Introduction
Cardiac hypertrophy occurs as a pathological process
associated with a number of cardiovascular diseases, including
hypertrophic cardiomyopathy (1),
hypertension (2) and myocardial
infarction (3). Unlike an
athlete's heart, pathological cardiac hypertrophy is the response
to stress or diseases, leading to increases in myocardial protein
synthesis and cell volume, instead of the enhancement of pumping
ability (4). Pathological cardiac
hypertrophy under long-term stress may develop into cardiomegaly,
heart failure, and even sudden cardiac mortality. Signaling
pathways involved in the pathogenesis of cardiac hypertrophy have
been investigated in recent studies. For example, inhibition of the
Raf-1/mitogen-activated protein kinase kinase
(MEK)/extracellular-signal-regulated kinase (ERK) cascade
suppresses protein synthesis and the expression of brain
natriuretic peptide in cardiac hypertrophy (5). Small GTP-binding proteins, protein
kinase C and signal transduction and activation of transcription-3
(STAT3) are also vital regulatory factors of cardiac
hypertrophy-associated genes (6–8).
The latest research has revealed significant roles
of microRNAs (miRNAs) in mediating cardiac hypertrophy. As small
non-coding RNAs, miRNAs regulate gene silencing
post-transcriptionally, suppressing translation and inducing
messenger RNA (mRNA) degradation, by binding to the 3′-untranslated
region (UTR) of mRNAs (9).
Overexpression of miR-208a in the heart induces cardiac hypertrophy
(10), the identical regulatory
pattern also being reported for miR-23a and miR-22 (11,12).
Several miRNAs exert anti-hypertrophic roles, such as miR-133, for
example, which targets the hypertrophic regulatory factors,
RhoA and Cdc42, to inhibit cardiac hypertrophy
(13), and miR-1, whose
attenuation evokes hypertrophy (14). Furthermore, miR-101 suppresses
hypertrophy in the rat heart via inhibition of its target gene,
Rab1a (15). In general,
miRNA-mediated gene expression and the regulation of further
downstream signaling events exert an appreciable influence on the
progression of cardiac hypertrophy, the details of which have yet
to be fully elucidated.
A comprehensive study has revealed multiple miRNAs
that are aberrantly expressed in cardiac hypertrophy, among which
miR-26a exhibits abundant expression in the normal heart (16). In this regard, it was hypothesized
that miR-26a may be an important regulator in cardiac hypertrophy,
and therefore, in the present study, a series of experiments in a
transverse abdominal aortic constriction (TAAC)-induced rat model
and angiotensin II (Ang II)-induced cardiomyocytes (CMs) isolated
from the neonatal rat heart were performed. miR-26a overexpression
and suppression were mediated by its mimic and inhibitor,
respectively. Atrial natriuretic factor (ANF) and β-myosin heavy
chain (β-MHC) were assessed as indicators of cardiac hypertrophy.
Furthermore, the present study aimed to confirm whether
GATA4 is the target of miR-26a via mutating the target sites
in the 3′-UTR of GATA4. By means of these experiments, the
present study aimed to elucidate the role of miR-26, and its
regulatory mechanism, in mediating cardiac hypertrophy, which may
provide leads for therapeutic targets in the treatment of cardiac
hypertrophy.
Materials and methods
Animals
All the animal experiments were performed following
the guidelines of our institute, and were approved by a local
ethics committee. Male Sprague Dawley (SD) rats (SPF grade; Vital
River Laboratories, Beijing, China) of 100–120 g were used for
inducing cardiac hypertrophy using the TAAC method. The rats were
maintained at 24°C with 50% humidity. A 12-h light/dark cycle was
used and they were allowed access to sufficient standard feed and
water. TAAC was performed on ten randomly chosen rat individuals
(the TAAC group) and another ten individuals (the Sham group),
which underwent the identical operation procedures except for TAAC.
Specifically, the rats were anesthetized by intraperitoneally
injecting 4% chloral hydrate solution (60 mg/kg). A vertical
incision of 2–3 cm was made under the center abdominal xiphoid
process to expose the surgical field. A ligation was made along the
abdominal aorta 3 mm above the branch point of the right renal
artery by ligating a 5-gauge needle with 4-0 sutures. Subsequently,
the needle was removed and the incision was closed. Following the
surgery, all the rats were treated with penicillin for 3 days. At 4
weeks post-surgery, the rats were sacrificed to examine the heart
weight (HW) and body weight (BW), and also the expression levels of
ANF, β-MHC and miR-26a.
Cells
SD rats (SPF grade, Vital River Laboratories) aged 2
days old were anesthetized by inhalation of 3% diethyl ether for 1
h and sacrificed for heart sampling. The heart samples were
immediately immersed in cold D-Hanks buffer (Sbjbio, Nanjing,
China), and the ventricular muscle was isolated and minced.
Subsequently, the minced tissues were incubated in trypsin (0.08%;
Sigma-Aldrich China, Inc., Shanghai, China) at 37°C for 5 min.
Following the termination of trypsin digestion, the cell suspension
was incubated in Dulbecco's minimum essential medium (DMEM;
Sigma-Aldrich China, Inc.) supplemented with 10% fetal calf serum
(FCS; Sigma-Aldrich China, Inc.) and cultured in an atmosphere of
5% CO2 at 37°C. The medium was changed every other day.
During the first 72 h of culture, 5-bromo-2′-deoxyuridine (0.1 mM;
Sigma-Aldrich China, Inc.) was added to the medium to inhibit the
growth of myocardial fibroblasts. The percentage of CMs present was
determined using a mouse monoclonal anti-heavy chain cardiac myosin
antibody (1:1,000; Abcam, Cambridge, UK; cat. no. ab207926) to be
>95%. After 24 h, the CMs were divided into two groups, namely
the Ang II-treated group (Ang II group) and the untreated group
(Control group), and the cells were cultured in serum-free medium.
Ang II (1 µM; Sigma-Aldrich China, Inc.) was added to cells
of the Ang II group and the cells were cultured for 48 h, with the
medium being changed every 8 h. Subsequently, the cells were
collected for further analyses.
Cell transfection and dual-luciferase
reporter assay
The target gene of miR-26a was predicted using
TargetScan (www.targetscan.org). The mutant
3′-UTR of GATA4 was synthesized using a QuikChange Multi
Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara,
CA, USA). The wild-type or mutant 3′-UTR of GATA4 was cloned
into the KpnI and BglII restriction sites downstream
of the open reading frame of Renilla luciferase in the
pGL3-promoter luciferase vector (Promega, Madison, WI, USA). CMs
cultured in 24-well plates (1.5×105 per well) were
transfected with miR-26a mimic (50 nM) or inhibitor (Sangon Biotech
Co., Ltd., Shanghai, China), and co-transfected with luciferase
reporter containing the wild-type or mutant 3′-UTR of GATA4
(200 ng) or Renilla luciferase reporter plasmid pRL-RSV (20
ng; Promega) using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
protocol. Overexpression of GATA4 was achieved by transfecting the
T vector (200 ng; Promega) containing the coding sequence of
GATA4. At 48 h post-transfection, the luciferase activity
was detected using the GloMax®-Multi Detection System (Promega)
normalized against Renilla luciferase gene activity.
Immunohistochemistry and
immunocytochemistry
Heart tissue from rats in the Control and TAAC
groups were embedded in paraffin and cut into 4 µm slices. A
total of 1×105 CMs were seeded onto the slide in 6-well
plates, and cultured and treated with Ang II as described above.
The cells were fixed with 4% paraformaldehyde for 15 min, and
transparentized with 0.5% Triton X-100 for 20 min. All the tissue
and cell slides were incubated in 3% H2O2 for
15 min at room temperature. The cells were blocked by incubation in
goat serum (Sigma-Aldrich China, Inc.) for 30 min, and subsequently
the cells were incubated with a mouse monoclonal anti-α-actinin
antibody (1:1,000; Abcam; cat. no. ab108198) at 4°C overnight, and
then with a goat anti-rabbit horseradish peroxidase
(HRP)-conjugated secondary antibody (1:2,000; Abcam; cat. no.
ab7090) for 30 min. Positive signals were developed using
diaminobenzidine (Solarbio, Beijing, China) staining, after which
the cells were counterstained with hematoxylin. The slides were
observed under a microscope (MM200; Nikon, Tokyo, Japan), and the
CM area was calculated using ImageJ version 1.49 (National
Institutes of Health, Bethesda, MD, USA).
Leucine incorporation assay
Leucine incorporation assay was performed to reflect
the protein synthesis in CMs. Following Ang II treatment,
3H-labeled leucine (1 µCi/ml) was added to the
cells for incubation at 37°C for 12 h. Subsequently, the cells were
washed three times with cold phosphate-buffered saline, and lysed
in lysis buffer. The [3H]leucine signal was detected by
liquid scintillation counting using Tri-Carb (PerkinElmer, Inc.,
Waltham, MA, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from CMs and heart tissues
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.).
First-strand complementary DNA (cDNA) was synthesized using a
PrimeScript™ 1st Strand cDNA Synthesis kit (Takara Bio, Dalian,
China). For miRNA extraction and reverse transcription, RNAiso for
Small RNA and a One Step PrimeScript miRNA cDNA Synthesis kit (both
from Takara Bio) were used. RT-qPCR was performed using a
LightCycler® 480 system (Roche, Basel, Switzerland) with
SYBR Green I master (Roche). Each reaction system consisted of 20
ng cDNA, and the specific primers used were as follows:
β-MHC [forward (F), 5′-CCT CGC AAT ATC AAG GGA AA-3′ and
reverse (R), 5′-TAC AGG TGC ATC AGC TCC AG-3′], ANF (F,
5′-GCC GGT AGA AGA TGA GGT CA-3′ and R, 5′-GGG CTC CAA TCC TGT CAA
TC-3′), GATA4 (F, 5′-GGG CGA GCC TGT TTG CAA TG-3′ and R,
5′-TGC TTG GAG CTG GCC TGT GA-3′) or miR-26 (F, 5′-ATG GCT TCA AGT
AAT CC-3′ and R, 5′-GTG CAG GGT CCG AGG T-3′). The thermocycling
conditions were as follows: 95°C for 10 min; 40 cycles of 95°C for
20 sec, 64°C for 30 sec and 72°C for 30 sec; and a melting curve of
95°C for 15 sec, 60°C for 1 min and 95°C for 15 sec. Data were
analyzed using the 2−ΔΔCq method.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; F, 5′-CGC
ATT GCC AGA CAT ATC AGC-3′ and R, 5′-AGG TGA AGC AGG CTC AAT
CAA-3′) and U6 (F, 5′-GCT TCG GCA GCA CAT ATA CTA AAA T-3′ and R,
5′-CGC TTC ACG AAT TTG CGT GTC AT-3′) were used as the internal
reference compounds.
Western blot analysis
CMs and heart tissues were lysed in cold
radioimmunoprecipitation assay (RIPA) buffer (Beyotime Institute of
Biotechnology, Shanghai, China), and the extracted protein samples
were quantified using the Bio-Rad protein assay kit (Bio-Rad,
Hercules, CA, USA). Identical quantities of protein samples were
separated using 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred on to a polyvinylidene fluoride
membrane (Roche). The membrane was blocked with 5% skimmed milk at
room temperature for 2 h, followed by an incubation with the
specific primary antibody against GATA4 (1:1,000; Abcam; cat. no.
ab86371) at 4°C overnight. GAPDH was used as the internal
reference. Following washing three times for 5 min each time in
Tris-buffered saline with Tween (TBST), the membrane was incubated
with the HRP-conjugated secondary antibody at room temperature for
2 h, and washed again in TBST. Subsequently, the positive bands
were developed using the enhanced chemiluminescence (ECL) Plus
Western Blotting substrate (Thermo Fisher Scientific, Inc.) and
analyzed using ImageJ 1.49 software.
Statistical analysis
All the experiments were performed at least in
triplicate, and results are expressed as the mean ± standard
deviation. Data were analyzed using one-way analysis of variance,
followed by an unpaired t-test with SPSS 19.0 software (IBM SPSS,
Armonk, NY, USA). P<0.05 was taken to indicate a statistically
significant difference.
Results
miR-26a is downregulated in the cardiac
hypertrophy rat model
Prior to analyzing the expression level of miR-26a
in the cardiac hypertrophy rat model, the wet weight of the heart
(in mg) compared with the BW (g) at 4 weeks post-TAAC surgery was
first determined. The results revealed a significantly larger
proportion of heart tissue in the TAAC group compared with the Sham
group (P<0.01; Fig. 1A). The
relative CM area, one of the characteristics of cardiac
hypertrophy, was also examined. By observing the tissue sections,
the CMs in the TAAC group were revealed to occupy larger areas
(P<0.01; Fig. 1B). These
results suggested that the TAAC surgery had successfully induced
cardiac hypertrophy in the rat model. Subsequently, two cardiac
hypertrophy-associated genes, ANF and β-MHC, were
analyzed in the Sham and TAAC groups to further verify the cardiac
hypertrophy rat model (Fig. 1C).
The mRNA expression levels of the two genes were significantly
promoted in the TAAC group compared with the Sham group
(P<0.01), which demonstrated that the rats in the TAAC group
were afflicted with cardiac hypertrophy. Following this
verification, the expression of miR-26a expression was detected,
which was shown to be significantly downregulated in the TAAC group
(P<0.05, Fig. 1D). This result
indicated that miR-26a was abnormally expressed in the heart tissue
of the cardiac hypertrophy rat model, suggesting that miR-26a may
serve as an important regulator in cardiac hypertrophy.
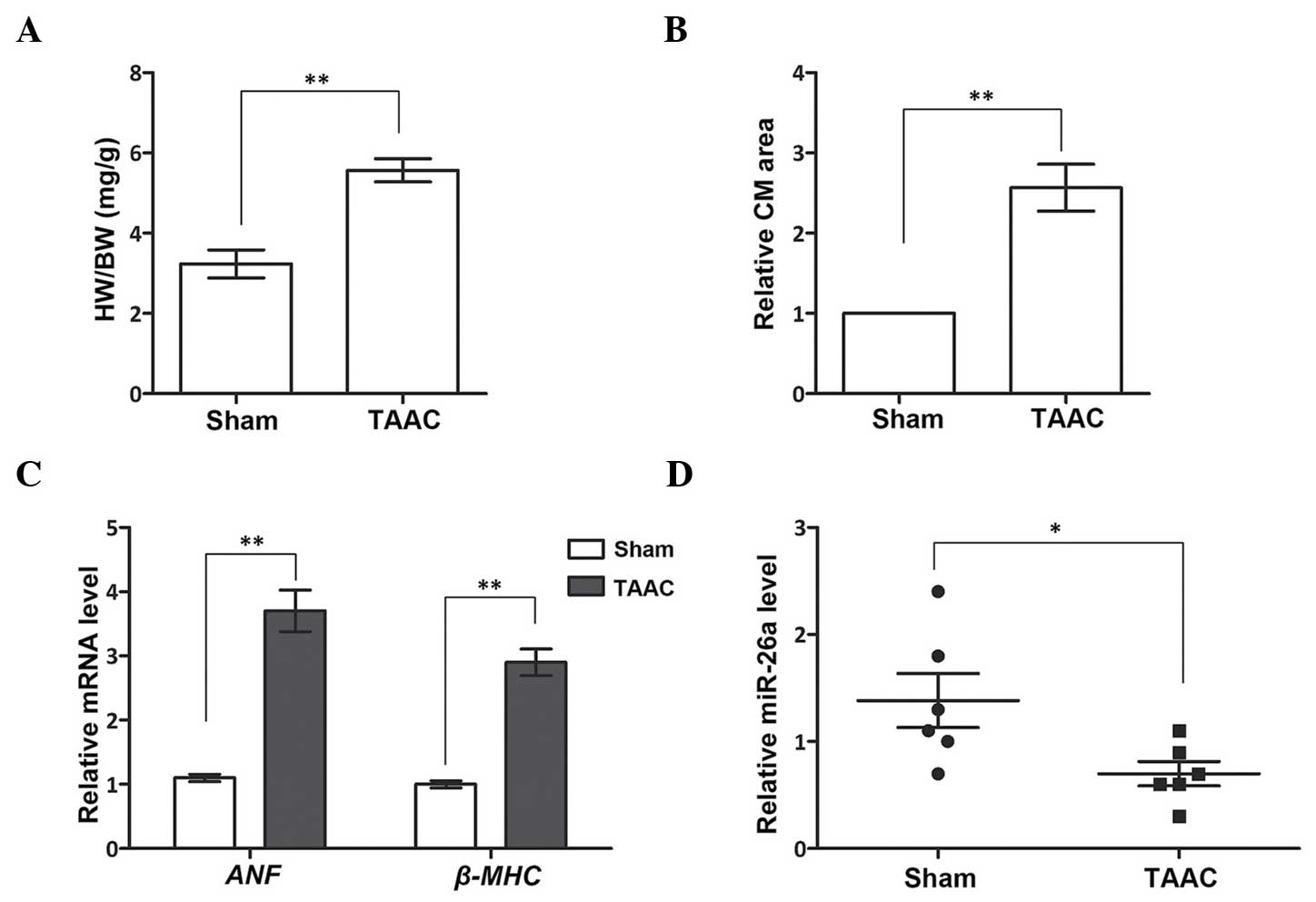 | Figure 1miR-26a inhibits cardiac hypertrophy
in a TAAC-induced rat model. (A) The HW proportion, indicated by
HW/BW, is larger in the TAAC group. (B) The relative CM area is
larger in the TAAC group. (C) mRNA expression of ANF and
β-MHC is upregulated in the TAAC group. (D) The miR-26a
level is inhibited in the TAAC group. *P<0.05;
**P<0.01 vs. Sham group. HW, heart weight. BW, body
weight; miR-26a, microRNA-26a; CM, cardiomyocyte; ANF, atrial
natriuretic factor; β-MHC, β-myosin heavy chain; TAAC, the cardiac
hypertrophy rat model induced by TAAC surgery; Sham, the rats
underwent the identical surgery procedures, except for TAAC; TAAC,
transverse abdominal aortic constriction. |
miR-26a inhibits cardiac hypertrophy in
cultured CMs
The cultured CMs from neonatal rats were treated
with Ang II to induce cardiac hypertrophy, after which it was
determined whether the induction had been effective by detecting
the expression of miR-26a, which was found in the above-mentioned
results to be downregulated in the heart of the cardiac hypertrophy
rat model. These results demonstrated that miR-26a was also
inhibited in the Ang II-treated CMs compared with the Control group
(P<0.01, Fig. 2A), indicating
that cardiac hypertrophy had been successfully induced in the
cultured CMs on Ang II treatment. Subsequently, miR-26a was
overexpressed to examine its influence on cardiac hypertrophic CMs.
The CM area was significantly enlarged, and the expression levels
of ANF and β-MHC mRNA were significantly upregulated,
on Ang II treatment (P<0.01; Fig.
2B and C), although these were restored to normal levels on
miR-26a overexpression (P<0.01). [3H]leucine
incorporation was also measured to assess the protein synthesis
rate in CMs, since an accelerated protein synthesis is another
characteristic of cardiac hypertrophy. These results demonstrated
that protein synthesis was enhanced in Ang II-treated CMs, although
this enhancement was diminished by miR-26a overexpression. Taken
together, these results indicated that CMs induced by Ang II
exhibited cardiac hypertrophic properties, such as a larger CM
area, accelerated protein synthesis and promotion of the expression
of cardiac hypertrophy-associated genes. However, overexpression of
miR-26a was able to significantly inhibit these effects, implying
that it exerts a role in inhibiting cardiac hypertrophy in cultured
CMs.
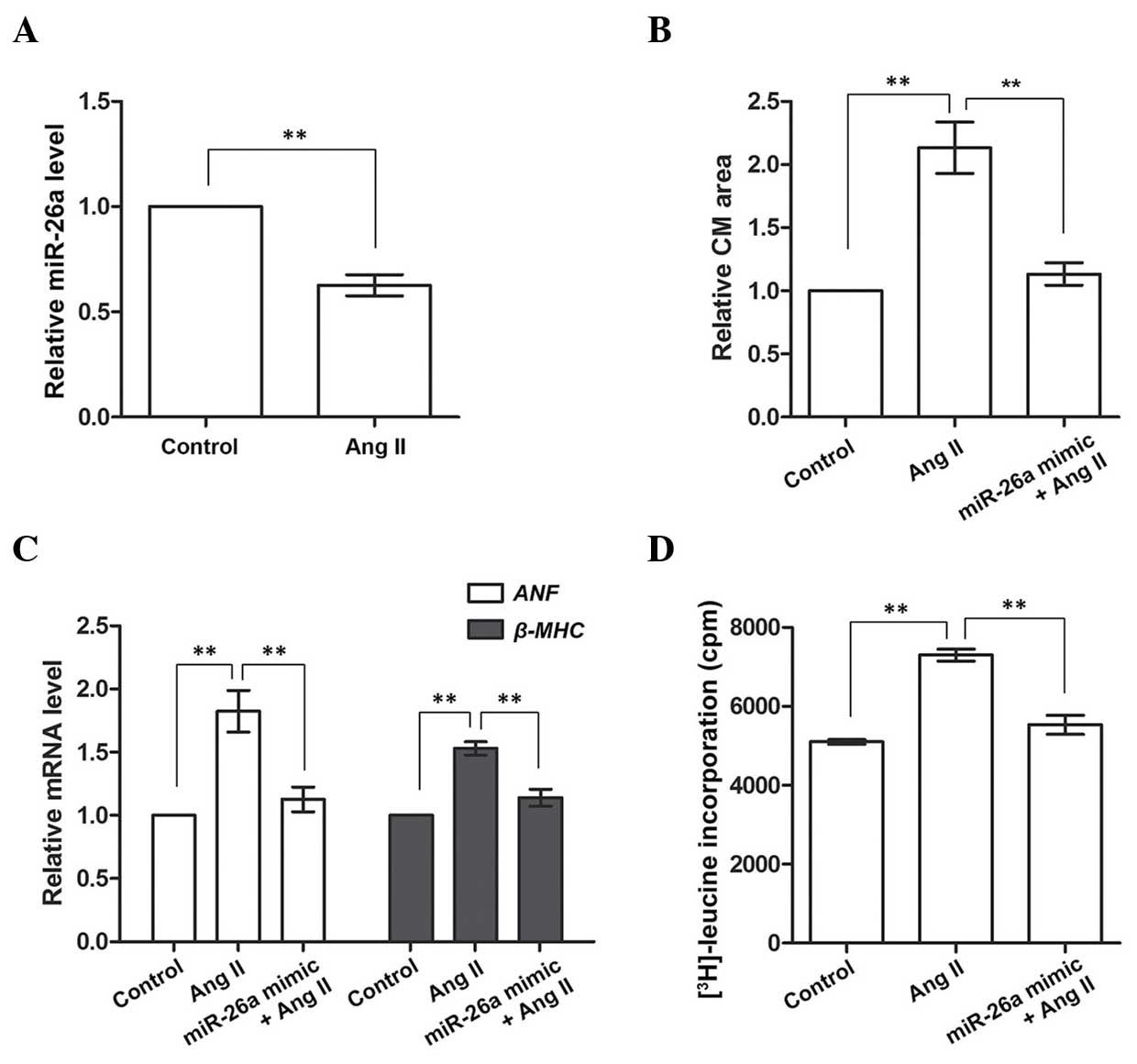 | Figure 2miR-26a inhibits cardiac hypertrophy
in Ang II-induced CMs. (A) miR-26a expression is inhibited in Ang
II-treated CMs. (B) The CM area is larger in Ang II-treated cells,
but decreases with miR-26a overexpression. (C) Expression of
ANF and β-MHC mRNA is upregulated in Ang II-treated
CMs, and downregulated by miR-26a overexpression. (D) Protein
synthesis, indicated by [3H]leucine incorporation, is
accelerated in Ang II-treated cells and inhibited by miR-26a
overexpression. **P<0.01 vs. the Ang II group.
Control group, CMs without Ang II treatment; Ang II group, CMs
treated with Ang II to induce cardiac hypertrophy. miR-26a mimic
group, CMs overexpressing miR-26a; CM, cardiomyocyte; Ang II,
angiotensin II; miR-26a, microRNA-26a; ANF, atrial natriuretic
factor. β-MHC, β-myosin heavy chain; cpm, counts per min. |
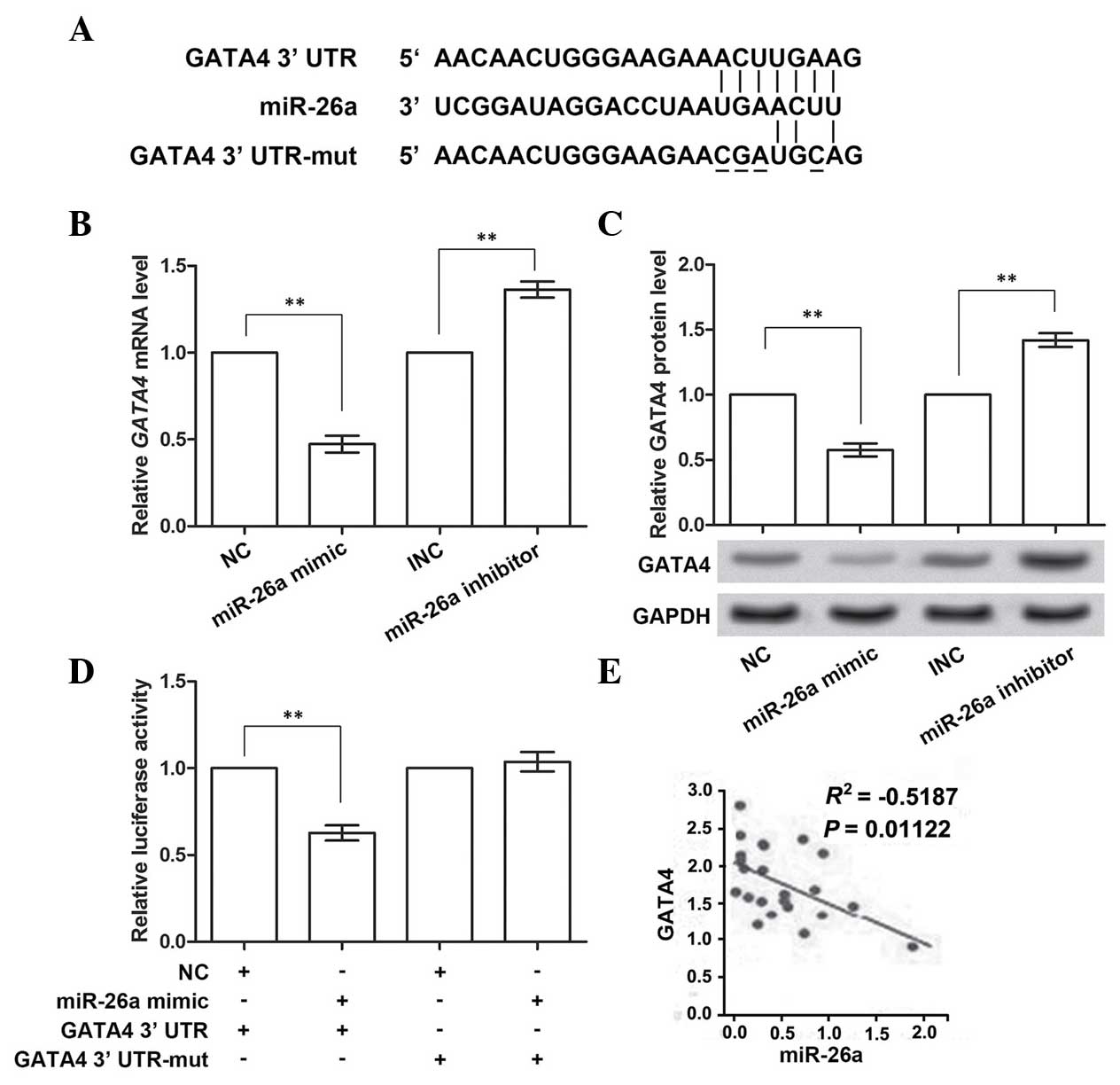
GATA4 is the direct target gene of
miR-26a
GATA4 was predicted to be the target gene of
miR-26a by TargetScan (Fig. 3A).
Therefore, the impact of miR-26a on GATA4 expression using the
miR-26a mimic and inhibitor was analyzed. The RT-qPCR results
demonstrated that the expression of GATA4 mRNA was inhibited
when overexpressing miR-26a (P<0.01; Fig. 3B), and was promoted when miR-26a
was inhibited (P<0.01), compared with the corresponding control
group. Similarly, the protein expression of GATA4 was also
downregulated or upregulated with the promotion or inhibition of
miR-26a expression, respectively (P<0.01; Fig. 3C). These data confirmed the
prediction that GATA4 was the target gene of miR-26a.
To investigate whether GATA4 was directly inhibited
by miR-26a, four sites in the predicted binding sites (positions
131–137 of 3′-UTR) in GATA4 were mutated (Fig. 3A). miR-26a mimic was co-transfected
with the wild-type or mutant GATA4 3′-UTR, and the
luciferase activities were measured (Fig. 3D). These results showed that, when
overexpressing wild-type GATA4 3′-UTR and miR-26a, the
activity of the luciferase reporter gene linked with the wild-type
GATA4 3′-UTR significantly decreased (P<0.01). However,
the activity of the mutant GATA4 3′-UTR was almost unchanged
on miR-26a overexpression. Multiple data groups were used for
analyzing the correlation between GATA4 and miR-26a
expression (Fig. 3E), and these
results revealed that there was a significant negative correlation.
Taken together, these data indicated that miR-26a was able to
directly bind to and suppress GATA4.
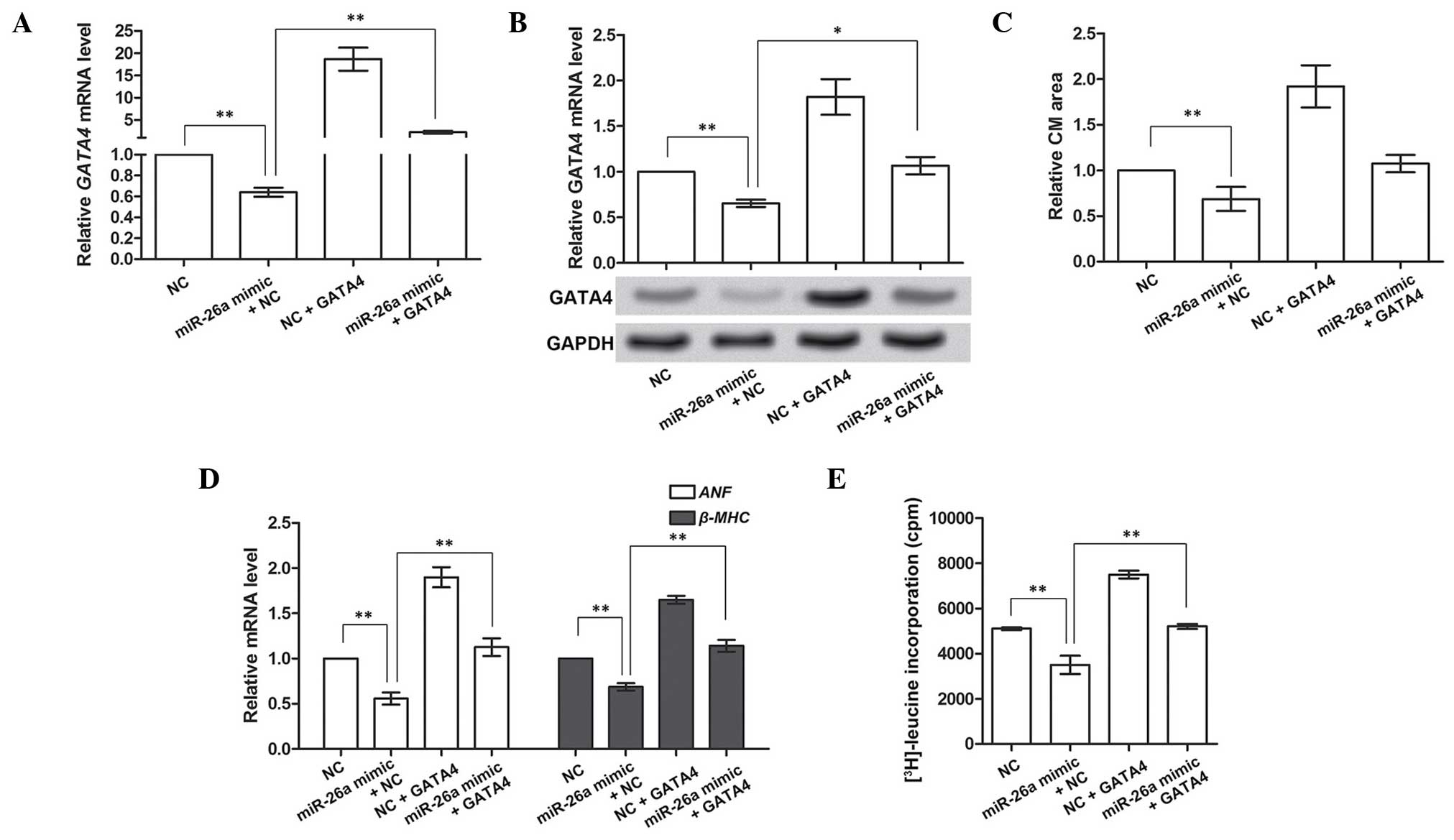
GATA4 compensates for the suppressive
roles of miR-26a in cardiac hypertrophy
In the above-mentioned experiments, GATA4 was
demonstrated to be the direct target gene of miR-26a. It was also
hypothesized that GATA4 may be involved in regulating cardiac
hypertrophy. The overexpression of GATA4 in CMs was therefore
investigated on the basis of miR-26a overexpression to investigate
its functions in cardiac hypertrophy. Prior to performing this
experiment, the overexpression of GATA4 was determined to be valid
(Fig. 4A and B). These results
indicated that miR-26a overexpression significantly inhibited the
mRNA and protein expression of GATA4 (P<0.01), and subsequent
overexpression of GATA4 was able to increase the inhibited GATA4
mRNA and protein levels caused by miR-26a (P<0.01 or P<0.05).
Therefore, the overexpression of GATA4 was shown to be effective
for further analyses.
Subsequently, the role of GATA4 in cardiac
hypertrophy was investigated by analyzing the CM area,
hypertrophy-associated gene expression and protein synthesis. The
CM area was decreased by miR-26a overexpression, and increased by
GATA4 overexpression alone (P<0.01). It also appeared to be
modestly enlarged by GATA4 overexpression when miR-26a was also
overexpressed, although not significantly so (P>0.05, Fig. 4C). Similarly, the expression of the
hypertrophy-associated genes, ANF and β-MHC, and the
protein synthesis rate, as determined by [3H]leucine
incorporation, were all inhibited by miR-26a, but promoted by GATA4
overexpression (P<0.01; Fig. 4D and
E), even with the overexpression of miR-26a. Taken together, as
the target gene inhibited by miR-26a, GATA4 overexpression could
compensate for the suppressive functions of miR-26a in cardiac
hypertrophy, suggesting that GATA4 may be a facilitator of cardiac
hypertrophy.
Discussion
In the present study, the anti-cardiac hypertrophic
roles of miR-26a were investigated. miR-26a is expressed at lower
levels in the heart of cardiac hypertrophy rat model, although its
overexpression, however, causes a significant protection against
cardiac hypertrophy in CMs via inhibition of GATA4. GATA4 was
demonstrated to be the direct target of miR-26a, exerting a
pro-hypertrophic role, since its overexpression leads to aggravated
cardiac hypertrophy in CMs.
miRNAs exert significant roles in the progression of
cardiac hypertrophy. A variety of miRNAs show differential and
temporal expression patterns during cardiac hypertrophy, such as
miR-1, miR-149 and miR-10a, amongst others, of which downregulation
of miR-26a may be detected at ~14 days post-TAAC (17). Thus, in the present study,
experiments were performed at 4 weeks post-TAAC in the rat model,
and miR-26a was revealed to be significantly inhibited, both in the
TAAC-induced rat model and in the cultured rat CMs following 48 h
of Ang II treatment. As elsewhere reported, miR-26a/b may perform
distinct functions in different diseases, not only oncogenic roles,
but also tumor-suppressive roles (18). However, miR-26b has been shown to
be able to inhibit GATA4 and suppress pressure-induced cardiac
hypertrophy (19). Additionally,
miR-26a/b has been revealed to be an anti-hypertrophy factor via
the suppression of glycogen synthase kinase 3β (20). In accordance with these findings,
the hypertrophy-suppressive roles of miR-26a identified in the
present study of the TAAC-induced rat model and Ang II-treated
cells disclosed a further signaling pathway involving miR-26a via
GATA4, revealing a novel regulatory mechanism for miR-26a.
In these experiments, HW/BW, the CM area, and
expression of hypertrophy-associated genes were used as indicators
of cardiac hypertrophy; protein synthesis, as measured by leucine
incorporation assay, was also included when monitoring changes in
CMs. Based on the aforementioned knowledge that cardiac hypertrophy
is usually manifested as larger volumes and accelerated protein
synthesis in CMs, the results of the present study revealed that
the TAAC-induced rat model exhibited larger hearts, with a higher
HW/BW ratio, and larger CMs, as indicated by their larger relative
area. The cultured CMs isolated from neonatal rats also possessed a
larger area, as well as accelerated protein synthesis, when treated
with Ang II, a factor that can increase protein synthesis and cause
hypertrophy of vascular smooth muscle cells (21,22).
ANF and β-MHC were used as indices of cardiac hypertrophy in the
present study for correlation of their expression with cardiac
hypertrophy, and upregulated expression levels were identified
compared with normal heart tissues (23). Switching from α-MHC to β-MHC causes
a higher expression level of β-MHC during hypertrophy (24). The levels of ANF and β-MHC are
mediated by various regulators and signaling pathways. Previous
studies have demonstrated the association between GATA4 and its
downstream factors, including ANF and β-MHC, which are activated by
GATA4 by binding of this protein at sites in their promoter region
(25,26). Similarly, in the present study,
overexpression of GATA4 could upregulate ANF and β-MHC in the
TAAC-induced rat model and Ang II-induced rat CMs, which indicated
that ANF and β-MHC were possible downstream targets of GATA4,
constituting a vital signaling pathway in cardiac hypertrophy
regulation.
As an important transcription factor closely
associated with heart development, GATA4 is the integrator of
several signaling pathways in regulating cardiac hypertrophy. In
addition to the above-mentioned ANF and β-MHC, GATA4 is also able
to regulate the expression of B-type natriuretic peptide in CMs,
possibly via the GATA4/nuclear factor of activated T-cells-3
signaling pathway (27). Several
factors regulating GATA4 in cardiac hypertrophy have been reported.
Transcription factor CHF-1/Hey2 binds GATA4 to inhibit the
regulation of GATA4 on the ANF promoter (28), whereas estrogen-related receptor γ
activates GATA4 to exacerbate cardiac hypertrophy (29). Other upstream mediators of GATA4
include the MEK1/ERK1/2 signaling pathway, where ERK2 directly
phosphorylates GATA4 (30), and
miRNAs such as miR-208a, which controls the expression of GATA4
during cardiac hypertrophy (10).
In the present study, miR-26a was shown to inhibit GATA4 by
targeting the binding sites in the 3′-UTR, since the mutated
GATA4 3′-UTR did not achieve successful regulation by
miR-26a. Taken together, GATA4 was able to regulate various
downstream factors, which were likely to be controlled by proteins
and miRNAs upstream, forming a part of the regulatory network
during cardiac hypertrophy.
In summary, the present study has identified a
protective role of miR-26a via suppression of GATA4, and, in turn,
further downstream factors in cardiac hypertrophy. These results
are potentially useful in terms of the possible use of miR-26a, and
therapeutic targets such as GATA4, in cardiac hypertrophy
treatment.
References
|
1
|
Senthil V, Chen SN, Tsybouleva N, Halder
T, Nagueh SF, Willerson JT, Roberts R and Marian AJ: Prevention of
cardiac hypertrophy by atorvastatin in a transgenic rabbit model of
human hypertrophic cardiomyopathy. Circ Res. 97:285–292. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Okere IC, Young ME, McElfresh TA, Chess
DJ, Sharov VG, Sabbah HN, Hoit BD, Ernsberger P, Chandler MP and
Stanley WC: Low carbohydrate/high-fat diet attenuates cardiac
hypertrophy, remodeling and altered gene expression in
hypertension. Hypertension. 48:1116–1123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Doyle B, Sorajja P, Hynes B, Kumar AH,
Araoz PA, Stalboerger PG, Miller D, Reed C, Schmeckpeper J, Wang S,
et al: Progenitor cell therapy in a porcine acute myocardial
infarction model induces cardiac hypertrophy, mediated by paracrine
secretion of cardiotrophic factors including TGFbeta1. Stem Cells
Dev. 17:941–951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weeks KL and McMullen JR: The athlete's
heart vs the failing heart: Can signaling explain the two distinct
outcomes? Physiology (Bethesda). 26:97–105. 2011. View Article : Google Scholar
|
|
5
|
Kodama H, Fukuda K, Pan J, Sano M,
Takahashi T, Kato T, Makino S, Manabe T, Murata M and Ogawa S:
Significance of ERK cascade compared with JAK/STAT and PI3-K
pathway in gp130-mediated cardiac hypertrophy. Am J Physiol Heart
Circ Physiol. 279:H1635–H1644. 2000.PubMed/NCBI
|
|
6
|
Lezoualc'h F, Metrich M, Hmitou I,
Duquesnes N and Morel E: Small GTP-binding proteins and their
regulators in cardiac hypertrophy. J Mol Cell Cardiol. 44:623–632.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pan J, Singh US, Takahashi T, Oka Y,
Palm-Leis A, Herbelin BS and Baker KM: PKC mediates cyclic
stretch-induced cardiac hypertrophy through Rho family GTPases and
mitogen-activated protein kinases in cardiomyocytes. J Cell
Physiol. 202:536–553. 2005. View Article : Google Scholar
|
|
8
|
Yue H, Li W, Desnoyer R and Karnik SS:
Role of nuclear unphosphorylated STAT3 in angiotensin II type 1
receptor-induced cardiac hypertrophy. Cardiovasc Res. 85:90–99.
2010. View Article : Google Scholar
|
|
9
|
Gladka MM, da Costa Martins PA and De
Windt LJ: Small changes can make a big difference - microRNA
regulation of cardiac hypertrophy. J Mol Cell Cardiol. 52:74–82.
2012. View Article : Google Scholar
|
|
10
|
Callis TE, Pandya K, Seok HY, Tang RH,
Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al:
MicroRNA-208a is a regulator of cardiac hypertrophy and conduction
in mice. J Clin Invest. 119:2772–2786. 2009. View Article : Google Scholar :
|
|
11
|
Wang K, Lin ZQ, Long B, Li JH, Zhou J and
Li PF: Cardiac hypertrophy is positively regulated by MicroRNA
miR-23a. J Biol Chem. 287:589–599. 2012. View Article : Google Scholar :
|
|
12
|
Huang ZP, Chen J, Seok HY, Zhang Z,
Kataoka M, Hu X and Wang DZ: MicroRNA-22 regulates cardiac
hypertrophy and remodeling in response to stress. Circ Res.
112:1234–1243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Care A, Catalucci D, Felicetti F, Bonci D,
Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, et al:
MicroRNA-133 controls cardiac hypertrophy. Nat Med. 13:613–618.
2007. View
Article : Google Scholar
|
|
14
|
Li Q, Song XW, Zou J, Wang GK, Kremneva E,
Li XQ, Zhu N, Sun T, Lappalainen P, Yuan WJ, et al: Attenuation of
microRNA-1 derepresses the cytoskeleton regulatory protein
twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci.
123:2444–2452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei L, Yuan M, Zhou R, Bai Q, Zhang W,
Zhang M, Huang Y and Shi L: MicroRNA-101 inhibits rat cardiac
hypertrophy by targeting Rab1a. J Cardiovasc Pharmacol. 65:357–363.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen
H, Dean DB and Zhang C: MicroRNAs are aberrantly expressed in
hypertrophic heart: Do they play a role in cardiac hypertrophy? Am
J Pathol. 170:1831–1840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sayed D, Hong C, Chen IY, Lypowy J and
Abdellatif M: MicroRNAs play an essential role in the development
of cardiac hypertrophy. Circ Res. 100:416–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao J and Liu QG: The role of miR-26 in
tumors and normal tissues (Review). Oncol Lett. 2:1019–1023.
2011.
|
|
19
|
Han M, Yang Z, Sayed D, He M, Gao S, Lin
L, Yoon S and Abdellatif M: GATA4 expression is primarily regulated
via a miR-26b-dependent post-transcriptional mechanism during
cardiac hypertrophy. Cardiovasc Res. 93:645–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang ZH, Li J, Liu BR, Luo CF, Dong Q,
Zhao LN, Zhong Y, Chen WY, Chen MS and Liu SM: MicroRNA-26 was
decreased in rat cardiac hypertrophy model and may be a promising
therapeutic target. J Cardiovasc Pharmacol. 62:312–319. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parmentier JH, Pavicevic Z and Malik KU:
Ang II stimulates phospholipase D through PKCzeta activation in
VSMC: Implications in adhesion, spreading and hypertrophy. Am J
Physiol Heart Circ Physiol. 290:H46–H54. 2006. View Article : Google Scholar
|
|
22
|
Li R, Xiao J, Qing X, Xing J, Xia Y, Qi J,
Liu X, Zhang S, Sheng X, Zhang X and Ji X: Sp1 mediates a
therapeutic role of MiR-7a/b in angiotensin II-induced cardiac
fibrosis via mechanism involving the TGF-β and MAPKs pathways in
cardiac fibroblasts. PLoS One. 10:e01255132015. View Article : Google Scholar
|
|
23
|
Garcia R, Thibault G and Cantin M:
Correlation between cardiac hypertrophy and plasma levels of atrial
natriuretic factor in non-spontaneous models of hypertension in the
rat. Biochem Biophys Res Commun. 145:532–541. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haddad F, Qin AX, Bodell PW, Zhang LY, Guo
H, Giger JM and Baldwin KM: Regulation of antisense RNA expression
during cardiac MHC gene switching in response to pressure overload.
Am J Physiol Heart Circ Physiol. 290:H2351–H2361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu X, Li T, Zhang C, Liu Y, Xu M, Wang W,
Jia Z, Ma K, Zhang Y and Zhou C: GATA4 regulates ANF expression
synergistically with Sp1 in a cardiac hypertrophy model. J Cell Mol
Med. 15:1865–1877. 2011. View Article : Google Scholar
|
|
26
|
Charron F, Paradis P, Bronchain O, Nemer G
and Nemer M: Cooperative interaction between GATA-4 and GATA-6
regulates myocardial gene expression. Mol Cell Biol. 19:4355–4365.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu YL, Huang CC, Chang CC, Chou CY, Lin
SY, Wang IK, Hsieh DJ, Jong GP, Huang CY and Wang CM:
Hyperphosphate-induced myocardial hypertrophy through the
GATA-4/NFAT-3 signaling pathway is attenuated by ERK inhibitor
treatment. Cardiorenal Med. 5:79–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiang F, Sakata Y, Cui L, Youngblood JM,
Nakagami H, Liao JK, Liao R and Chin MT: Transcription factor
CHF1/Hey2 suppresses cardiac hypertrophy through an inhibitory
interaction with GATA4. Am J Physiol Heart Circ Physiol.
290:H1997–H2006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwon DH, Eom GH, Kee HJ, Nam YS, Cho YK,
Kim DK, Koo JY, Kim HS, Nam KI, Kim KK, et al: Estrogen-related
receptor gamma induces cardiac hypertrophy by activating GATA4. J
Mol Cell Cardiol. 65:88–97. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang Q, Wiese RJ, Bueno OF, Dai YS,
Markham BE and Molkentin JD: The transcription factor GATA4 is
activated by extracellular signal-regulated kinase 1- and
2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol
Cell Biol. 21:7460–7469. 2001. View Article : Google Scholar : PubMed/NCBI
|