrated disproportional short stature with short limbs:
Height, 84 cm (−3 standard deviations); arm span, 83 cm; upper
segment/lower segment ratio, 1.51. Moderate generalized
brachydactyly of the hand was observed and extension of the
shoulders and elbows was restricted. He additionally presented with
pectus carinatum. His intelligence was normal. The radiographic
images presented in Fig. 1F
exhibited clinical features of PSACH disease. The patient had
similar radiographic characteristics to that of patient 2. His
parents and relatives were healthy with no similar features.
Sequence analysis
Screening was conducted for mutations in COMP in all
patients and their parents using PCR followed by direct sequence
analysis. The sequencing identified that five patients carried
heterozygous missense mutations of COMP and one carried a
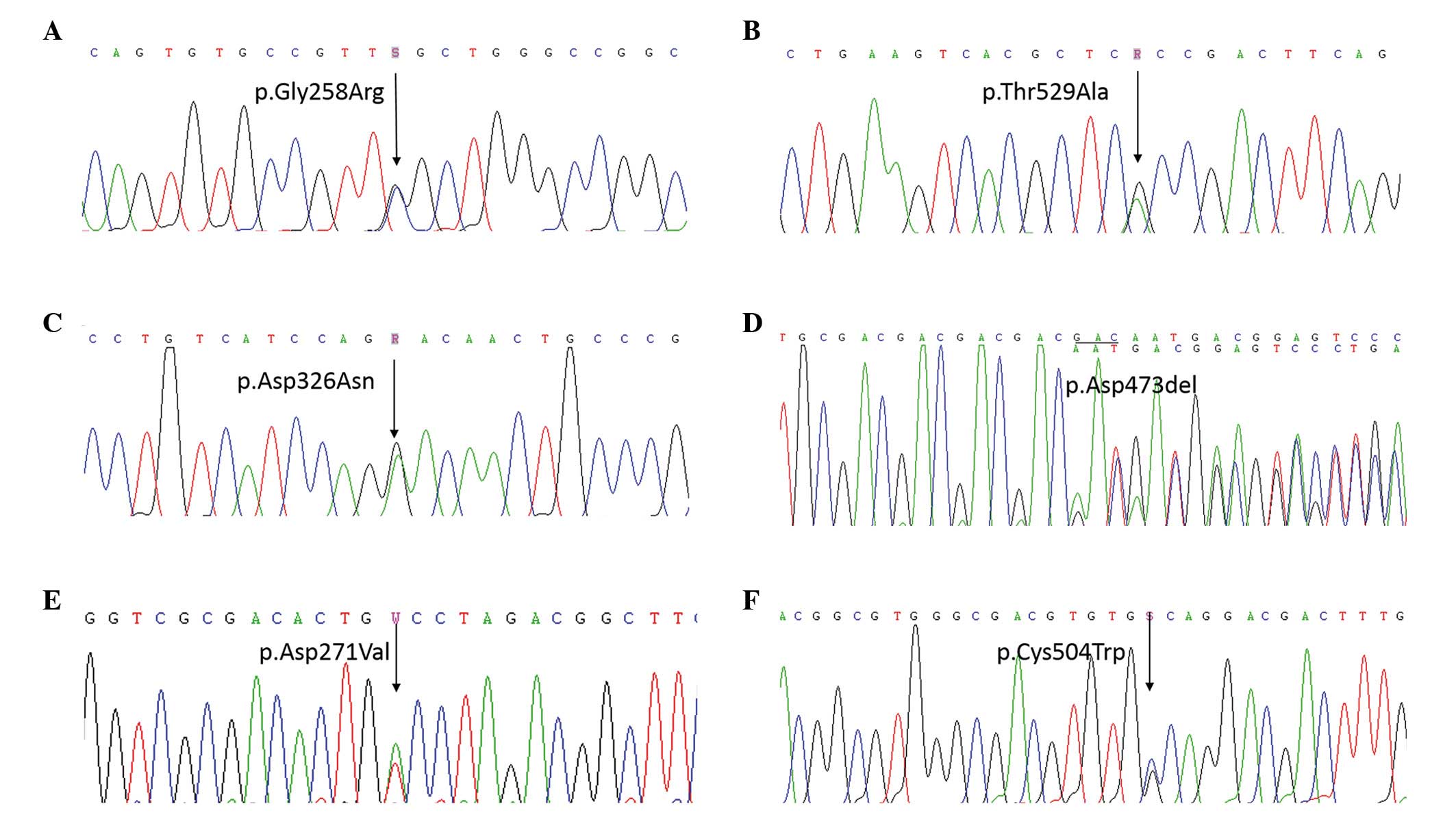
heterozygous in-frame deletion in exon 13 of COMP: c.772G>C
(p.Gly258Arg) in exon 8 of patient 1 (Fig. 2A), c.1585A>G (p.Thr529Ala) in
exon 14 of patient 2 (Fig. 2B),
c.976G>A (p.Asp326Asn) in exon 10 of patient 3 (Fig. 2C), c.1417_1419delGAC (p.Asp473del)
in exon 13 of patient 4 (Fig. 2D),
c.812A>T (p.Asp271Val) in exon 8 of patient 5 (Fig. 2E), and c.1512C>G (p.Cys504Trp)
in exon 14 of patient 6 (Fig. 2F).
Based on DNA sequence analysis, all the mutations were de
novo, and were not identified in DNA samples from healthy
parents and 250 healthy volunteers. Among these mutations,
c.976G>A (p.Asp326Asn) and c.1585A>G (p.Thr529Ala) are, to
the best of our knowledge, novel. p.Asp326Asn and p.Thr529Ala were
non-conservative, evolutionarily highly conserved amino acids from
fish to mammals (Fig. 3A and B),
and were predicted in silico by PROVEAN to be of pathogenic
relevance (p.Asp326Asn, PROVEAN score of −4.542; p.Thr529Ala,
PROVEAN score of −4.610).
Discussion
Although PSACH is an unambiguous skeletal dysplasia
easily diagnosed by physicians, two major entities should be
considered in the differential diagnosis. Firstly, the body
proportions of patients with PSACH resemble those of patients with
achondroplasia at the second year of life or later. However,
patients with PSACH appear normal at birth and their faces are
normal, while the characteristics of achondroplasia are recognized
at birth with characteristic frontal bossing (14). In addition, there is gradual
narrowing of the lumbar interpedicular distances in the lower
lumbar spine in achondroplasia, which is absent in PSACH. Secondly,
mucopolysaccharidosis IV, also termed Morquio syndrome, may be
confused with PSACH, due to the fact that both disorders present
with anterior beaking of the vertebral bodies (15). However, the anterior beaking of the
vertebral bodies is seen exclusively during childhood in patients
with PSACH, and resolves with age. In addition, pointed proximal
2nd, 3rd, 4th and 5th metacarpals, which are unique to
mucopolysaccharidosis IV, are not observed in PSACH.
Mucopolysaccharide analysis of urine will confirm diagnosis.
Mutation analysis is, however, a simple way of confirming the
diagnosis of PSACH.
Previous ultrastructural studies on cartilage from
patients with PSACH demonstrated abnormal accumulation of
electron-dense and light material within the chondrocyte rough
endoplasmic reticulum (16,17).
Biochemical and histochemical studies have additionally suggested
that PSACH was a generalized cartilage disorder involving
abnormalities of proteoglycans (18,19).
Cartilage consists of chondrocytes and abundant extracellular
matrix components including glycoproteins, aggrecans and collagens.
COMP is a pentameric glycoprotein of the cartilage extracellular
matrix that is localized predominantly to the territorial matrix
surrounding the chondrocytes. In vitro studies have reported
that chondrocyte-specific intracellular trafficking defects were
observed for the p.Asp469del mutant COMP (20,21).
Furthermore, different COMP mutations in the type 3 repeat domain
have been identified to produce variable effects on intracellular
transport, which correlate with clinical severity (22). Mutations in COMP have additionally
been reported to affect the secretion of type IX collagen and
matrilin 3, however not the secretion of aggrecan and type II
collagen (23). Furthermore, a
recent study using the COMP p.Asp469del mutant mouse model
suggested that chondrocyte stress triggered by the expression of
mutant COMP was central to the pathogenesis of PSACH (24).
The key observation of the present study was the
identification of two novel mutations in COMP: c.976G>A
(p.Asp326Asn) and c.1585A>G (p.Thr529Ala). Notably, previous
studies reported that p.Asp326Tyr resulted in PSACH while
p.Asp326Gly led to MED (25,26).
In the current study, p.Asp326Asn in COMP was observed to result in
a PSACH phenotype in a Chinese girl. This patient manifested with
severe epiphyseal deformities and clear delay in linear growth.
Furthermore, the radiographic images indicated clear platyspondyly
and anterior beaking of the vertebral bodies. These features can
help differentiate between PSACH from MED. It is suggested that
different substitutions at the same residue of p.Asp326 lead to
different phenotypes. It is also possible that genetic modifiers of
phenotypic severity are likely to influence disease severity
(5). This data may be useful for
further analysis of correlations between genotype and phenotype. In
addition, a notable hotspot, c.1417_1419delGAC (p.Asp473del), was
identified. This mutation had been previously reported in 49 cases
of PSACH and was identified as the most common mutation (1).
In summary, the current study reported the molecular
and clinical observations of six Chinese patients with PSACH. Two
novel mutations in the COMP gene were identified. The present study
expanded upon the mutation spectrum in the COMP gene, and
contributed to the understanding of the phenotype/genotype of
COMP-associated diseases.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant nos. 81370978 and
81170803 to Professor Zhen-Lin Zhang and grant no. 81200646), the
National Basic Research Program of China (grant no. 2014CB942903),
Shanghai Leading Talents Award (grant no. 051 to Professor Zhen-Lin
Zhang), the Science and Technology Commission of Shanghai
Municipality (grant no. 14JC140500 to Professor Zhen-Lin Zhang),
Shanghai Municipal Commission of Health and Family Planning (grant
no. 2014ZYJB0009), and the Science and Technology Commission of
Chongqing Municipality (grant no. CSTC2013jcyjC00009 to Professor
Zhen-Lin Zhang).
References
|
1
|
Briggs MD and Chapman KL:
Pseudoachondroplasia and multiple epiphyseal dysplasia: Mutation
review, molecular interactions, and genotype to phenotype
correlations. Hum Mutat. 19:465–478. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Briggs MD, Hoffman SM, King LM, Olsen AS,
Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines
ES, et al: Pseudoachondroplasia and multiple epiphyseal dysplasia
due to mutations in the cartilage oligomeric matrix protein gene.
Nat Genet. 10:330–336. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hecht JT, Nelson LD, Crowder E, Wang Y,
Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere
M, et al: Mutations in exon 17B of cartilage oligomeric matrix
protein (COMP) cause pseudoachondroplasia. Nat Genet. 10:325–329.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Newton G, Weremowicz S, Morton CC,
Copeland NG, Gilbert DJ, Jenkins NA and Lawler J: Characterization
of human and mouse cartilage oligomeric matrix protein. Genomics.
24:435–439. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Briggs MD, Brock J, Ramsden SC and Bell
PA: Genotype to phenotype correlations in cartilage oligomeric
matrix protein associated chondrodysplasias. Eur J Hum Genet.
22:1278–1282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie X, Liao L, Gao J and Luo X: A novel
COMP mutation in a Chinese patient with pseudoachondroplasia. Gene.
522:102–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai L, Xie L, Wang Y, Mao M, Li N, Zhu J,
Kim C and Zhang Y: A novel COMP mutation in a pseudoachondroplasia
family of Chinese origin. BMC Med Genet. 12:722011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao LH, Wang LB, Wang SS, Ma HW, Ji CY and
Luo Y: Identification of novel and recurrent mutations in the
calcium binding type III repeats of cartilage oligomeric matrix
protein in patients with pseudoachondroplasia. Genet Mol Res.
10:955–963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu CT, Guo L, Zahng ZH, Lin WX, Song YZ
and Feng L: Clinical features and COMP gene mutation in a family
with a pseudoachondroplasia child. Zhongguo Dang Dai Er Ke Za Zhi.
15:937–941. 2013.In Chinese. PubMed/NCBI
|
|
10
|
Liu FX, Li ZL, Wei ZJ, Meng Y, Ren CA,
Zhang XD, Yu MX and Huang SZ: Genetic analysis and serum level of
cartilage oligomeric matrix protein in patients with
pseudoachondroplasia. Chin Med J (Engl). 123:2181–2184. 2010.
|
|
11
|
Yue H, Yu JB, He JW, Zhang Z, Fu WZ, Zhang
H, Wang C, Hu WW, Gu JM, Hu YQ, et al: Identification of two novel
mutations in the PHEX gene in Chinese patients with
hypophosphatemic rickets/osteomalacia. PLoS One. 9:e978302014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, He JW, Fu WZ, Zhang CQ and Zhang
ZL: Mutations in the SLCO2A1 gene and primary hypertrophic
osteoarthropathy: A clinical and biochemical characterization. J
Clin Endocrinol Metab. 98:E923–E933. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7:e466882012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hunter AG, Hecht JT and Scott CI Jr:
Standard weight for height curves in achondroplasia. Am J Med
Genet. 62:255–261. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Northover H, Cowie RA and Wraith JE:
Mucopolysaccharidosis type IVA (Morquio syndrome): A clinical
review. J Inherit Metab Dis. 19:357–365. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maynard JA, Cooper RR and Ponseti IV: A
unique rough surfaced endoplasmic reticulum inclusion in
pseudoachondroplasia. Lab Invest. 26:40–44. 1972.PubMed/NCBI
|
|
17
|
Merritt TM, Bick R, Poindexter BJ, Alcorn
JL and Hecht JT: Unique matrix structure in the rough endoplasmic
reticulum cisternae of pseudoachondroplasia chondrocytes. Am J
Pathol. 170:293–300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pedrini Mille A, Maynard JA and Pedrini
VA: Pseudoachondroplasia: Biochemical and histochemical studies of
cartilage. J Bone Joint Surg Am. 66:1408–1414. 1984.PubMed/NCBI
|
|
19
|
Stanescu V, Maroteaux P and Stanescu R:
The biochemical defect of pseudoachondroplasia. Eur J Pediatr.
138:221–255. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dinser R, Zaucke F, Kreppel F, Hultenby K,
Kochanek S, Paulsson M and Maurer P: Pseudoachondroplasia is caused
through both intra- and extracellular pathogenic pathways. J Clin
Invest. 110:505–513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen TL, Stevens JW, Cole WG, Hecht JT and
Vertel BM: Cell-type specific trafficking of expressed mutant COMP
in a cell culture model for PSACH. Matrix Biol. 23:433–444. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen TL, Posey KL, Hecht JT and Vertel BM:
COMP mutations: Domain-dependent relationship between abnormal
chondrocyte trafficking and clinical PSACH and MED phenotypes. J
Cell Biochem. 103:778–787. 2008. View Article : Google Scholar
|
|
23
|
Hecht JT, Hayes E, Haynes R and Cole WG:
COMP mutations, chondrocyte function and cartilage matrix. Matrix
Biol. 23:525–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suleman F, Gualeni B, Gregson HJ, Leighton
MP, Piróg KA, Edwards S, Holden P, Boot-Handford RP and Briggs MD:
A novel form of chondrocyte stress is triggered by a COMP mutation
causing pseudoachondroplasia. Hum Mutat. 33:218–231. 2012.
View Article : Google Scholar :
|
|
25
|
Jackson GC, Mittaz-Crettol L, Taylor JA,
Mortier GR, Spranger J, Zabel B, Le Merrer M, Cormier-Daire V, Hall
CM, Offiah A, et al: Pseudoachondroplasia and multiple epiphyseal
dysplasia: A 7-year comprehensive analysis of the known disease
genes identify novel and recurrent mutations and provides an
accurate assessment of their relative contribution. Hum Mutat.
33:144–157. 2012. View Article : Google Scholar :
|
|
26
|
Zankl A, Jackson GC, Crettol LM, Taylor J,
Elles R, Mortier GR, Spranger J, Zabel B, Unger S, Merrer ML, et
al: Preselection of cases through expert clinical and radiological
review significantly increases mutation detection rate in multiple
epiphyseal dysplasia. Eur J Hum Genet. 15:150–154. 2007. View Article : Google Scholar
|