Introduction
Dengue is an important mosquito borne diseases in
tropical and sub-tropical regions. In recent decades, the infection
has spread significantly, reaching greater than 50 million cases
per year (1). Dengue disease is
caused by the dengue virus (DENV), which belongs to the
flaviviridae family, and is an enveloped virus formed by a single
stranded RNA genome and includes 4 serotypically different viruses
(DENV-1 to 4) (2). Infection with
any of the four serotypes results in similar pathological
manifestations, which range from sub-clinical discomfort in
approximately 30% of cases, dengue fever in 70% of cases to dengue
hemorrhagic fever and significant mortality in 1% of cases
(3,4). The pathogenesis of DENV infection
remains to be fully understood.
There is no specific therapy to treat the disease,
and clinical management is limited to supportive care. Certain
reports have indicated that cholesterol and lipid synthesis
pathways are associated with DENV infection, and in addition, DENV
susceptibility to cholesterol depleting treatments has been
reported (5). Furthermore, in
vitro studies have demonstrated the importance of the
cholesterol pathway in viral fusion, replication and assembly.
However, conflicting reports have indicated that the addition of
cholesterol upon viral binding and attachment blocks the entry of
the flavivirus into the cell and impairs viral replication
(6,7). It remains unclear how viruses use
cellular machinery and molecules including cholesterol, to support
viral production. It has been reported that genetic inhibition of
cholesterol biosynthesis (siRNA against mevalonate decarboxylase)
reduced DENV replication in an in vitro replicon (8). Another previous study demonstrated
that cholesterol present in the viral envelope is necessary for
successful infection, however, cell membrane cholesterol depletion
had no effect on DENV infectivity towards several animal and human
cell lines (9). These apparently
conflicting results suggest that DENV binding and attachment is
independent of cell membrane cholesterol but dependent on viral
particle cholesterol levels.
Alternatively, genetic and pharmacological
inhibition of cholesterol synthesis has been indicated to impair
the late stages of the DENV cycle, including viral replication,
particle assembly and viral budding (10,11).
However, the mechanisms of DENV interaction with cholesterol rich
compartments in the cell remain to be fully understood. The
inhibitory properties of cholesterol modifying agents towards DENV
appear to be orchestrated by additional mechanisms involving the
modulation of host genes. Statins are a group of drugs that were
initially developed to reduce lipid levels. However, previous
reports have raised considerable interest in the additional
properties of statins, including anti-inflammatory effects at the
endothelium and antiviral effects against dengue and hepatitis C
viruses (12). It is possible that
such antiviral effects may additionally be mediated by the
regulation of cellular metabolic pathways, such as the antiviral
profile of the cell. Previous studies have connected pathogens,
cholesterol metabolism and cell antiviral profile (13–15).
Therefore, the aim of the present study was to elucidate the
mechanisms involved in the negative regulation of DENV replication
induced by agents that reduce intracellular cholesterol levels.
Materials and methods
Cell culture and viral strains
The Huh7 hepatocyte derived carcinoma cells were
used for the inhibition experiments. Cells were grown in advanced
Dulbecco's modified Eagle medium (ADMEM; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 2 mM glutamine, 5% fetal
bovine serum (FBS), penicillin (5×104 U/ml)-streptomycin
(50 µg/ml), 1 ml/l amphotericin B, non-essential amino
acids, at 37°C in a 5% CO2 atmosphere. Additionally,
baby hamster kidney [BHK-21; American Type Culture Collection
(ATCC); Manassas, VA, USA] cells were used for the DENV titer
assessment. BHK-21 cells were cultured on minimum essential medium
(MEM; Sigma-Aldrich, St. Louis, MO, USA), 5% FBS, penicillin
(5×104 U/ml)-streptomycin (50 µg/ml), 1 ml/l
anphotericin B and 2 mM glutamine, non-essential amino-acids, at
37°C in a 5% CO2 atmosphere. C6/36 HT cells (from
Aedes albopictus and adapted to grow at 34°C), were grown at
34°C in MEM (Thermo Fisher Scientific, Inc., Waltham, MA, UAS),
supplemented with 7% FBS, non-essential amino acids, MEM vitamin
solution (Sigma-Aldrich), 0.370 g/l sodium bicarbonate, 50 U/ml of
penicillin and 50 µg/ml of streptomycin. The dengue-2 New
Guinea C virus (GenBank M29095) was propagated in C6/36 insect
cells (ATCC CRL-1660™). Briefly, C6/36 HT cells grown in a 175
mm2 flask to ~85% confluence at 34°C, were infected at a
multiplicity of infection (MOI) of 1 for 2 h at 34°C. Subsequently,
fresh medium was added and the infection was permitted until
cytopathic effects were observable at 34°C. Cells were then lysed
by 20 strokes using a glass pestle and tube at 4°C, and lysates
were centrifuged at 1,000 × g at 4°C to eliminate cellular debris.
DENV particles were then precipitated in a 1 in 4 dilution of
polyethylene glycol in 2 M NaCl overnight. The solution was
centrifuged at 6,000 rpm for 1 h to separate DENV particles, prior
to re-suspension in GNTE buffer (200 mM glycine, 100 mM NaCl, 1 mM
EDTA, 50 mM Tris-base at pH 7.5) and aliquoted and stored at −70°C
until use. For DENV ultraviolet (UV) light inactivation, viral
preparations with a titer of 1×106 plaque forming units
(PFU)/ml were placed on ice and irradiated with a UV lamp for 1 h.
The irradiated virus preparations were titrated by plaque assay to
confirm viral inactivation.
Dengue virus titration
Dengue virus titers were determined by plaque assays
in confluent monolayers of BHK-21 cells grown at 80,000 cells/well.
BHK-21 cells were inoculated with 10-fold serial dilutions of
supernatants from each experimental condition [lovastatin (LOV),
pravastatin (PRA), atorvastatin (ATO), fluvastatin (FLU) and
simvastatin (SIM) purchased from Sigma Aldrich, and HMGCR RNAi]
when monolayers reached 80–90% confluence. Following 2 h of viral
adsorption, the monolayers were overlaid with 1 ml of MEM
containing 1% carboximethil-cellulose (Sigma Aldrich), 0.5% FBS and
2 mM L glutamine. The cultures were incubated at 37°C for 5 days
and the the PFU counted following staining with 0.5%
naphtol-blue-black (Sigma Aldrich).
Cytotoxicity assay
The cytotoxicity of statins was measured using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Roche Diagnostics, Basel, Switzerland) based on the
reduction of the tetrazolium salt, MTT, in the presence of an
electron-coupling reagent. Cells grown in a 96-well plate at 20,000
cells/well, the tissue culture plate were treated for the indicated
periods and then incubated with the MTT solution for ~4 h.
Following this incubation period, water-insoluble formazan crystals
were formed, they were solubilized using 10% SDS in 0.01 M HCl, the
formazan dye was quantified using a scanning multi-well plate
reader at 585 nm.
Pharmacological inhibition of DENV
infection
For all antiviral profile experiments, cells were
treated for 48 h in the same conditions Huh-7 cells were incubated
and 24 h later were infected with DENV-2 at an MOI of 1. Following
12 h of infection, the cells were treated with LOV (20 µM),
PRA (50 µM), ATO (10 µM), FLU (10 µM) and SIM
(20 µM) (Sigma-Aldrich) at indicated concentrations, and
then incubated for 48 h. For the different treatments, Huh7 cells
were plated, and one day later the media was replaced and the cells
were treated with each statin, with this considered as time zero.
At the end of the incubation, the supernatants were collected to
measure the PFU for each treatment in order to quantify the DENV
particles and the cholesterol levels using Cayman's Cholesterol
Fluorimetric Assay kit (Cayman Chemical, Ann Arbor, MI, USA)
according to the manufacturer's instructions. All determinations
were performed in triplicate.
RNA interference (RNAi) assay to inhibit
3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) expression
A pre-validated heterogeneous mixture of 21–22 base
pair short interfering RNAs (siRNA) that specifically inhibits
HMGCR expression (cat. no. AM51331; Ambion; Thermo Fisher
Scinetific, Inc.), were used according to the manufacturer's
instruction. Huh7 cells (5×105) were seeded on 12-well
plates in ADMEM containing 2% FBS, 1% nonessential amino acids, 100
U/ml penicillin G and 100 mg/ml streptomycin. Following seeding,
the cells were transfected with serum-free DMEM and 100 mM siRNA
directed against HMGCR (siRNA-HMGCR). Cells were at 50–60%
confluence at the time of transfection and 24 h later were
incubated in the presence or absence of DENV-2, for 2 h. siRNAs
were transfected at a final concentration of 100 nM, using siPORT
Lipid Transfection Agent (Ambion; Thermo Fisher Scientific, Inc.).
Silencer negative control siRNA (100 nM; cat. no. AM4611; Ambion;
Thermo Fisher Scientific, Inc.) and untransfected cells containing
siPORT Lipid Agent alone were used as negative controls, Select
Silencer GAPDH Positive Control siRNA (cat no. AM4390849; Ambion;
Thermo Fisher Scientifc, Inc.) was also used. Cells were incubated
and the infection was allowed to progress for 48 h. The statin
concentrations were selected based on previous toxicity assessment.
Statins stock solutions were diluted with dimethyl sufloxide (DMSO)
and adjusted to 0.1% final concentration for all statins. DMSO was
used as control for the statin treatment and UV-inactivated DENV2
for the mock infection. Following each incubation, total RNA was
extracted and reverse transcribed to cDNA using a high-capacity
cDNA archive kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). From 100 ng of cDNA, quantitative polymerase chain reaction
(qPCR) was performed to quantify HMGCR-mRNA levels, using
the following primers: P1: 5′-GACGCAACCTTTATATCCGTTT-3′ and P2:
5′-TTTTGAAAGTGCTTTCTCTGTACC-3′. For each PCR reaction, 10 µl
of SYBR Green PCR-Master mix (Invitrogen; Thermo Fisher Scientific,
Inc.), 1 µl of each primer 10 µM, and 5 µl of
cDNA diluted in RNase-free water were added. Thermal cycling
conditions were as follows: Initial step at 50°C for 2 min, then
95°C for 10 min, followed by 35 cycles of 95°C for 15 sec and 60°C
for 60 sec. Fluorescence was monitored at the annealing step, and
amplification plots were generated. GAPDH expression was used to
normalize the RNA concentration. For GAPDH-RNA quantification, a
GAPDH (20X) assay was used (Applied Biosystems; Thermo Fisher
Scientific, Inc.) according to the manufacturer's specifications.
In parallel, the supernatant was used to measure the PFUs to
quantify the DENV particles. HMGCR-mRNA levels were normalized
based on the ratio of HMGCR/ACT mRNA. Data are expressed as the
relative fold levels of HMGCR-RNA to the cells transfected with
silencer negative control siRNA, which was set at 1.0. The mRNA
levles were calculated using the 2−ΔΔCq method.
Cellular cholesterol determination
Huh-7 cells grown in 6 well plates were infected and
treated as described above. Subsequently, cells were washed 3 times
with phosphate-buffered saline and lysed with 250 µl lysis
buffer RSB-NP40 (1.5 mM MgCl2, 10 mM Tris-HCl pH 7.5, 10 mM NaCl,
1% IGEPAL) at 48 h. Cells were homogenized on ice by 10 strokes in
a glass pestle and tube. Subsequently, the cholesterol levels were
measured using Fluorimetric Cholesterol assay kit (Cayman Chemical
Company, Ann Arbor, MI, USA). Determinations were performed
according to the manufacturer's instructions.
RT2 profiler PCR arrays
RT2 Profiler PCR Arrays for antiviral
pathway assessment (cat. no. PAHS-122Z) were used to analyze RNA
expression (Qiagen GmbH, Hilden, Germany). The RT2
Profiler PCR Arrays in 96-well plates contained primer assays for
84 antiviral pathway genes and 5 housekeeping genes. In addition,
one well contains a genomic DNA control, 3 wells contain
reverse-transcription controls, and 3 wells contain positive PCR
controls. Total RNA from samples was extracted using an RNeasy Plus
Mini kit (Qiagen GmbH) following the manufacturer's instructions.
Briefly, samples were first lysed and homogenized in a highly
denaturing guanidine-isothiocyanate-containing buffer, which
immediately inactivates RNases to ensure isolation of intact RNA.
The lysate was then passed through a gDNA eliminator spin column.
This column, in combination with the optimized high-salt buffer,
allows efficient removal of genomic DNA. Ethanol was added to the
flow-through to provide appropriate binding conditions for RNA, and
the sample was then applied to an RNeasy spin column, where total
RNA binds to the membrane and contaminants were efficiently washed
away. High-quality RNA was then eluted in 30 µl water.
Subsequently, the experimental RNA samples were converted into
first-strand cDNA using the RT2 First Strand kit (Qiagen
GmbH). The cDNA was then mixed with the RT2 SYBR Green
Mastermix (Qiagen GmbH). PCR was performed according to the
manufacturer's protocol and the relative expression was determined
using the 2−∆∆Cq method. An Applied Biosystem 7500
Real-Time Cycler (Thermo Fisher Scientific, Inc) was used, and qPCR
7500 software, version 2.0.1 (Thermo Fisher Scientific, Inc.) and
RT2 Profiler PCR Array Data Analysis software, version
3.5 (Qiagen GmbH) were used for data analysis.
Statistical analysis
All variables were tested in triplicate, and
experiments were repeated a minimum of three times. Values are
expressed as the mean ± standard deviation. One-way analysis of
variance was used to test for differences between groups, followed
by Tukey's honest significant difference test. All data analysis
was performed using SPSS (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Viability of hepatoma Huh7 cells treated
with different statins
First, it was determined whether statins are able to
induce a cytotoxic effect on treated cells. Huh7 cells were exposed
to four different concentrations of each statin, LOV, PRA, ATO, FLU
and SIM (10–1,000 µM) and incubated for 24–72 h, following
which the total cell count and viability determinations were
performed using an MTT assay. Fig.
1A indicates that following 72 h of treatment, no significant
difference in cell viability was observed between the untreated
(100% viability) and treated cell lines at concentrations below 50
µM for each statin (85–95%; P>0.05; Fig. 1A). However, cells treated with
higher concentrations (over 100 µM) showed lower cell
survival following 72 h of exposure with the exception of PRA,
which did not lead to a reduction in cell survival at that
concentration. Based on these results, concentrations lower than 50
µM were selected for each statin treatment in the subsequent
experiments.
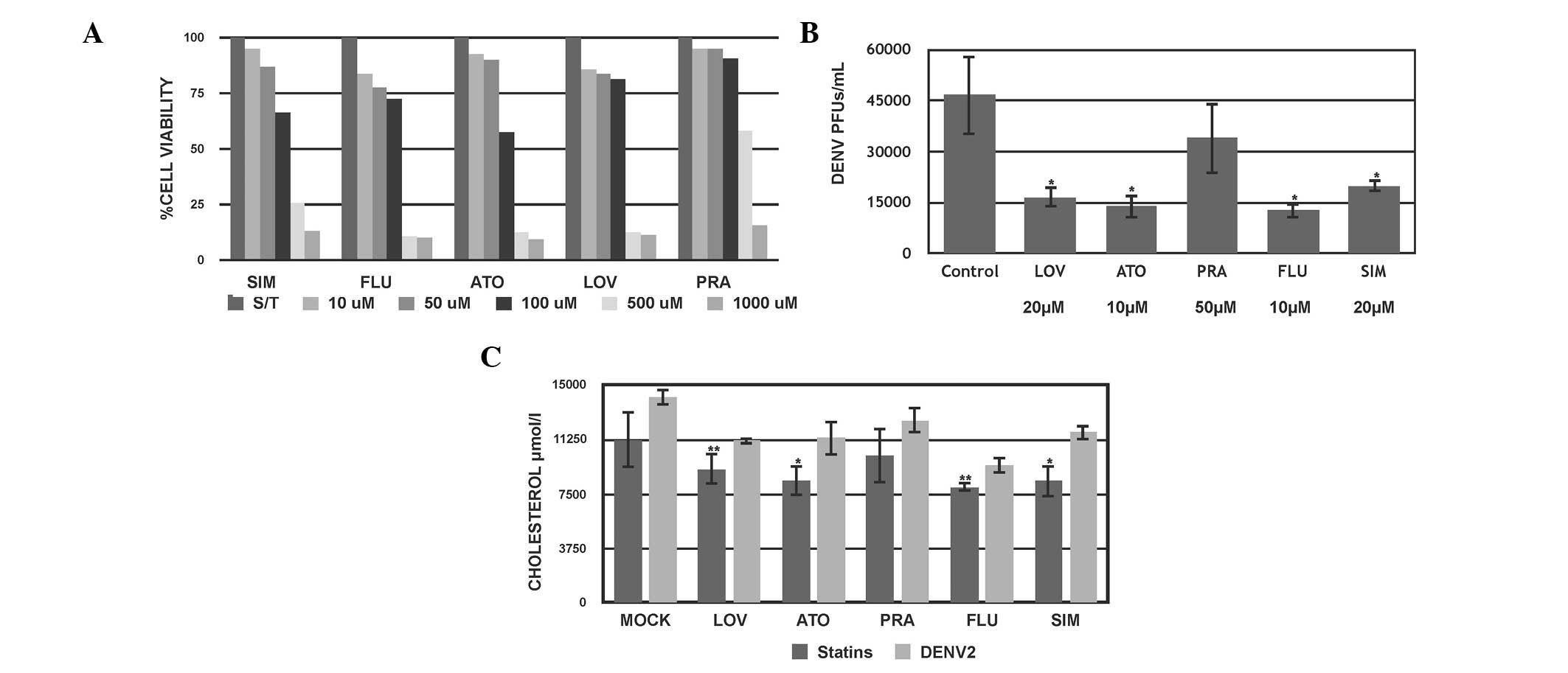 | Figure 1Effect of the pharmacological
inhibition of cholesterol on DENV2 infection in HuH-7 cells. (A)
Cytotoxic assessment of the effect of 72 h treatment with different
concentrations of statins on Huh-7 cells, evaluated by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
(B) Viral titer was evaluated by counting PFUs in the supernatant
from each condition. (C) Intracellular cholesterol levels from cell
extracts. *P<0.05; **P<0.01. DENV2,
dengue virus type 2; PFU, plaque forming units; LOV, lovastatin;
ATO, atorvastatin; PRA, pravastatin; FLU, fluvastatin; SIM,
simvastatin. |
Statins exert differential in vitro
antiviral effects against DENV
The present study sought to evaluate whether the
modulation of the cholesterol synthetic pathway would have an
impact on DENV 2 infection. Statins target the HMGCR enzyme, which
is the rate-controlling enzyme of the cholesterol synthesis
pathway. To evaluate the effect of the five different statins on
the DENV viral cycle, Huh-7 cells were infected with DENV-2 (New
Guinea strain) at MOI 1 for 24 h. Subsequently, the infected cells
were treated for 48 h with 20 µM LOV, 50 µM PRA, 10
µM ATO, 10 µM FLU and 20 µM SIM. Following
treatment, the media supernatant was collected and used to evaluate
the viral titer using a PFU assay, and cells were observed for the
assessment of the cytopathic effect. It was observed that the
majority of the statins reduced the DENV titer at 48 h compared
with the untreated cells (P<0.001; Fig. 1B). In addition, a differential
capacity to reduce the DENV titer was observed between the tested
statins. FLU exerted greater than 75% inhibition on the mature
infectious viruses released into the supernatants, followed by ATO
with 72% and LOV with 68% inhibition compared with the untreated
infected cells. SIM and PRA exhibited a moderate effect on the
viral titer at the selected concentrations (Fig. 1B). These results indicate that
statins exert significant inhibition on DENV2 titers suggesting
that the dengue viral cycle is cholesterol dependent. At their
maximum non-cytotoxic concentrations, the statins had different
antiviral profiles against DENV2 in the cell culture system used
(Fig. 1B). The results for the
cytopathic effect assessment on LOV, ATO and PRA treated cells
supported those of the DENV2 viral titer (data not shown).
DENV2 infection upregulates cholesterol
synthesis
It has been reported that cholesterol is associated
with sub-cellular compartments in which viral replication and
maturation occur, however, previous studies have reported that
certain stages of cellular DENV infection are independent of
cholesterol (16). Therefore, the
present study aimed to investigate whether the observed antiviral
properties of statins correlated with cholesterol levels in
infected cells. A flourometric assay was performed to quantify the
inhibition of cholesterol by statins on infected and uninfected
Huh-7 cells. Uninfected and infected cells with DENV-2 (MOI 1) were
cultured for 24 h and then treated with the statins at the
previously indicated concentrations. Cells were then harvested at
0, 24 and 48 h following treatment and the intracellular
cholesterol levels were measured. A moderate effect of the statins
in reducing cholesterol levels was observed in the treated
uninfected cells (LOV, 30% ATO, 15% PRA, 35% FLU and 25% SIM
compared with the untreated cells) (P<0.05; Fig. 1C) with a greater effect at 48 h
upon treatment. Contrary to expectations, the antiviral effect of
statins may be contributed to both lowering of cholesterol and
unknown cellular mechanisms.
Notably, it was observed that the cholesterol levels
were greater in the DENV-infected cells compared with the
un-infected cells, suggesting that DENV replication is able to
modulate and increase cholesterol production (P<0.01; Fig. 1C). Also, it was observed that in
DENV-infected cells treated with the different statins the
cholesterol levels were increased, compared with the untreated
infected cells.
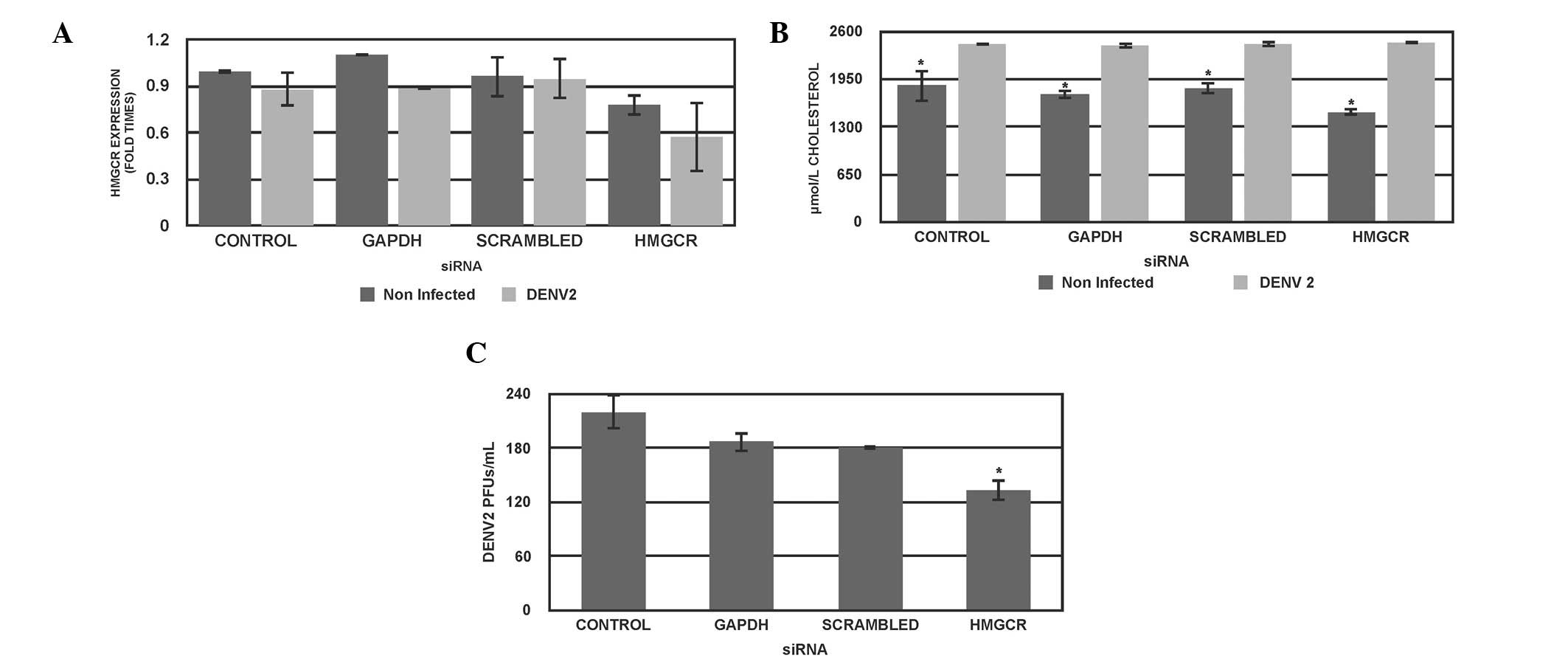
HMGCR-specific RNAi reduces DENV
titer
To further investigate whether the inhibition of
cholesterol is associated with a reduction of the DENV titer, the
effects of HMGCR-specific siRNA were examined in Huh7 cells in the
presence or absence of DENV-2 infection. HMGCR-specific siRNA was
transfected into Huh7 cells, incubated for 24 h, then cells were
infected with DENV-2 for 1 h and incubated for 48 h further.
Subsequently, the cells and supernatant were collected and
HMGCR-mRNA levels, cholesterol and DENV titer were quantified. This
indicated that the introduction of the HMGCR specific siRNA reduced
the expression of HMGCR-mRNA to ~45% in Huh7-infected cells
(P<0.05; Fig. 2A). In addition,
the cholesterol levels were reduced in the uninfected cells at 48 h
post-infection in all conditions (P<0.05; Fig. 2B). Regarding the effect of HMGCR
siRNA on the DENV titer, genetic inhibition of HMGCR reduced the
viral titer by 45% compared with the control cells (P<0.05;
Fig. 2C).
A control siRNA directed against the GAPDH gene was
observed to reduce GAPDH-mRNA (data not shown) and did not have any
significant effects on the expression levels of HMGCR-mRNA
(Fig. 2A), DENV titer (Fig. 2B) cholesterol levels (Fig. 2C) in uninfected and infected cells.
A scrambled siRNA sequence was used as a control of specificity
(Fig. 2A). It is of note that
partial knockdown of HMGCR expression was able to significantly
reduce cholesterol levels only in uninfected cells; however, in
DENV-infected cells reduction in cholesterol levels was not
significant. As aforementioned, DENV may be inducing unknown
mechanisms, which lead to the increase of cholesterol levels.
Together these results suggest that partial genetic inactivation of
HMGCR reduced DENV titer; however did not necessarily modify
cholesterol in DENV-infected cells.
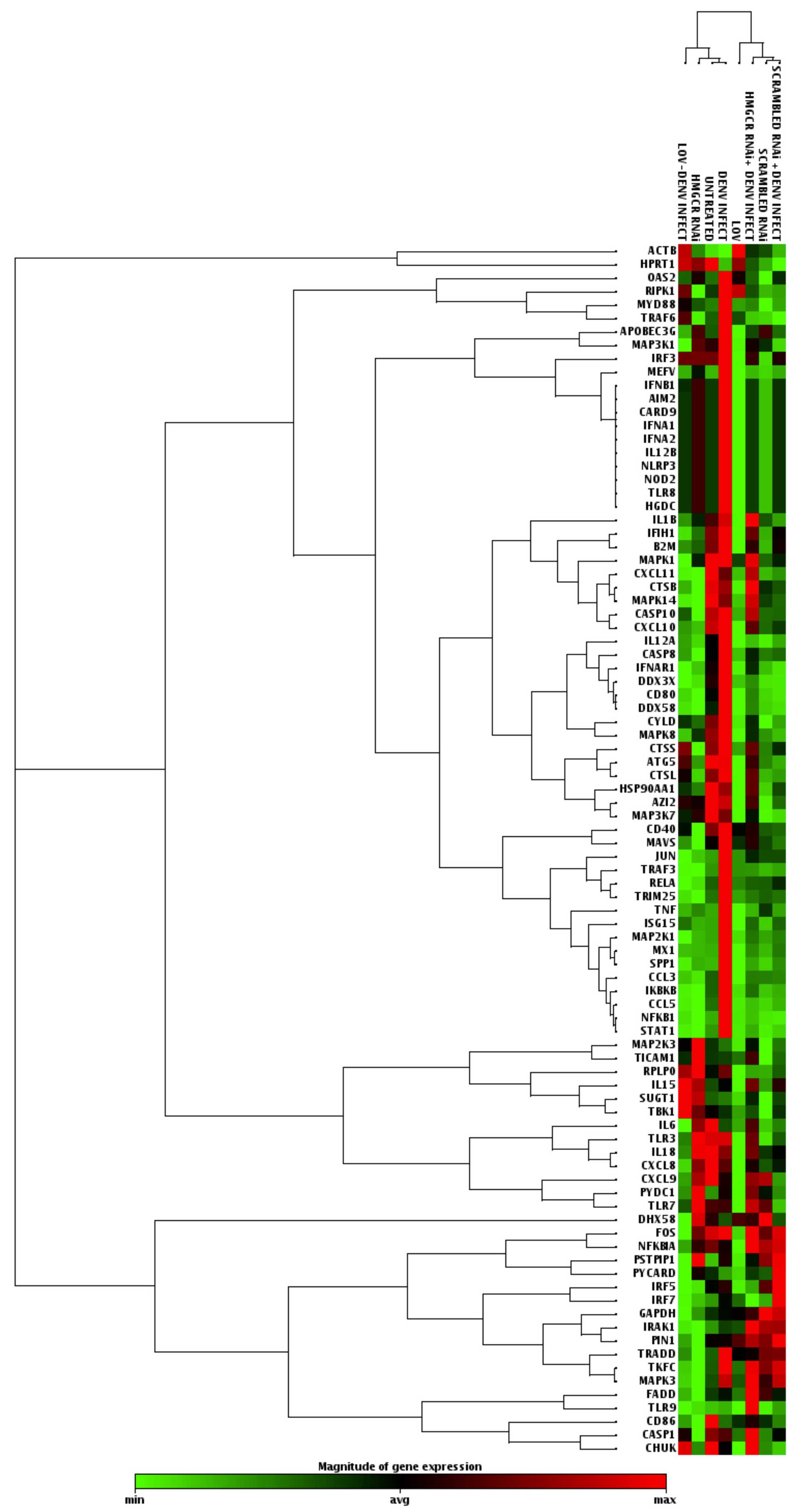
LOV and HMGCR-specific siRNA treatment
modify the antiviral cell response in DENV2-infected cells
Although these results suggest that negative
regulation of DENV replication mediated by statins and HMGCR
genetic blockage, cholesterol levels were not affected in infected
cells; therefore, it is likely that other mechanisms are also
involved in this phenomenon. To further investigate these
mechanisms, the cellular antiviral response profile of
statin-treated and DENV-infected cells was assessed. Huh-7 cells
were infected with DENV2 at MOI 1, then treated with 20 µM
LOV for 24 h, washed and the infection left to progress for 48 h.
Un-infected untreated Huh-7 cells were used as a control.
Total RNA was extracted and reverse transcribed to
cDNA for PCR-array analysis using a Qiagen PCR-Array platform. A
3-fold alteration in gene expression was considered significant.
The results indicate that following infection with DENV2, a number
of genes associated with the cellular antiviral response were
upregulated on infection compared with the uninfected Huh-7 cells.
Table I presents all the altered
genes in each condition compared with the uninfected untreated
cells and Fig. 3 shows a heat map
representation for the genes modulated in each condition. For
example, CCL3, CCL5 and ISG15 (an interferon stimulated gene) were
upregulated in DENV2-infected cells. In addition, NFKB1, STAT1,
TNF, TRADD and TRAF3, well known as pro-inflammatory genes, were
upregulated in the infected cells.
 | Table IGenes differentially expressed
following HMGCR knockdown or LOV treatment in DENV-infected
cells. |
Table I
Genes differentially expressed
following HMGCR knockdown or LOV treatment in DENV-infected
cells.
A, HMGCR knockdown
|
|---|
| Symbol | Fold change
|
|---|
| DENV2+ | HMGCR+ | HMGCR+/DENV2+ | Nonsense+ | Nonsense+/DENV2+ |
|---|
| APOBEC3G | 2.5913 | 1.6792 | 1.2715 | 2.1757 | 1.0884 |
| CASP1 | 0.8947 |
0.1482 | 1.4944 | 0.5314 | 0.8266 |
| CASP10 | 1.1208 |
0.2921 | 1.1763 | 0.6859 | 0.6429 |
| CASP8 | 1.8980 |
0.1283 | 1.0563 | 0.7463 | 0.7720 |
| CCL3 | 2.8558 |
0.2629 | 0.9965 | 1.0633 | 0.9265 |
| CCL5 | 3.4334 |
0.0615 | 0.5424 |
0.3904 | 0.6373 |
| CXCL10 | 1.1153 |
0.2406 | 0.9339 | 0.5363 | 0.6072 |
| CXCL11 | 0.7471 |
0.1264 | 1.036 |
0.3158 |
0.3702 |
| CXCL9 | 0.6312 | 0.8372 | 0.9695 | 1.1206 |
0.3793 |
| DAK | 2.5950 |
0.2479 | 2.9312 | 2.5551 | 2.8154 |
| IL6 |
0.4649 | 0.7981 | 0.8367 |
0.3276 |
0.4456 |
| IRAK1 | 1.6561 |
0.2766 | 4.5517 | 4.1872 | 3.7730 |
| IRF7 | 1.6136 | 0.7975 | 0.7024 | 1.1784 | 3.1306 |
| ISG15 | 2.7561 | 0.9373 | 1.4947 | 1.0859 | 1.5254 |
| JUN | 3.7323 | 0.7412 | 2.1547 | 2.0135 | 1.8654 |
| MAP2K1 | 3.4680 | 0.8739 | 1.5281 | 1.3052 | 1.4556 |
| MX1 | 2.6361 | 0.9405 | 1.3409 | 1.2384 | 1.4264 |
| MYD88 | 2.5260 | 1.1246 | 1.1519 | 0.6599 | 1.0037 |
| NFKB1 | 3.0587 | 0.6116 | 1.0658 | 0.9385 | 0.8300 |
| NLRP3 | 2.0192 | 1.3297 | 1.1711 | 0.7204 | 1.2132 |
| NOD2 | 2.0192 | 1.3297 | 1.1711 | 0.7204 | 1.2132 |
| OAS2 | 2.0192 | 1.3297 | 1.2084 | 0.7808 | 1.4042 |
| SPP1 | 6.0050 | 1.0592 | 1.4141 | 0.8863 | 1.7468 |
| STAT1 | 2.4066 | 0.6361 | 1.0925 | 0.8951 | 0.9486 |
| TLR8 | 2.0192 | 1.3297 | 1.1711 | 0.7204 | 1.2132 |
| TLR9 | 1.1410 | 0.7514 | 5.3502 | 0.8110 | 1.4939 |
| TNF | 4.2555 | 1.2103 | 1.0659 | 2.4258 | 1.2316 |
| TRADD | 2.4880 |
0.3227 | 1.715 | 2.4017 | 2.2466 |
| TRAF3 | 2.3467 | 0.6484 | 1.1828 | 1.1444 | 1.1295 |
| TRIM25 | 2.4894 |
0.435 | 1.153 | 1.3754 | 1.1828 |
B, Lovastatin
treatment
|
|---|
| Symbol | Fold change
|
|---|
| DENV2+ | LOV+ | LOV+/DENV2+ |
|---|
| APOBEC3G | 2.5913 |
0.3222 | 0.5959 |
| CASP8 | 1.8980 |
0.4229 |
0.4556 |
| CCL3 | 2.8558 |
0.2094 |
0.4366 |
| CCL5 | 3.4334 |
0.2222 |
0.1507 |
| CXCL10 | 1.1153 |
0.1709 |
0.3245 |
| CXCL11 | 0.7471 |
0.2969 |
0.1469 |
| CXCL9 | 0.6312 |
0.2115 |
0.3182 |
| ISG15 | 2.7561 | 0.8487 | 1.2345 |
| JUN | 3.7323 | 1.4106 |
0.4214 |
| MAP2K1 | 3.4680 | 0.8560 | 0.5121 |
| MX1 | 2.6361 | 0.9190 | 0.9391 |
| MYD88 | 2.526 | 1.1338 | 1.5763 |
| NFKB1 | 3.0587 | 0.9303 | 0.7462 |
| NLRP3 | 2.0192 |
0.4765 | 1.0178 |
| NOD2 | 2.0192 |
0.4765 | 1.0178 |
| OAS2 | 2.0192 | 1.7578 | 1.0373 |
| SPP1 | 6.0050 |
0.2858 |
0.3619 |
| STAT1 | 2.4066 | 0.9093 | 0.7034 |
| TLR8 | 2.0192 |
0.4765 | 1.0178 |
| TNF | 4.2555 |
0.4747 | 0.9264 |
| TRADD | 2.4880 | 1.8014 | 0.8012 |
| TRAF3 | 2.3467 | 1.3013 | 0.6837 |
| TRIM25 | 2.4894 | 1.0330 | 0.5596 |
| ACTB | 0.9643 | 2.4916 | 1.8144 |
Regarding the effect of LOV on uninfected Huh-7
cells, notable results were observed, with several genes up- and
downregulated in the antiviral pathway. In contrast with the
previously described results in infected cells without treatment,
in LOV treated infected-cells, CCL5 and CCL3 were downregulated
following statin treatment, while OAS2, a gene that encodes a
protein involved in the innate immune response to viral infection,
was upregulated greater than 3-fold.
Additionally, the effect of LOV treatment on Huh-7
cells infected with DENV2 was investigated. Notably, it was
observed that in LOV treated cells, the majority of genes involved
in the cellular antiviral response remained unaffected or were
downregulated compared with the untreated DENV2-infected cells.
Among the few upregulated genes was IL-15, which is involved in
cell proliferation and the induction of natural killer cells.
However, CCL5 and the interferon induced CXCL11 gene, which are
involved in cellular pro-inflammatory responses and the chemotaxis
of activated T cells, respectively, were downregulated.
Furthermore, using the same approach the effect of
HMGCR-RNAi on the cellular antiviral profile of Huh-7
DENV2-infected cells was assessed. A similar effect was observed as
in LOV treated cells, in particular, TLR9, known as a
double-stranded RNA sensor, was upregulated greater than 4-fold
compared with untreated DENV2-infected cells. Additionally, it was
observed that the SPP1, TNF-α and CCL5 genes were downregulated,
similar to LOV-treated cells.
Discussion
A systematic review of a number of studies
evaluating the role of cholesterol and lipids in DENV infections
raised the idea that the effect of cholesterol modulation upon DENV
infection may be influenced by the nature of the inhibitory drug,
the period of treatment, the cell line and the serotype or viral
strain. Despite these variations, some assumptions can be
summarized. In the present study, the effect of five different
statins were investigated in order to observe their particular
antiviral profiles. It was observed that four of the five statins,
LOV, ATO, FLU and SIM, possess antiviral activity against DENV2.
PRA was the least effective statin against DENV, which may be
attributed to its poor liposolubility, which delays its entrance
into cells.
To further clarify the DENV infection mechanism,
cholesterol synthesis was genetically inhibited using HMGCR-siRNA
in infected cells. This showed that enzyme blockage slightly
reduced the viral titer, and indicates that DENV entry into the
cells is complex and flexible, involving other host proteins and
mechanisms.
A marked increase in intracellular cholesterol
levels was observed in infected cells compared with the uninfected
cells at 48 h post infection with DENV2, however, further analysis
is required to clarify whether this alteration is due to a
stimulation of cholesterol transport or its biosynthesis. These
data support previous studies in which stimulation with HMGCR and
cholesterol transport has been reported in DENV-infected cells
(17,18). For DENV, it has been reported that
internal membrane rearrangement such as endoplasmic reticulum
expansion and the formation of replication micro-domains, occurs in
the early stages of the infection through the modulation of lipid
and cholesterol transport, by the expression of viral genes
(18). DENV protein expression
specifically increased lipid raft formation and total cholesterol
levels at the early stages of infection by upregulating low-density
lipoprotein particle uptake and promoting HMGCR activity (19).
In addition, the present study evaluated the
modifications of the antiviral profile of Huh-7 cells as an
alternative mechanism by which statins exert their antiviral
activity. As expected, the cellular antiviral profile was altered
by DENV2 infection. When the Huh-7 infected were compared with the
uninfected cells, a number of genes were observed to be altered.
The CCL3 and CCL5 genes were observed to be upregulated in the
infected cells. In a previous study that evaluated DENV infection
in mice, increased levels of these two chemokines were detected in
the spleen and liver, and were indicative of severe dengue.
Furthermore, in the same study, downregulation of the IL-6 gene was
associated with a milder presentation of the disease (20). However in the present study, IL-6
was reduced in infected cells compared with the uninfected cells.
In addition, ISG15, an interferon stimulated gene was observed to
be upregulated in DENV2 infected cells, and this gene has been
implicated in specific anti-DENV function via protein ISGylation,
and its expression facilitates cellular antiviral responses to
viral infection (14). In LOV
treated cells, CCL5 and CCL3 were downregulated following treatment
while OAS2, a gene that encodes a protein involved in the innate
immune response to viral infection, was upregulated greater than
3-fold. In addition, TRADD, which encodes an apoptotic inhibitor
protein, was upregulated in treated cells.
The results of the present study suggest that
pharmacological and genetic inhibition of cholesterol may
alleviate, to some extent, the pro-inflammatory state of the Huh7
hepatoma cells. To evaluate if an alternative pathway not
associated with cholesterol was responsible for the antiviral
effects of the statins and RNAi, infected cells were treated with
LOV and HMGCR-siRNA. IL-15, involved in cell proliferation and the
induction of natural killer cells, was upregulated by LOV
treatment, however, this gene has been reported not to be
stimulated by DENV infection, with only LOV treatment resulting in
increased production (21).
However, in uninfected LOV treated cells this gene was not altered
and further studies are required to clarify its involvement in the
DENV2 antiviral cellular response. By contrast, CCL5, SPP1 and TNF
genes were downregulated by both LOV and HMGCR-siRNA treatments.
Additionally, in contrast to what was expected, a number of genes
in the cellular antiviral pathway were downregulated following LOV
and RNAi treatment. This may render cells vulnerable to infection,
and mean that cholesterol inhibition may reduce viral titer and
attenuate antiviral gene expression. Another explanation for the
downregulation of several antiviral genes in cholesterol diminished
cells may be that this molecule participates in the synthesis and
processing of these antiviral proteins, and that the blockade of
cholesterol synthesis impairs their production.
In conclusion, a differential downregulation of DENV
titer was observed to be induced by different statins. DENV2
infection resulted in a sustained increase in the levels of
intracellular cholesterol following infection. However, whilst
treatment with statins and HMGCR-siRNA reduced the viral titer,
they did not do so via an effect on cholesterol levels. An
alternative inhibitory mechanism was evaluated determining the
antiviral profile modifications of the Huh7 cells following
infection with DENV2, and LOV and HMGCR-siRNA treatment. It is not
clear whether LOV and HMGCR-siRNA attenuation of cellular antiviral
gene expression is due to DENV reduction through another
undiscovered mechanism or via the stimulation of additional
antiviral genes not evaluated in the present study. More detailed
assessments are required to clarify the association between DENV
replication and the role of cholesterol availability and cellular
signaling pathways in infected cells.
Abbreviations:
|
DENV
|
dengue virus
|
|
FLU
|
fluvastatin
|
|
ATO
|
atorvastatine
|
|
LOV
|
lovastatin
|
|
PRE
|
pravastatin
|
|
SIM
|
simvastatin
|
|
MOI
|
multiplicity of infection
|
|
RT-PCR
|
reverse transcription polymerase chain
reaction
|
|
PFU
|
plaque forming units
|
Acknowledgments
The current study was funded by the Conacyt (grant
no. BASICA-CB2010-01-155082, SALUD-2012-16933 and
SALUD-2010-C01-86996).
References
|
1
|
Guzman MG, Halstead SB, Artsob H, Buchy P,
Farrar J, Gubler DJ, Hunsperger E, Kroeger A, Margolis HS, Martínez
E, et al: Dengue: A continuing global threat. Nat Rev Microbiol.
8(12 Suppl): S7–S16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henchal EA and Putnak JR: The dengue
viruses. Clin Microbiol Rev. 3:376–396. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Halstead SB, Nimmannitya S and Cohen SN:
Observations related to pathogenesis of dengue haemorrhagic fever.
IV Relation of disease severity to antibody response and virus
recovered. Yale J Biol Med. 42:311–328. 1970.PubMed/NCBI
|
|
4
|
Gibbons RV, Kalanarooj S, Jarman RG,
Nisalak A, Vaughn DW, Endy TP, Mammen MP Jr and Srikiatkhachorn A:
Analysis of repeat hospital admissions for dengue to estimate the
frequency of third or fourth dengue infections resulting in
admissions and dengue hemorrhagic fever, and serotype sequences. Am
J Trop Med Hyg. 77:910–913. 2007.PubMed/NCBI
|
|
5
|
Poh MK, Shui G, Xie X, Shi PY, Wenk MR and
Gu F: U18666A, an intra-cellular cholesterol transport inhibitor,
inhibits dengue virus entry and replication. Antiviral Res.
93:191–198. 2012. View Article : Google Scholar
|
|
6
|
Lee CJ, Lin HR, Liao CL and Lin YL:
Cholesterol effectively blocks entry of flavivirus. J Virol.
82:6470–6480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chazal N and Gerlier D: Virus entry,
assembly, budding and membrane rafts. Microbiol Mol Biol Rev.
67:226–237. 2003. View Article : Google Scholar :
|
|
8
|
Rothwell C, Lebreton A, Young Ng C, Lim
JY, Liu W, Vasudevan S, Labow M, Gu F and Gaither LA: Cholesterol
biosynthesis modulation regulates dengue viral replication.
Virology. 389:8–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carro AC and Damonte EB: Requirement of
cholesterol in the viral envelop for dengue virus infection. Virus
Res. 174:78–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martínez-Gutierrez M, Castellanos JE and
Gallego-Gómez J: Statins reduce dengue virus production via
decreased virion assembly. Intervirology. 54:202–216. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Umashankar M, Sánchez-San Martín C, Liao
M, Reilly B, Guo A, Taylor G and Kielian M: Differential
cholesterol binding by class II fusion proteins determines membrane
fusion properties. J Virol. 82:9245–9253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ali N, Allam H, Bader T, May R,
Basalingappa KM, Berry WL, Chandrakesan P, Qu D, Weygant N, Bronze
MS, et al: Fluvastatin interferes with hepatitis C virus
replication via microtubule bundling an a doublecortin-like
kinase-mediated mechanism. PloS One. 8:e803042013. View Article : Google Scholar
|
|
13
|
Liu SY, Aliyari R, Chikere K, Li G,
Marsden MD, Pernet O, Guo H, Nusbaum R, Zack JA, et al:
Interferon-inducible cholesterol-25-hydroxylase broadly inhibits
viral entry by production of 25-hydroxycholesterol. Immunity.
38:92–105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai J, Pan W and Wang P: ISG15 facilitates
cellular antiviral response to dengue and west nile virus infection
in vitro. Virol J. 8:4682011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Blanc M, Hsieh WY, Robertson KA, Kropp KA,
Forster T, Shui G, Lacaze P, Watterson S, Grifitts SJ, Spann NJ, et
al: The transcription factor STAT-1 couples macrophage synthesis of
25-hydroxycholesterol to the interferon antiviral response.
Immunity. 38:106–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reyes-Del Valle J, Chávez-Salinas S,
Medina F and Del Angel RM: Heat shock protein 90 and heat shock
protein 70 are components of dengue virus receptor complex in human
cells. J Virol. 79:4557–4567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soto-Acosta R, Mosso C, Cervantes-Salazar
M, Puerta-Guardo H, Medina F, Favari L, Ludert JE and del Angel RM:
The increase in cholesterol levels at early stages after dengue
virus infection correlates with an augment in LDL particle uptake
and HMG-CoA reductase activity. Virology. 442:132–147. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peña J and Harris E: Early dengue virus
protein synthesis induces extensive rearrangement of the
endoplasmic reticulum independent of the UPR and SREBP-2 pathway.
PloS One. 7:e382022012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gudleski-O'Regan N, Greco TM, Cristea IM
and Shenk T: Increased expression of LDL receptor-related protein 1
during human cytomegalovirus infection reduces virion cholesterol
and infectivity. Cell Host Microbe. 12:86–96. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guabiraba R, Marques RE, Besnard AG,
Fagundes CT, Souza DG, Ryffel B and Teixeira MM: Role of the
chemokine receptors CCR1, CCR2 and CCR4 in the pathogenesis of
experimental dengue infection in mice. PloS One. 5:e156802010.
View Article : Google Scholar
|
|
21
|
Reis SR, Sampaio AL, Henriques Md, Gandini
M, Azeredo EL and Kubelka CF: An in vitro model for dengue virus
infection that exhibits human monocyte infection, multiple cytokine
production and dexamethasone immunomodulation. Mem Inst Oswaldo
Cruz. 102:983–990. 2007. View Article : Google Scholar
|