Introduction
Spinocerebellar ataxia (SCA) is a progressive,
degenerative, genetic and neurodegenerative disease with multiple
types, and currently no known effective treatment or cure (1). More than 60 different types of SCA
that have been identified to date, which are diagnosed via autopsy
(2). SCA3 is an autosomal dominant
neurodegenerative disorder with numerous clinical features,
including ataxia, ophthalmoplegia, pyramidal signs, basal ganglia
symptoms and peripheral neuropathy (1). The causative gene, ataxin 3 (ATXN3)
has been mapped to chromosome 14q32.1 (2). Nakano et al (2) first reported SCA3 in 1972 in the
Machado family, who were Portuguese immigrants living in
Massachusetts. In 1976, Rosenberg et al (3) identified SCA3 in the Joseph family.
Subsequently, it was demonstrated that SCA3 is the result of
expansion of CAG trinucleotide-repeats in the ATXN3 gene. Sequences
of healthy individuals contain 14–40 CAG-repeats; however,
sequences of patients with SCA3 contain 72–86 CAG-repeats (4). The numbers of expanded repeats vary
between generations, resulting in significant phenotypic
variations; patients with increased numbers of triplet-repeats
suffer from a greater disease severity and earlier onset (5). The triplet repeat primed polymerase
chain reaction (TP-PCR) method was developed to screen for expanded
alleles.
In 1997, Zhou et al (4) confirmed the expansion of CAG-repeats
in the ATXN3 gene in Chinese patients with SCA3. However, to date,
few cases of SCA3 have been reported in the Chinese population. In
the present study, a Chinese family with SCA3 was identified, and
the genetic and clinical characteristics of these patients
reported. The results of the present study provide insight that may
improve the accuracy of the clinical diagnosis for SCA3.
Materials and methods
Patients and diagnoses
Blood samples from the patient and his siblings were
collected and treated with an anticoagulant,
ethylenediaminetetraacetic acid. The diagnosis of SCA was based on
established criteria (6).
Interviews with the patient and his family were performed to obtain
information on family history. The study was approved by the ethics
committee of Wenzhou People's Hospital (Wenzhou, China). Informed
consent was obtained from all subjects prior to blood sample
collection. Clinical assessments, including regular neurological
tests, magnetic resonance imaging (MRI) and ocular examinations
were conducted at the Wenzhou People's Hospital.
Sample analysis and PCR
DNA was isolated from peripheral blood lymphocytes
using the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany).
SCA1, SCA2, SCA3, SCA6, SCA7 and dentatorubropallidoluysian atrophy
(DRPLA) loci were amplified by PCR using
5′-carboxyfluores-cein-labeled primers and AmpliTaq
Gold® DNA polymerase (catalog no. N8080247; Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) as
previously described (1,7–10).
PCR products were analyzed with capillary electrophoresis according
to the previous study (4) using
the ABI 3130xl system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Standard PCR was performed with a reaction
volume of 50 µl, containing 100 ng genomic DNA, 2 pM each
primers and 25 µl 2X Taq PCR Master Mix (Biotake GmbH,
Glashütten, Germany). The PCR process was performed using a thermal
cycler platform (Applied Biosystems; Thermo Fisher Scientific,
Inc.). TP PCR assay was performed in a reaction volume of 25
µl, which contained 200 ng DNA, 1.5 mmol/l MgCl2,
10 mmol/l Tris, 50 mmol/l KCl, 0.8 µmol/l primers, 200
µmol/l dNTPs each and 2 U Taq polymerase (Eppendorf AG;
Hamburg, Germany).
Results
A case of SCA3 was diagnosed in a Chinese family by
identification of CAG expansion at the SCA3 locus using PCR. The
proband patient (III-3) was a 40-year-old male who presented with
coughing and expectoration and was with bedridden with mobility
limitation. The patient first noticed symptoms, including
difficulty walking in a straight line and a tendency to fall, about
20 years previously. Five years prior to the present study, the
patient began to experience severe neurological problems, including
choking and dribbling while drinking. Disease symptoms worsened
approximately one year ago, as the patient became incontinent and
unable to feed himself. Three days prior to the present study, the
patient became unable to walk and was experiencing coughing and
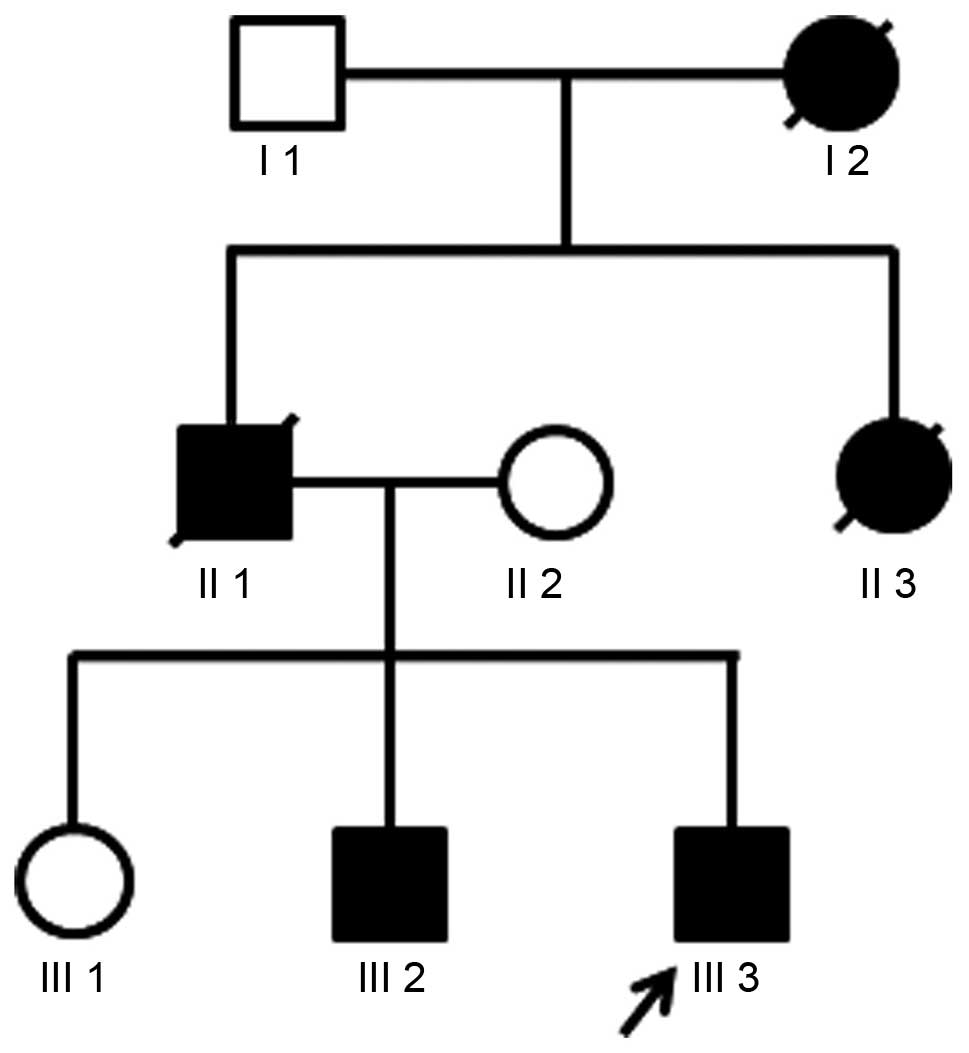
expectoration. Among the other members of his family, the father of
the patient died of SCA and his elder brother walked unsteadily.
However, his mother and elder sister were healthy (Fig. 1).
Clinical examinations demonstrated that the patient
was alert and fully oriented with a blood pressure of 110/60 mmHg
and a resting heart rate of 72 bpm. The patient exhibited neck
abnormalities, dysarthria, and moderate dysmetria as assessed by
the finger-to-nose and heel-knee-shin test. Pupils were symmetrical
and sensitive to light with signs of nystagmus and restricted eye
movement. In addition, the patient was suffering from dysphagia and
amyotrophy of the tongue. Other physiological measurements of the
patients were as follows: Glutamic-oxalacetic transaminease, 165
U/l; creatine kinase, 2419 U/l; and creatine kinase isoenzyme, 45
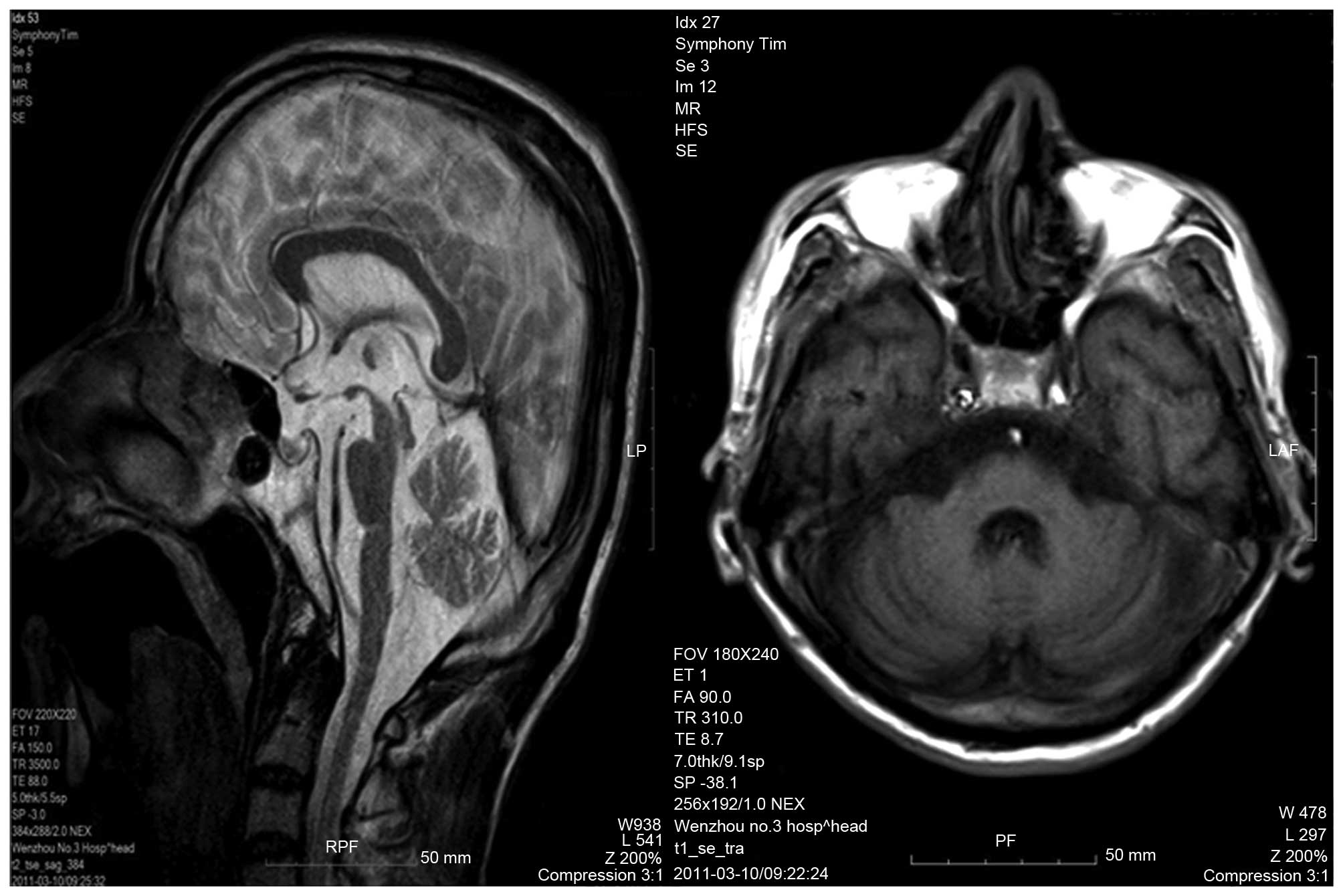
U/l. An MRI scan of the head identified brain, cerebellar and brain
stem atrophy, typical of patients with SCA (Fig. 2).
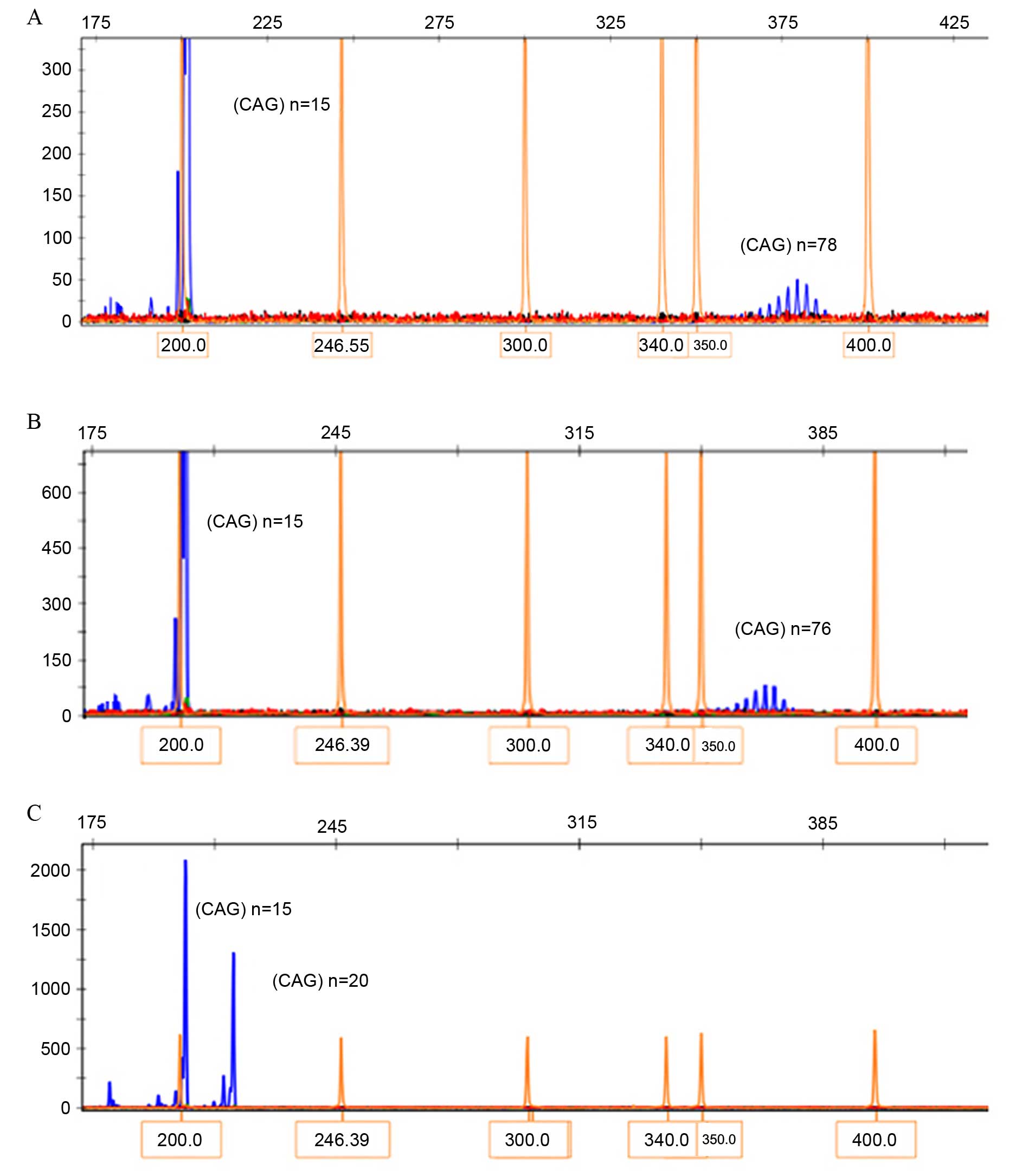
The genotyping results of blood samples collected
from family members are presented in Fig. 3. PCR amplification and capillary
electrophoresis analysis identified a CAG-repeat length of 15 in
the normal allele and an expanded 78 CAG-repeat in the patient
III-3. A 76 CAG-repeat was identified in his brother III-2, while
20 CAG-repeats were identified in his sister III-1. The CAG-repeats
within the SCA1, SCA2, SCA6, SCA7, SCA12 and DRPLA genes of the
patient were within the normal range.
Discussion
SCA diseases have been classified into three groups
by Harding et al (6),
according to clinical features. These classification criteria
remain the guidelines for genetic diagnosis of SCA. SCA3 belongs to
type I autosomal dominant cerebellar ataxia (ADCA), which includes
ataxia accompanied by optic atrophy, ophthalmoplegia,
extrapyramidal signs, neuropathy and cognitive impairment. The
subject of the present study demonstrated signs of slurred speech,
moderate dysmetria as assessed by the finger-to-nose and
heel-knee-shin test, dysphagia, amyotrophy of the tongue,
symmetrical pupils, sensitivity to light and restrictive movement
of eyes, which are typical of type I ADCA. However, symptoms
overlapping the three types of SCA are frequently observed in
patients. SCA3 results from mutations involving CAG
trinucleotide-repeat expansions in the coding regions of the ATXN3
gene. Healthy individuals have a small and stable number of
CAG-repeats (14–40). However, disease occurs when the CAG-repeats
exceed a certain size (4). A
feature of SCA resulting from CAG-repeat expansion is genetic
anticipation, as the number of CAG-repeats typically increases with
each subsequent generation. This increase in the numbers of
CAG-repeats leads to a greater disease severity and earlier onset
(5). While normal alleles are
stably transmitted without modification, mutated alleles are
unstable and further expansion of CAG-repeats may occur during
transmission. However, the underlying mechanism by which CAG-repeat
expansion occurs remains to be fully elucidated. Typically, disease
severity correlates with the extent of CAG-repeat expansion: The
greater the number of CAG-repeats, the greater the severity of the
disease and the earlier its onset. In the present study, the
patient harboring 78 CAG-repeats at the disease gene demonstrated
an earlier onset of SCA3 compared with his brother, who had fewer
CAG-repeats. These results are consistent with the observations of
our previous study (11).
Due to somatic mosaicism, variable repeat sizes in
different tissues of the same patient may influence disease onset
and severity. ATXN3 exerts greater toxicity in the nucleus compared
with the cytoplasm. Previous studies have demonstrated that ATXN3
interacts with various transcription regulators to modulate the
cellular stress response, and directly regulates the ubiquitin
proteasome system. CAG-repeat expansion in ATXN3 altered its
affinity for other regulators, which may explain the pathogenesis
of SCA3 (12,13).
Short tandem repeat analysis based on capillary
electrophoresis is a simple and reliable method for the detection
of CAG-repeats. However, PCR using flanking primers only allows
amplification up to ~100 CAG-repeats; amplification of PCR
templates over this size is unreliable. Triplet-repeat primed PCR
(TP-PCR), by contrast, allows rapid identification of large
pathogenetic CAG-repeats that may not be amplified using standard
PCR (14). Therefore, TP-PCR was
used to confirm the presence of large expansions in the present
study when only one allele was identified by standard PCR (data not
shown).
In conclusion, the present study reported the
clinical symptoms and genetic characteristics of a Chinese family
with SCA3. These observations provide insight into the clinical
diagnosis and genetic typing of patients with SCA3, which may
benefit future patients.
Acknowledgments
The authors would like to thank the patient and his
family for their participation in the present study. The present
study was supported by the Zhejiang Provincial Natural Science
Foundation of China (grant no. Y13H040023; to Dr Jiayong Zheng) and
the Wenzhou Science and Technology Foundation (grant no. Y20140408;
awarded to Dr Yanhui Jin, The First Affiliated Hospital of Wenzhou
Medical University, Wenzhou, China).
References
|
1
|
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa
M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M,
Akiguchi I, et al: CAG expansions in a novel gene for
Machado-Joseph disease at chromosome 14q32.1. Nat Genet. 8:221–228.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakano KK, Dawson DM and Spence A: Machado
disease. A hereditary ataxia in Portuguese emigrants to
Massachusetts. Neurology. 22:49–55. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosenberg RN, Nyhan WL, Bay C and Shore P:
Autosomal dominant striatonigral degeneration. A clinical,
pathologic, and biochemical study of a new genetic disorder.
Neurology. 26:703–714. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou YX, Takiyama Y, Igarashi S, Li YF,
Zhou BY, Gui DC, Endo K, Tanaka H, Chen ZH, Zhou LS, et al:
Machado-Joseph disease in four Chinese pedigrees: Molecular
analysis of 15 patients including two juvenile cases and clinical
correlations. Neurology. 48:482–485. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maciel P, Gaspar C, DeStefano AL, Silveira
I, Coutinho P, Radvany J, Dawson DM, Sudarsky L, Guimarães J,
Loureiro JE, et al: Correlation between CAG repeat length and
clinical features in Machado-Joseph disease. Am J Hum Genet.
57:54–61. 1995.PubMed/NCBI
|
|
6
|
Harding AE: Clinical features and
classification of inherited ataxias. Adv Neurol. 61:1–14.
1993.PubMed/NCBI
|
|
7
|
Zhuchenko O, Bailey J, Bonnen P, Ashizawa
T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY and Lee
CC: Autosomal dominant cerebellar ataxia (SCA6) associated with
small polyglutamine expansions in the alpha 1A-voltage-dependent
calcium channel. Nat Genet. 15:62–69. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stevanin G, Giunti P, Belal GD, Dürr A,
Ruberg M, Wood N and Brice A: De novo expansion of intermediate
alleles in spinocerebellar ataxia 7. Hum Mol Genet. 7:1809–1813.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanpei K, Takano H, Igarashi S, Sato T,
Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, et
al: Identification of the spinocerebellar ataxia type 2 gene using
a direct identification of repeat expansion and cloning technique,
DIRECT. Nat Genet. 14:277–284. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koide R, Ikeuchi T, Onodera O, Tanaka H,
Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, et
al: Unstable expansion of CAG repeat in hereditary
dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet. 6:9–13.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin Y, Zheng JY, Jin YH, Xie YC and Jin
ZB: Trinucleotide expansions in the SCA7 gene in a large family
with spinocerebellar ataxia and craniocervical dystonia. Neurosci
Lett. 434:230–233. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Todi SV, Scaglione KM, Blount JR, Basrur
V, Conlon KP, Pastore A, Elenitoba-Johnson K and Paulson HL:
Activity and cellular functions of the deubiquitinating enzyme and
polyglutamine disease protein ataxin-3 are regulated by
ubiquitination at lysine 117. J Biol Chem. 285:39303–39313. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reina CP, Zhong X and Pittman RN:
Proteotoxic stress increases nuclear localization of ataxin-3. Hum
Mol Genet. 19:235–249. 2010. View Article : Google Scholar
|
|
14
|
Warner JP, Barron LH, Goudie D, Kelly K,
Dow D, Fitzpatrick DR and Brock DJ: A general method for the
detection of large CAG repeat expansions by fluorescent PCR. J Med
Genet. 33:1022–1026. 1996. View Article : Google Scholar : PubMed/NCBI
|