Introduction
Even if first-stage resuscitation is successful
following cardiac arrest (CA) and cardiopulmonary resuscitation
(CPR), the body is subjected to systemic ischemia/reperfusion (I/R)
injury due to the return of spontaneous circulation (ROSC), thereby
leading to multiple organ dysfunction syndrome (MODS) (i.e.
post-resuscitation syndrome). Acute kidney injury (AKI) is one of
the features of post-resuscitation syndrome. AKI is most commonly
caused by systemic hypoperfusion and can lead to acute renal
failure (1,2). AKI is common in the survivors of CA
with a reported incidence of 23.2%. Incomplete recovery of renal
function from AKI may cause excessive long-term morbidity and
mortality and result in a higher risk of chronic kidney disease
(3–5). Following ROSC, focus is placed on the
condition of the brain and heart; however, prevention against AKI
is also important and requires investigation.
Toll-like receptors (TLRs) are a family of signal
transduction molecules that are implicated in the induction of
innate and adaptive immunity. Recently, accumulating evidence has
demonstrated that Toll-like receptor 4 (TLR4) is activated by
endogenous proteins released from damaged tissues and participates
in mediating renal injury following I/R (6,7).
However, the majority of previous studies have employed regional
I/R models generated by clamping renal pedicles, which are
different from the global I/R models induced by CA/CPR. In
addition, it remains unclear whether TLR4 is causal within the
kidney after CA/CPR.
It is well known that larger animal models that
allow for whole body hypoperfusion, lack access to the full toolset
of genetic manipulation possible in the mouse. However, recently a
mouse model of CA/CPR has emerged, which can be adapted to model
AKI (8–10). This model is reliable to reproduce
physiological, functional and histological outcomes observed in
clinical AKI. In the present study, a mouse model of CA/CPR was
used to test the hypothesis that TLR4 activation contributes to
renal injury following resuscitation.
Materials and methods
Animals
C57BL/6 male mice (n=40; age, 10–12 weeks) were
obtained from the Animal Resource Center of Wuhan University
(Wuhan, China). TLR4 mutant male mice (C3H/HeJ; n=20; age, 10–12
weeks) were purchased from Shanghai SLAC Animal Center (Shanghai,
China). In these mice, the intracellular region of TLR4 amino acids
had a mutation at the 712 site from proline to histidine, which
resulted in no response of TLR4 to its ligand, lipopolysaccharide
(11). TLR4 wild-type male mice
(C3H/HeN; n=20; age, 10–12 weeks) were purchased from Beijing
Vitalriver Experimental Animal Center (Beijing, China). All animals
were housed in an animal facility with a specific-pathogen free
environment. The mice were kept in a dry, ventilated and clean
environment. The room temperature was maintained at ~25°C and
humidity was maintained at 40–70%. The mice were allowed ad
libitum access to food and water under automatic day/night
control (12:12 h). Animal protocols were approved by the Laboratory
Animal Ethics Committee of Huazhong University of Science and
Technology (Huazhong, China).
Experimental protocols
Experiment 1: Assessing the role of
TLR4 in renal injury as the duration of CA increases
C57BL/6 mice were randomly assigned to 4 groups. In
the sham group, the animals underwent surgical preparation without
CPR. In the CA 3 min, CA 5 min and CA 8 min groups, resuscitation
was started after 3, 5 and 8 min CA, respectively (n=10, per
group).
Experiment 2: Assessing the
inflammatory response to CA/CPR in TLR4-mutant mice
Animals were randomly assigned to 4 groups (n=10,
per group) which were the C3H/HeN sham group (sham-en), C3H/HeJ
sham group (sham-ej), C3H/HeN CPR group (model-en) and C3H/HeJ CPR
group (model-ej). Chest compressions and mechanical ventilation
were started after 5 min of CA.
Surgical preparation
Mice were anesthetized with 40 mg/kg body weight of
1% pentobarbitol sodium (Merck & Co., Inc., Whitehouse Station,
NJ, USA) delivered by intraperitoneal injection. The mice were
immobilized by taping their four legs in the supine position, and
the rectal temperature was controlled at close to 37°C during
surgery with a heating pad and lamp. The mice were immobilized 4
extremities by tape in a supine position on a heating pad. Rectal
temperature was controlled at ~37°C during surgery with a heating
pad and lamp. The mice were orally intubated with a 22-gauge
catheter, connected to a mouse ventilator (ALC-V8S, Shanghai Alcbio
Company, Shanghai, China) set to a respiratory rate of 130
breaths/min. Needle electrodes were placed subcutaneously on the
chest for electrocardiogram monitoring throughout the experimental
procedures. A middle incision was made in the neck of the mice and
the external jugular vein was carefully separated and a catheter
was inserted.
CA/CPR procedure
CA was induced by injection of 0.08 mg/g body weight
KCl via the jugular catheter, and confirmed by the appearance of
asystole on the electrocardiography monitor and no spontaneous
breathing (12,13). At this time, mechanical ventilation
was interrupted for the length of CA duration. After different CA
durations, CPR was begun by injection of 0.4 μg/g
epinephrine followed by chest compressions at a rate of ~300
beats/min and ventilation with 100% oxygen at a respiratory rate of
160 breaths/min. As soon as ROSC was achieved, defined as
electrocardiographic activity with visible cardiac contractions,
chest compressions were stopped (12,13).
If ROSC could not be achieved within 10 min of CPR, resuscitation
was stopped. Sham animals underwent anesthesia, oral intubation,
mechanical ventilation, surgical preparation and insertion of
vascular catheters. An equivalent volume of isotonic saline was
given as a placebo control of KCl and epinephrine in the sham
groups. All mice were alive in the sham groups after 3 days.
However, in experiment 1, 9 and 8 mice survived in the CA 3 min and
CA 5 min groups, respectively. While only 6 mice survived for 72 h
in the CA 8 min group. In experiment 2, 7 and 8 mice survived in
the C3H/HeN and C3H/HeJ CPR groups after ROSC 72 h,
respectively.
Measurement of blood biochemical
parameters
After ROSC (72 h), the surviving mice were
sacrificed by cervical dislocation, and blood samples were
collected from the heart. After centrifugation (4,000 × g/min, 20
min), the supernatant was stored at −80°C. Plasma serum creatinine
(Scr) and blood urea nitrogen (BUN) levels were measured by an
automatic biochemical analyzer (BECKMAN LX20, Beckman Coulter,
Brea, CA, USA).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The left kidneys were taken from the surviving mice
and preserved at −80°C. Total RNA was extracted from 100 mg kidney
tissue using TRIzol reagent (Invitrogen, Thermo Fisher, Waltham,
MA, USA) in accordance with the manufacturer's instructions. The
concentration and purity of RNA were tested using an ultraviolet
spectrophotometer (UV-2802, Unico Co., Dayton, NJ, USA). The
reverse transcription of RNA to cDNA was performed using a reverse
transcription kit (Takara Bio Inc., Shiga, Japan). The primers used
were: TLR4 sense, 5′-TGAGGACTGGGAGAAATGAGC-3′ and antisense,
5′-CTGCCATGTTTGCAATCTCAT-3′; and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) sense, 5′-CTCTGATTTGGTCGTATTGGG-3′ and
antisense, 5′-CTGGAAGATGATGGGAT-3′. The PCR reaction (using an
LY96G PCR instrument; Hangzhou LongYang Scientific Instruments Co.,
Ltd., Hangzhou, China) was conducted in a thermal cycler with
initial 4 min denaturation at 95°C, followed by 30 (TLR4) or 28
cycles (GAPDH) of denaturation at 94°C for 50 sec, annealing at
58°C for 40 sec (TLR4) or 56°C for 30 sec (GAPDH), extension at
72°C for 60 sec (TLR4) or 50 sec (GAPDH), and a final extension at
72°C for 7 min (TLR4) or 10 min (GAPDH). The target bands were
quantitatively determined by the relative intensity of TLR4 gene
compared with that of GAPDH by using the GeneSnap gel image
acquisition system (GeneGenius; Syngene UK, Cambridge, UK) and
GeneTools gel image analysis software (version 2.0; Syngene UK)
(14).
Protein preparation and western
blotting
Proteins from the left kidney tissues were prepared
using radioimmunoprecipitation assay buffer. All protein samples
were subjected to concentration determination with the
bicinchoninic acid assay method. Protein samples were separated
using 10% SDS/PAGE and transferred onto polyvinylidene fluoride
membranes (Merck Millipore, Billerica, MA, USA). Membranes were
blocked for 40 min with a 5% bovine serum albumin solution at room
temperature, and subsequently incubated with the appropriate
antibodies overnight at 4°C, and then the membranes were washed
three times with Tris-buffered saline/Tween 20 (TBST), followed by
incubation with a secondary antibody. Subsequently, the membranes
were washed four times with TBST. The indicated antibodies
included: Rabbit anti-mouse TLR4 polyclonal antibody (1:500, cat.
no. ab13867, Abcam, Cambridge, UK), rabbit anti-mouse intercellular
adhesion molecule-1 (ICAM-1) polyclonal antibody (1:500, cat. no.
ab7815, Abcam) and rabbit anti-mouse growth-related gene product β
(GRO-β) polyclonal antibody (1:500, cat. no. ab9950, Abcam, UK).
Goat anti-mouse β-actin antibody (1:5,000, cat. no. ANT009,
Antgene, Wuhan, China) was used as an internal control, and the
secondary antibodies included goat anti-rabbit polyclonal antibody
(1:5,000; cat. no. BA1003, Boster Biological Technology, Wuhan,
China) and mouse anti-goat polyclonal antibody (1:5,000, cat. no.
BA1006, Boster Biological Technology). The membranes were
visualized using a Kodak Image Station 4000 MM imaging system
(Kodak, Tokyo, UK) in accordance with the manufacturer's
instructions. Semi-quantitative analysis was conducted on strips
with Quantity one software software (version 4.6.2; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Morphological studies
In experiment 1 and 2, the mice were sacrificed by
cervical dislocation and the right kidneys were fixed in 4%
paraformaldehyde and embedded in paraffin. Sections (5 μm)
were stained with hematoxylin and eosin according to standard
protocols. The changes in renal histopathology were observed with
electron microscopy. Jablonski grading was used to analyze the
injury severity of renal tubules (0–4 levels) (15). In experiment 2, tissue samples for
transmission electron microscopy were obtained from the left
kidney. The cortex and outer section of the outer medulla were
separated and cut into pieces of ~1 mm3. The tissue
sections were prefixed with 4% glutaraldehyde, stored at 4°C until
processed and then post-fixed with 1% osmium tetroxide. The
specimens were immersed in propylene oxide after dehydration with a
graded series of ethanol, embedded with epoxy resin and cut into
ultrathin sections (0.1 μm). The sections were stained
subsequently with lead-uranium and the changes in ultra-structural
organization were then observed by a transmission electron
microscope.
Myeloperoxidase (MPO) activity
assay
Tissues from the left kidney were homogenized and
centrifuged (12,000 × g, 20 min). Then, the supernatant was
harvested and stored at −80°C. The MPO activity in the supernatant
was detected using an enzyme-linked immunosorbent assay kit
according to the manufacturer's instructions (NanJing JianCheng
Bioengineering Institute, Nanjing, China).
Statistical analysis
Data are presented as the mean ± standard deviation
for each experiment. The data were analyzed by SPSS 17.0
statistical software (SPSS Inc., Chicago, IL, USA) as appropriate.
Two-group comparisons were analyzed using Student's t-test, whereas
multiple-group comparisons were conducted using analysis of
variance followed by Bonferroni's multiple comparisons test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Indicators of kidney function
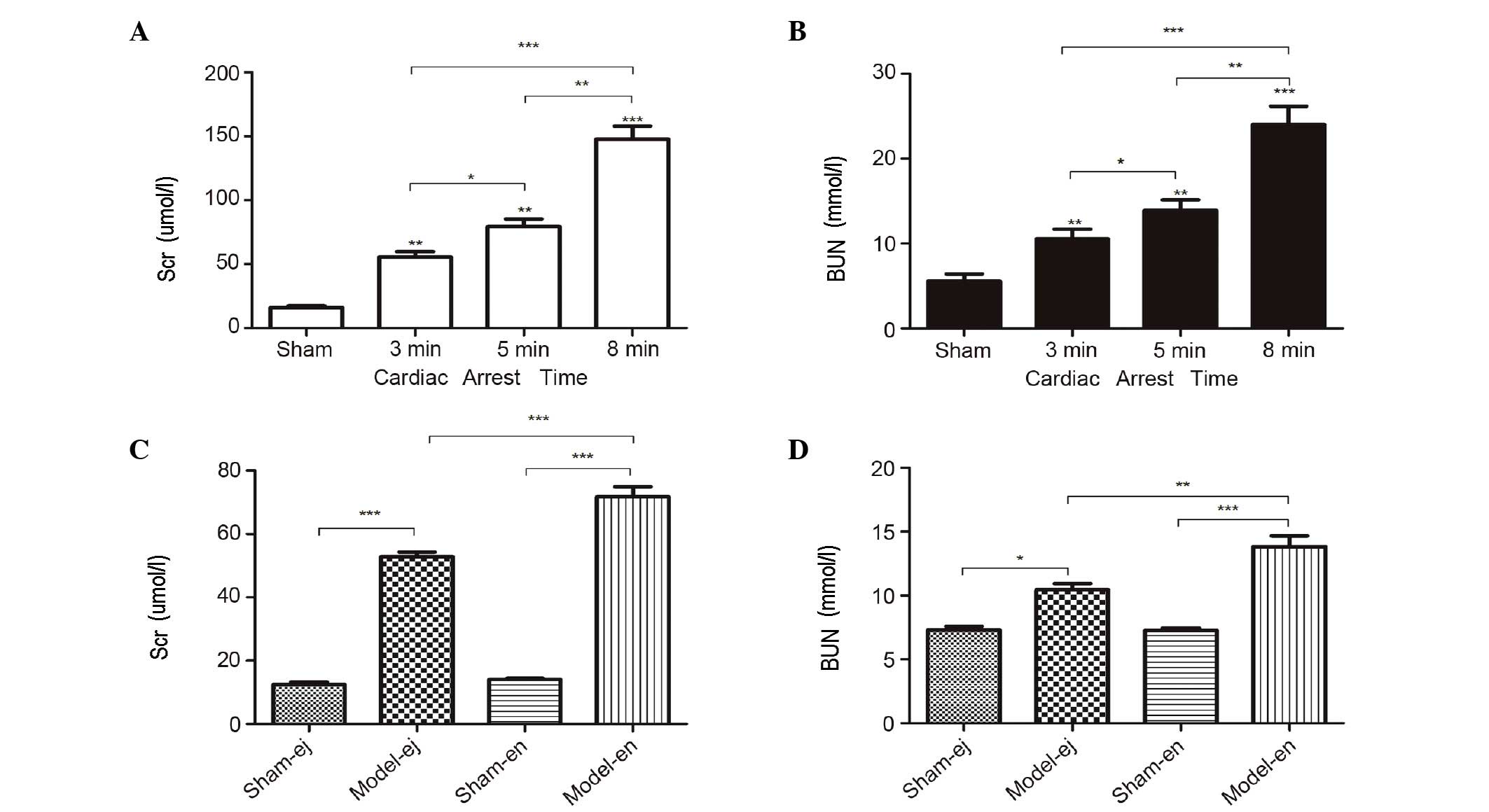
In experiment 1, the plasma concentration of Scr and
BUN were markedly increased after CA/CPR. Moreover, the levels of
Scr and BUN increased significantly with the increasing duration of
CA, suggesting that the length of CA duration may be important in
AKI following CPR (Fig. 1A and B).
In experiment 2, there were also higher plasma concentrations of
Scr and BUN following CPR in C3H/HeJ and C3H/HeN mice. However, the
levels of Scr and BUN in the model-ej group were significantly
lower than those in model-en group, indicating that TLR4-mutant
mice exhibited minor renal dysfunction following CPR (Fig. 1C and D).
TLR4 mRNA and TLR4 protein
expression
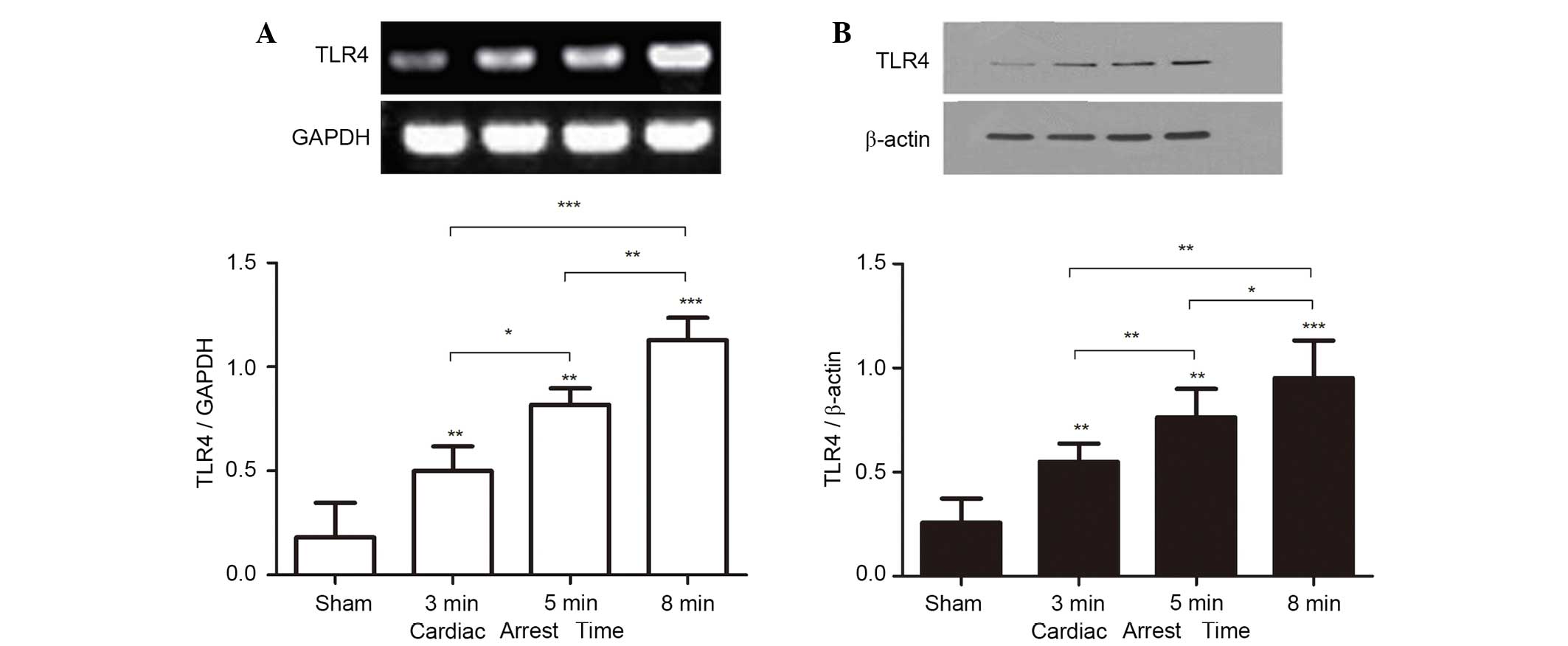
In experiment 1, to address whether CA/CPR
stimulates TLR4 activation, the mRNA and protein expression of TLR4
in the kidney were measured on day 3 after ROSC. Normal renal
tissue expressed TLR4 at a basal level. However, TLR4 mRNA and
protein levels were significantly increased following CA/CPR, and
increased in a CA-time dependent manner (Fig. 2A and B).
ICAM-1 and GRO-β protein expression
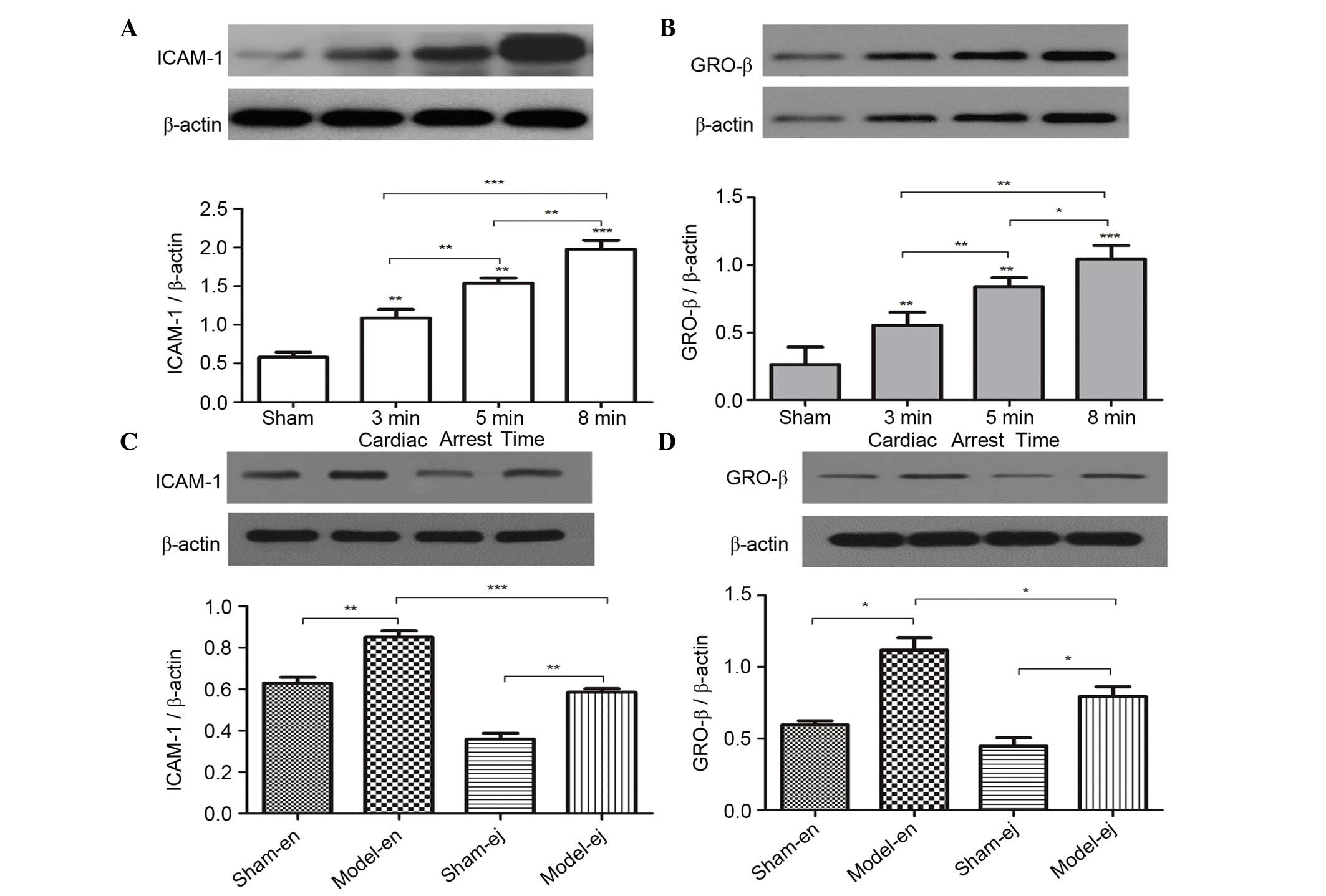
To determine the effect of CA/CPR on the expression
of the downstream factors regulated by TLR4 signaling the protein
expression of ICAM-1 and GRO-β, which are classically known for
their role in neutrophil infiltration and aggregation were
measured. In experiment 1, ICAM-1 and GRO-β levels were
significantly increased with the extension of CA duration (Fig. 3A and B). In experiment 2 it was
also demonstrated that CPR induced an increase in ICAM-1 and GRO-β
expression. However, the expression of ICAM-1 and GRO-β in C3H/HeJ
mice was significantly lower than that in the C3H/HeN mice,
indicating that TLR4 mutation reduced neutrophil infiltration into
renal tissues following CPR (Fig. 3C
and D).
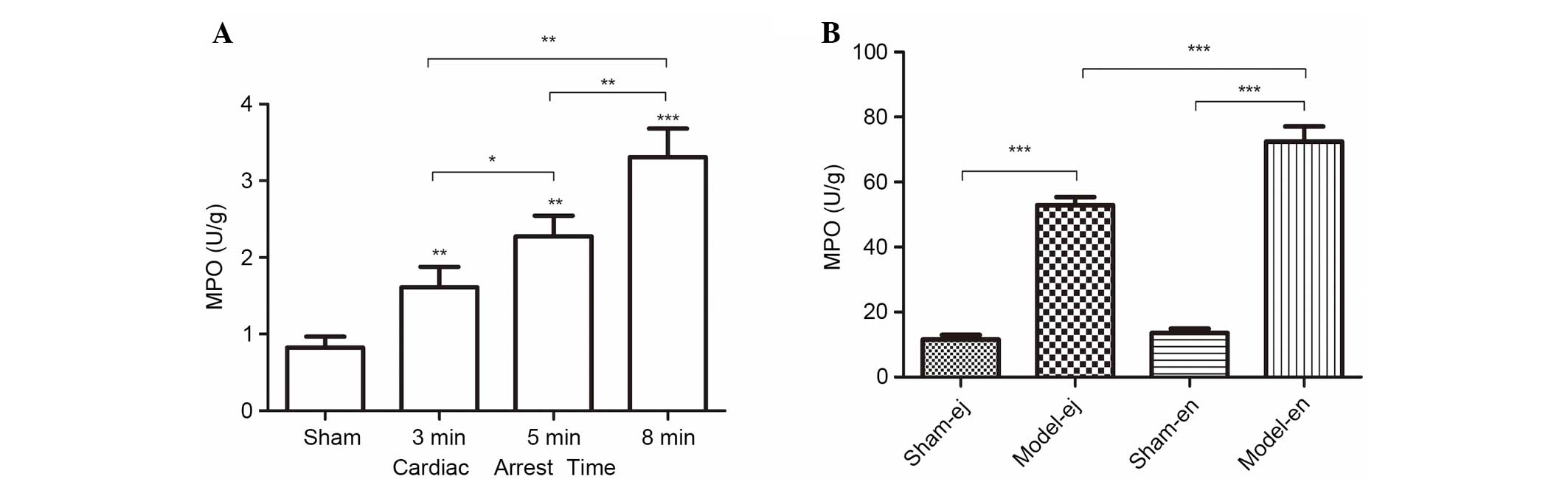
Activity of MPO
In the two experiments, CPR contributed to the
elevation of MPO activity in the renal tissues, a biochemical
marker of neutrophil infiltration. As the length of CA duration was
prolonged, the activity of MPO gradually increased (Fig. 4A). However, in experiment 2 there
was a reduction in MPO activity in the model-ej group compared with
that in the model-en group, suggesting that TLR4 may be required
for neutrophil infiltration into renal tissues after CPR (Fig. 4B).
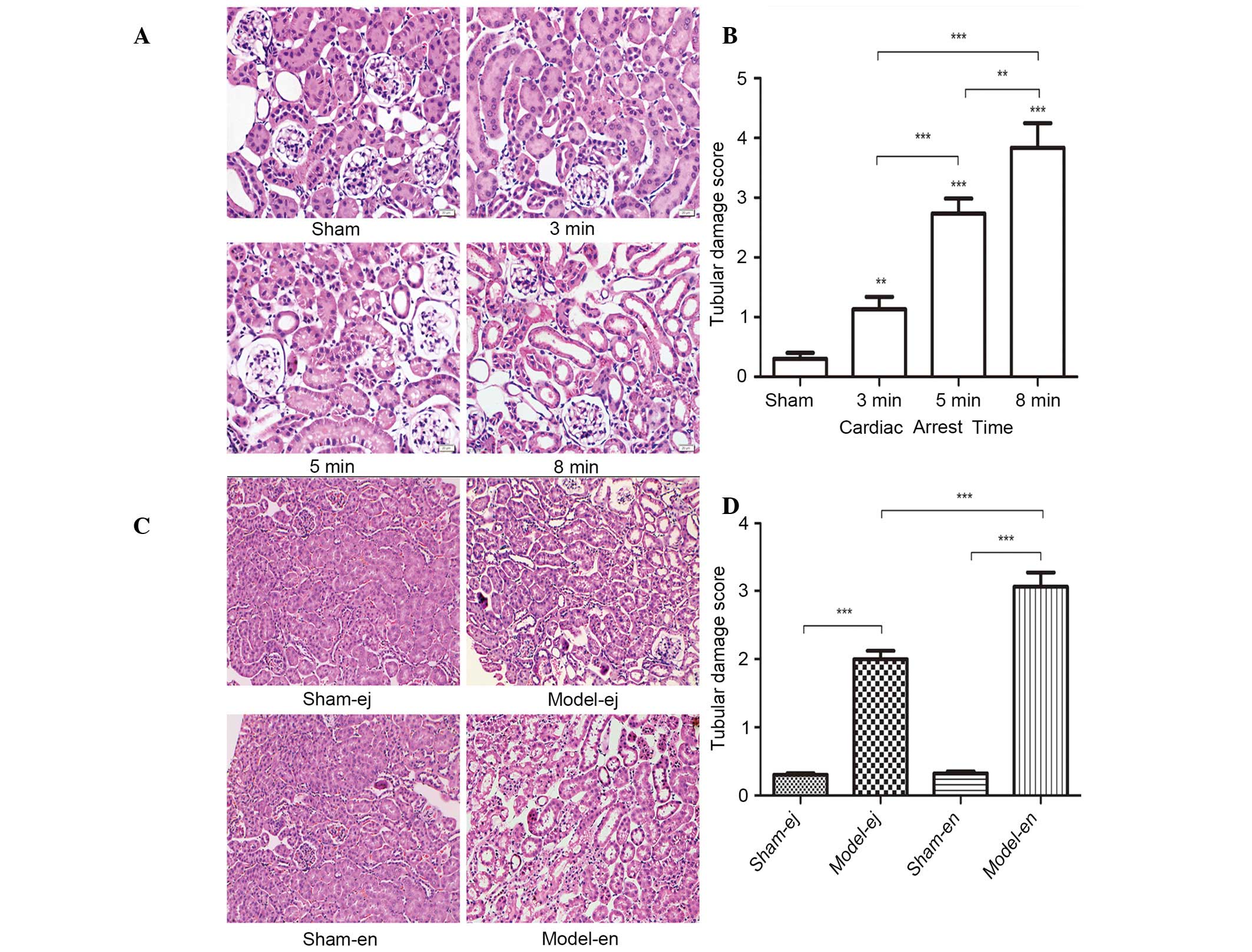
Renal histopathology
Examination by light microscopy revealed that in
experiment 1, the glomerular and tubular structures were clear and
intact in the sham group. However, histological abnormalities that
reflected renal tissue damage were observed after CPR, including
atrophy of tubules, interstitial edema, denaturation, swelling,
vacuolation of tubule epithelium and tubular necrosis. The renal
abnormalities were most severe in the CA 8 min group as shown by
the highest renal damage scores (Fig.
5A and B). In experiment 2, the epithelium of the tubules
showed signs of denaturation, swelling and vacuolation in the
model-ej and model-en groups. However, the renal damage score was
significantly lower in model-ej group than that in model-en group,
suggesting that TLR4-mutant mice were more resistant to renal
injury after CPR (Fig. 5C and
D).
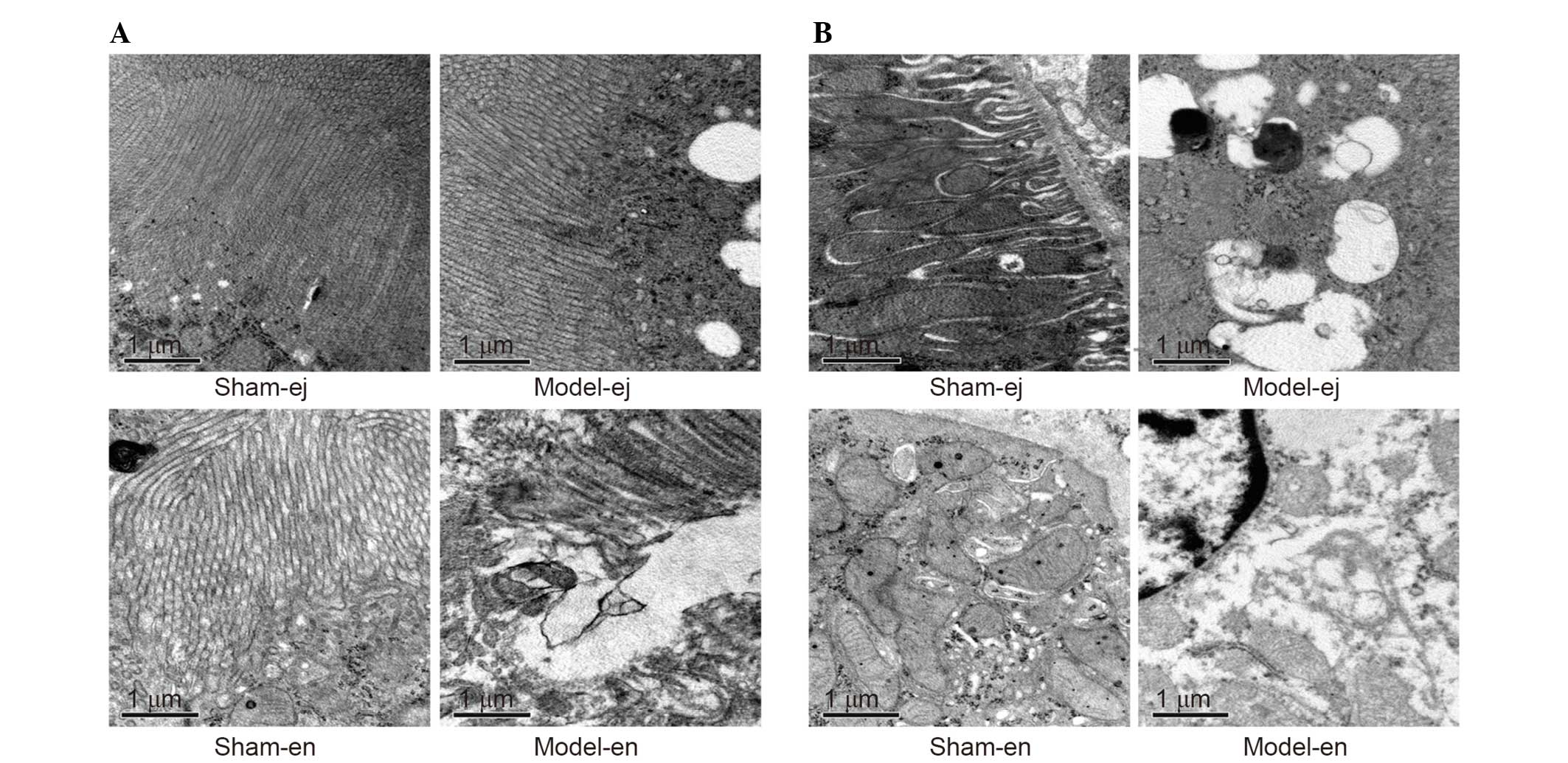
Examination by electron microscopy revealed that the
membranes of the microvilli remained intact and the brush borders
were closely arrayed in the sham-ej and sham-en groups. However,
the loss of microvilli and increased cell vacuolization were shown
in the model-ej and model-en groups. The ultrastructure
abnormalities of the microvilli were more severe in the model-en
group than that in the model-ej group (Fig. 6A). In addition, it was demonstrated
that the glomerular epithelium in the model-en group presented more
evident swollen endoplasmic reticulum and mitochondria with broken
or absent cristae, some of which appeared to be vacuolized when
compared with the model-ej group (Fig.
6B).
Discussion
The present study demonstrated that the mice
suffered from AKI after successful CPR and severe AKI occurred in
mice with prolonged CA. Notably, the results showed that CA/CPR
increased the expression of TLR4, ICAM-1, GRO-β and MPO in kidney
tissues in a CA-duration dependent manner. However, TLR4-mutant
mice were protected from renal I/R injury as shown by the minor
changes in renal function and histology, suggesting that
TLR4-mediated inflammatory responses may be involved in AKI
triggered by CA/CPR.
Post-resuscitation syndrome is one of the leading
causes of mortality in patients following ROSC (16). Renal damage can cause metabolic
acidosis and hyperkalemia, which are associated with cardiovascular
events (such as ventricular fibrillation), threatening the life of
critical patients. Incomplete recovery of renal function following
AKI may cause excessive long-term morbidity and mortality and may
be associated with a higher risk of chronic kidney disease. A
number of survivors require renal replacement therapy during the
advanced life-support phase. Thus, it is worth paying more
attention to prevention of AKI following CPR.
Recently, it has been demonstrated that injection of
KCl is a feasible method to induce immediate CA and allow
successful resuscitation in a high fraction of animals (9). In the present study, the results
demonstrated successful reproducibility of the AKI model after
CA/CPR as judged by the changes in renal histology and function. In
addition, AKI became more severe with prolonged CA duration. The
results were consistent with clinical studies, which showed that
transient impaired renal function is common in patients surviving
CA (17). Duration of CA,
pre-existing impaired renal function and blood pressure at
admission were not independent risk factors associated with renal
outcome (18). Moreover, a recent
study demonstrated that AKI may not just be a consequence of CA but
of the time without spontaneous circulation (19). In experiment 1, it was demonstrated
that there was a relatively high survival rate with notable renal
histopathological changes after CPR in the CA 5 min group.
Therefore 5 min duration of CA was selected in experiment 2.
The mechanism of AKI after CPR is hypothesized to be
associated with I/R injury, which is now shown to be involved in
inflammation. TLR4, is a pattern recognition receptor that
recognizes exogenous microbial or endogenous ligands resulting in
the induction of natural immune and inflammatory responses
(20). I/R rapidly activates
innate immune responses. TLR4 has been shown to be upregulated in
kidney I/R by clamping the renal pedicles (6,7).
However, it is unclear whether TLR4 exhibits a causal role within
the kidney after the global I/R induced by CA/CPR. In the present
study, it was demonstrated that TLR4 was activated by CA/CPR as
shown by the significant increase in TLR4, which occurred in a
CA-time dependent manner. Moreover, the present study aimed to
demonstrate the critical role for TLR4 in the pathophysiology of
AKI using TLR4 genetically mutant mice. Notably, C3H/HeJ mice were
resistant to CPR-induced AKI as shown by the protection associated
with a concomitant decrease in Scr and BUN levels and attenuation
in histological changes, suggesting that TLR4 may contribute to AKI
following CPR.
It has been suggested that endogenous TLR ligands,
such as high-mobility group box 1 were released from damaged or
necrotic cells in response to I/R (21). TLR4 engagement by its ligands
triggers multiple downstream effects, including the activation and
expression of chemokines responsible for neutrophil accumulation,
which are also the features of pathological changes of AKI. MPO is
one of the principal enzymes released from neutrophil azurophilic
granules, and MPO activity was evaluated as an index of neutrophil
accumulation (22). The results
showed that MPO activity was increased in C3H/HeN and C3H/HeJ mice
after resuscitation, indicating that neutrophils were recruited
within the kidney following CPR. ICAM-1 is predominantly expressed
on the surface of endothelial cells, involved in regulating a
variety of effector cells to migrate to areas of inflammation. In
addition, ICAM-1-deficient mice are protected against ischemic
renal injury (23), indicating
that ICAM-1 is a key mediator of acute ischemic renal failure
likely acting via potentiation of neutrophil-endothelial
interactions. Similar to ICAM-1, GRO-β can also induce inflammatory
cells, such as neutrophils, mono-nuclear cells and lymphocytes, to
migrate towards I/R injury regions and exacerbate inflammatory
reactions. The inhibition of neutrophil migration by genetic
deletion of GRO-β receptor or its ligand suppresses I/R-induced
kidney injury (24,25). In the present study, the expression
of ICAM-1 and GRO-β were elevated in kidney tissues after CPR,
which are associated with the upregulation of TLR4 in C3H/HeN mice.
This suggests that TLR4 and its downstream signaling events that
promote neutrophil infiltration via ICAM-1 and GRO-β may be
important in mediating inflammatory responses to renal injury
following CPR.
It is noteworthy that expression of renal ICAM-1,
GRO-β and MPO were also significantly increased following CPR in
C3H/HeJ mice, albeit less so than in C3H/HeN mice. This implies
that there is a response to CPR-induced I/R through
TLR4-independent pathways. One of the possible explanations may be
that renal TLR2, which is predominantly expressed by tubular cells,
also mediates I/R injury in the kidney. Genetic absence or
knockdown of TLR2 was previously shown to reduce cytokine and
chemokine production, reduce leukocyte infiltration, and protect
against kidney dysfunction and tubular damage (26,27).
The present study had certain limitations. It is
better to use a mechanical compressor instead of manual chest
compressions after CA. However, as far as we know, a mechanical
compressor for small animals was not commercially available when
the experiments were conducted. In addition, the immediate
reperfusion period following ROSC is characterized by an abrupt
increase in the plasma tumor necrosis factor-α concentration
(28). In the present study, the
levels of inflammatory cytokines were not measured following ROSC.
In addition, it is better to observe the dynamic changes in
inflammatory cytokines after resuscitation. Furthermore, patients
treated with CPR generally have a clinical disease; however, the
mice in the present study were healthy. Therefore, the outcome of
this study in a mouse model of CPR remains to be demonstrated in
large-animal and clinical studies.
In conclusion, the results documented that AKI after
successful CPR is not rare and the duration of CA is associated
with the renal outcome. In addition, renal TLR4 is crucial in
mediating CPR-induced AKI, via systemic cytokine release and
subsequent intrarenal events, such as renal neutrophil
infiltration. Our study suggests that TLR4 has a potential
therapeutic application for acute kidney injury after
cardiopulmonary resuscitation.
Acknowledgments
The present study was supported by grants from the
National Nature Science Foundation of China (grant nos. 81201444
and 81101401).
References
|
1
|
Joannidis M, Druml W, Forni LG, Groeneveld
AB, Honore P, Oudemans-van Straaten HM, Ronco C, Schetz MR and
Woittiez AJ; Critical Care Nephrology Working Group of the European
Society of Intensive Care Medicine: Prevention of acute kidney
injury and protection of renal function in the intensive care unit.
Expert opinion of the Working Group for Nephrology, ESICM.
Intensive Care Med. 36:392–411. 2010. View Article : Google Scholar
|
|
2
|
Poukkanen M, Vaara ST, Reinikainen M,
Selander T, Nisula S, Karlsson S, Parviainen I, Koskenkari J and
Pettilä V; FINNAKI Study Group: Predicting one-year mortality of
critically ill patients with early acute kidney injury: Data from
the prospective multicenter FINNAKI study. Crit Care. 19:1252015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Susantitaphong P, Cruz DN, Cerda J,
Abulfaraj M, Alqahtani F, Koulouridis I and Jaber BL; Acute Kidney
Injury Advisory Group of the American Society of Nephrology: World
incidence of AKI: A meta-analysis. Clin J Am Soc Nephrol.
8:1482–1493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coca SG, Yusuf B, Shlipak MG, Garg AX and
Parikh CR: Long-term risk of mortality and other adverse outcomes
after acute kidney injury: A systematic review and meta-analysis.
Am J Kidney Dis. 53:961–973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wald R, Quinn RR, Luo J, Li P, Scales DC,
Mamdani MM and Ray JG; University of Toronto Acute Kidney Injury
Research Group: Chronic dialysis and death among survivors of acute
kidney injury requiring dialysis. JAMA. 302:1179–1185. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu H, Chen G, Wyburn KR, Yin J, Bertolino
P, Eris JM, Alexander SI, Sharland AF and Chadban SJ: TLR4
activation mediates kidney ischemia/reperfusion injury. J Clin
Invest. 117:2847–2859. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu H, Ma J, Wang P, Corpuz TM,
Panchapakesan U, Wyburn KR and Chadban SJ: HMGB1 contributes to
kidney ischemia reperfusion injury. J Am Soc Nephrol. 21:1878–1890.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burne-Taney MJ, Kofler J, Yokota N,
Weisfeldt M, Traystman RJ and Rabb H: Acute renal failure after
whole body ischemia is characterized by inflammation and T
cell-mediated injury. Am J Physiol Renal Physiol. 285:F87–F94.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menzebach A, Bergt S, von Waldthausen P,
Dinu C, Nöldge-Schomburg G and Vollmar B: A comprehensive study of
survival, tissue damage, and neurological dysfunction in a murine
model of cardiopulmonary resuscitation after potassium-induced
cardiac arrest. Shock. 33:189–196. 2010. View Article : Google Scholar
|
|
10
|
Hutchens MP, Traystman RJ, Fujiyoshi T,
Nakayama S and Herson PS: Normothermic cardiac arrest and
cardiopulmonary resuscitation: A mouse model of
ischemia-reperfusion injury. J Vis Exp. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poltorak A, He X, Smirnova I, Liu MY, Van
Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al:
Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations
in Tlr4 gene. Science. 282:2085–2088. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng G, Yonchek JC, Quillinan N, Strnad
FA, Exo J, Herson PS and Traystman RJ: A novel mouse model of
pediatric cardiac arrest and cardiopulmonary resuscitation reveals
age-dependent neuronal sensitivities to ischemic injury. J Neurosci
Methods. 222:34–41. 2014. View Article : Google Scholar :
|
|
13
|
Wang QY, Sun P, Zhang Q and Yao SL:
Minocycline attenuates microglial response and reduces neuronal
death after cardiac arrest and cardiopulmonary resuscitation in
mice. J Huazhong Univ Sci Technolog Med Sci. 35:225–229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun P, Zhang Q, Han JY, Tian Y and Zhang
JH: TLR4 signaling induced TLR2 expression in the process of mimic
cerebral ischemia/reperfusion in vitro. Sci China Life Sci.
53:223–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jablonski P, Howden BO, Rae DA, Birrell
CS, Marshall VC and Tange J: An experimental model for assessment
of renal recovery from warm ischemia. Transplantation. 35:198–204.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Neumar RW, Nolan JP, Adrie C, Aibiki M,
Berg RA, Böttiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC,
et al: Post-cardiac arrest syndrome: Epidemiology, pathophysiology,
treatment, and prognostication A consensus statement from the
international liaison committee on resuscitation (American heart
association Australian and New Zealand council on resuscitation,
European resuscitation council, heart and stroke foundation of
Canada, InterAmerican heart foundation, resuscitation council of
Asia, and the resuscitation council of Southern Africa); the
American heart association emergency cardiovascular care committee;
the council on cardiovascular surgery and Anesthesia; the council
on cardiopulmonary, perioperative, and critical care; the council
on clinical cardiology; and the stroke council. Circulation.
118:2452–2483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Domanovits H, Müllner M, Sterz F,
Schillinger M, Klösch C, Paulis M, Hirschl MM and Laggner AN:
Impairment of renal function in patients resuscitated from cardiac
arrest: Frequency, determinants and impact on outcome. Wien Klin
Wochenschr. 112:157–161. 2000.PubMed/NCBI
|
|
18
|
Domanovits H, Schillinger M, Müllner M,
Thoennissen J, Sterz F, Zeiner A and Druml W: Acute renal failure
after successful cardiopulmonary resuscitation. Intensive Care Med.
27:1194–1199. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chua HR, Glassford N and Bellomo R: Acute
kidney injury after cardiac arrest. Resuscitation. 83:721–727.
2012. View Article : Google Scholar
|
|
20
|
De Nardo D: Toll-like receptors:
Activation, signalling and transcriptional modulation. Cytokine.
74:181–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Doi K, Ishizu T, Tsukamoto-Sumida M,
Hiruma T, Yamashita T, Ogasawara E, Hamasaki Y, Yahagi N, Nangaku M
and Noiri E: The high-mobility group protein B1-Toll-like receptor
4 pathway contributes to the acute lung injury induced by bilateral
nephrectomy. Kidney Int. 86:316–326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Voudris KV, Chanin J, Feldman DN and
Charitakis K: Novel inflammatory biomarkers in coronary artery
disease: Potential therapeutic approaches. Curr Med Chem.
22:2680–2689. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kelly KJ, Williams WW Jr, Colvin RB,
Meehan SM, Springer TA, Gutierrez-Ramos JC and Bonventre JV:
Intercellular adhesion molecule-1-deficient mice are protected
against ischemic renal injury. J Clin Invest. 97:1056–1063. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Huang L, Sung SS, Vergis AL, Rosin
DL, Rose CE Jr, Lobo PI and Okusa MD: The chemokine receptors CCR2
and CX3CR1 mediate monocyte/macrophage trafficking in kidney
ischemia-reperfusion injury. Kidney Int. 74:1526–1537. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahuja N, Andres-Hernando A, Altmann C,
Bhargava R, Bacalja J, Webb RG, He Z, Edelstein CL and Faubel S:
Circulating IL-6 mediates lung injury via CXCL1 production after
acute kidney injury in mice. Am J Physiol Renal Physiol.
303:F864–F872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rusai K, Sollinger D, Baumann M, Wagner B,
Strobl M, Schmaderer C, Roos M, Kirschning C, Heemann U and Lutz J:
Toll-like receptors 2 and 4 in renal ischemia/reperfusion injury.
Pediatr Nephrol. 25:853–860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim HJ, Park SJ, Koo S, Cha HJ, Lee JS,
Kwon B and Cho HR: Inhibition of kidney ischemia-reperfusion injury
through local infusion of a TLR2 blocker. J Immunol Methods.
407:146–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niemann JT, Rosborough JP, Youngquist S,
Shah AP, Lewis RJ, Phan QT and Filler SG: Cardiac function and the
proinflammatory cytokine response after recovery from cardiac
arrest in swine. J Interferon Cytokine Res. 29:749–758. 2009.
View Article : Google Scholar : PubMed/NCBI
|