Introduction
Rheumatoid arthritis (RA) is a common chronic
inflammatory joint disease, which is characterized by inflammation,
hyperplasia and destruction of joints (1,2).
Hypertrophic synovial tissue in RA is composed of fibroblast-like
synoviocytes (FLSs) and inflammatory cells, which secrete several
inflammatory cytokines (3).
Previous studies have demonstrated that tumor necrosis factor
(TNF)-α, which stimulates FLS proliferation, is critical in the
pathogenesis of RA (4–6). TNF-α also activates a broad array of
intracellular signaling mechanisms, including the nuclear factor
(NF)-κB pathway (7).
NF-κB is important in inflammatory regulation, and
several studies have suggested the role of NF-κB activation during
the development of RA (8–10). TNF-α stimulation activates NF-κB
signaling in FLSs and leads to the production of cytokines,
including TNF-α, interleukin (IL)6 and IL8 (11). In addition, there is a positive
feedback loop between NF-κB and TNF-α (12), which is critical during the
pathogenesis of RA. The inhibition of NF-κB signaling, particularly
antibody-mediated inhibition of TNF-α function, has been found to
significantly suppresses the progression of inflammation (13). These findings suggest that the
inhibition of NF-κB is an important therapeutic approach for the
treatment of RA (14–16).
Heparin is a sulfated glycosaminoglycan, which is
widely applied as an injectable anticoagulant (17,18).
Although used principally in medicine for anticoagulation, other
biological and physiological roles of heparin, and its underlying
mechanisms remain to be fully elucidated. Previous reports suggest
that heparin exerts inhibitory effects on NF-κB signaling in human
endothelial cells, suggesting it may function as an inhibitor of
FLS proliferation and lead to the progression of RA (19,20).
In the present study, the effects of heparin on the proliferation
and activation of NF-κB in TNF-α-stimulated FLSs were investigated.
The present study aimed to determine the importance of heparin in
FLSs, which may provide insights into the clinical impacts of this
anticoagulant in patients with RA.
Materials and methods
Reagents and antibodies
Recombinant TNF-α was obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). RPMI-1640, fetal bovine serum
(FBS), antibiotics, trypsin, phosphate-buffered saline (PBS) and
other products for cell culture were purchased from Invitrogen;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Phosphorylated
(p-) Akt, Akt, IL-6, IL-8, p-p65, p65, p-IκB, IκB and β-actin
antibodies were purchased from Cell Signaling Technology, Inc.
Heparin sodium for injection was purchased from Shanghai No. 1
Biochemistry & Pharmaceutical Co., Ltd. (Shanghai, China).
Isolation and culture of FLSs
The present study was approved by the First
Affiliated Hospital and Shengjing Hospital of China Medical
University (Shenyang, China). Informed consent was obtained from
all patients. The FLSs were isolated from primary synovial tissues
obtained from three patients with RA who had undergone joint
replacement surgery or synovectomy between 20013 and January 2014.
The synovial tissue was sectioned and digested with 0.25% trypsin
to isolate the synoviocytes, which were cultured in 5%
CO2 at 37°C. Following overnight culture, the
non-adherent cells were removed, and the adherent cells were
cultivated in RPMI-1640 supplemented with 10% FBS. The FLSs were
passaged following 0.25% trypsin treatment. FLSs in passages 3–8
were used in subsequent experiments. The cells were observed using
a light microscope and were morphologically homogeneous and
exhibited a typical bipolar configuration.
Treatment of the cells with TNF-α and
heparin
FLS cells were cultured in plates. When 80%
confluence was achieved, cells were treated with 10 ng/ml TNF-α
alone and TNF-α (10 ng/ml) with different concentrations of heparin
(0, 0.1, 1 and 5 U/ml) for 24, 48 and 72 h.
MTT assay
The cells were plated in 96-well plates (~3,000
cells per well) and cultured for 5 days. MTT solution (20 μl
of 5 mg/ml MTT) was added to each well. Following incubation for 4
h at 37°C, the medium was removed and the remaining MTT formazan
was dissolved in 150 μl DMSO. The absorbance of the solution
was measured at 490 nm using an absorbance microplate reader.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from fresh tissue samples
and cells with TRIzol reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNA was reversed to
cDNA using PrimeScript RT Master Mix (Takara, Biotechnology Co.,
Ltd., Dalian, China). A total 10 μl of reverse-transcription
reaction solution was prepared using 2 μl of 5X RT Master
Mix and 500 ng RNA. Reverse-transcription reaction was performed by
incubating the samples at 37°C for 15 min and then 85°C for 5 sec.
qPCR was performed as follows using 9 μl water, 1 μl
cDNA and 10 μl SYBR Green master mix (Thermo Fisher
Scientific, Inc.): 50°C for 2 min, 95°C for 10 min, 40 cycles of
95°C for 15 sec, 60°C for 60 sec. The RT-qPCR analysis was
performed using a 7900HT Real-Time PCR system (Thermo Fisher
Scientific, Inc.). β-actin was used as the reference gene. The
relative expression levels of target genes were calculated as ΔCq =
Cq gene - Cq reference, and the fold change in target gene
expression was calculated using the 2−ΔΔCq method
(21). Experiments were repeated
in triplicate. The primer sequences were as followers: TNF-α,
forward 5′-TGACTGTCGCCCGCAGTACG-3′ and reverse
5′-CGGCAATTTAGTGACACGCG-3′; IL-6, forward
5′-ACCGTCATCATGTCTGACCA-3′ and reverse 5′-TGGAACACCCTGTCTTTGAC-3′;
IL-8, forward 5′-ACCGTCATCATGTCTGACCA-3′ and reverse
5′-TGGAACACCCTGTCTTTGAC-3′; cyclin D1, forward
5′-GCTGGAGGTCTGCGAGGA-3′ and reverse 5′-ACAGGAAGCGGTCCAGGTAGT-3′;
β-actin, forward 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′and reverse
5′-CACCTTCTACAATGAGCTGCGTGTG-3′.
Western blot analysis
Total proteins from the cells were extracted in
lysis buffer and quantified using the Bradford method. A 30 mg
quantity of protein was separated by 10% SDS-PAGE. The samples were
then transferred onto PVDF membranes (EMD Millipore, Billerica, MA,
USA) and incubated overnight at 4°C with primary antibodies against
TNF-α (cat. no. 6945), IL-6 (cat. no. 12153) and cyclin D1 (cat.
no. 2978; 1:900; all from Cell Signaling Technology, Inc., Danvers,
MA, USA), IL-8 (cat. no. 7922; 1:500; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and actin (cat. no. 47778; 1:1,000; Santa
Cruz Biotechnology, Inc.). This was followed by incubation with
peroxidase-coupled anti-mouse or rabbit IgG antibody (1:1,000
dilution; Cell Signaling Technology, Inc.) at 37°C for 2 h. Target
proteins on the PVDF membrane were visualized using a Pierce ECL
kit (Pierce, Rockford, IL, USA) and images were captured using a
DNR Bio-Imaging system (DNR Bio-Imaging systems, Ltd., Jerusalem,
Israel).
Immunofluorescence staining
Following washing with cold PBS, The FLSs were fixed
with 4% formaldehyde in PBS for 10 min. Subsequently, the cells
were treated with 0.25% Triton X-100 (Sigma-Aldrich, St. Louis, MO,
USA). The cells were then washed three times in PBS for 5 min each,
followed by blocking for 10 min with 5% goat serum. The cells were
then incubated with TNF-α primary antibody diluted in PBS with 3%
bovine serum albumin (1:500; Beyotime Institute of Biotechnology,
Shanghai, China) overnight at 4°C. The cells were then incubated
with AlexaFluro 488 conjugated-goat anti-rabbit IgG for 2 h at room
temperature. Immunofluorescence was visualized under a fluorescence
microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analysis was performed using SPSS 12.0 software (SPSS,
Inc., Chicago, IL, USA). The results were compared using one-way
analysis of variance with post-hoc tests. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effect of heparin on the proliferation
rate of FLSs
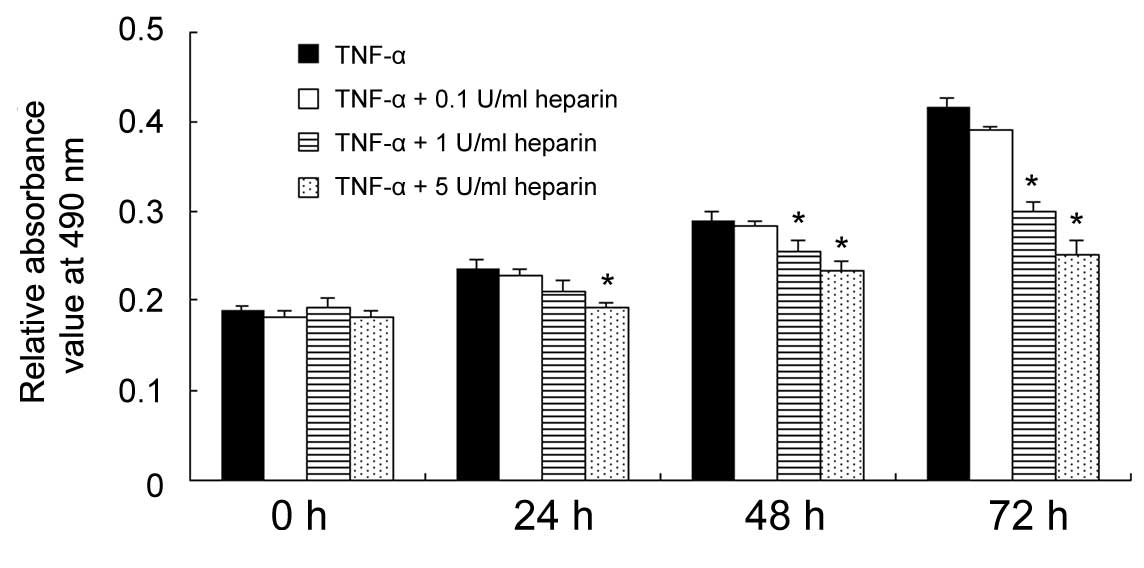
The effect of heparin on FLS growth rate was
determined using an MTT assay. The cells were treated with TNF-α
(10 ng/ml) and different concentrations of heparin (0, 0.1, 1 and 5
U/ml) for 24, 48 and 72 h. As shown in Fig. 1, heparin treatment, particularly at
concentrations of 1 and 5 U/ml, significantly inhibited the
proliferation rate of the FLSs induced by TNF-α treatment
(P<0.05; 5 U/ml group at 24 h; 1 and 5 U/ml groups at 48 h; 1
and 5 U/ml groups at 72 h).
Effect of heparin on TNF-α-induced mRNA
expression levels of the TNF-α, IL-6, IL-8 and cyclin D1 NF-κB
target genes in FLSs
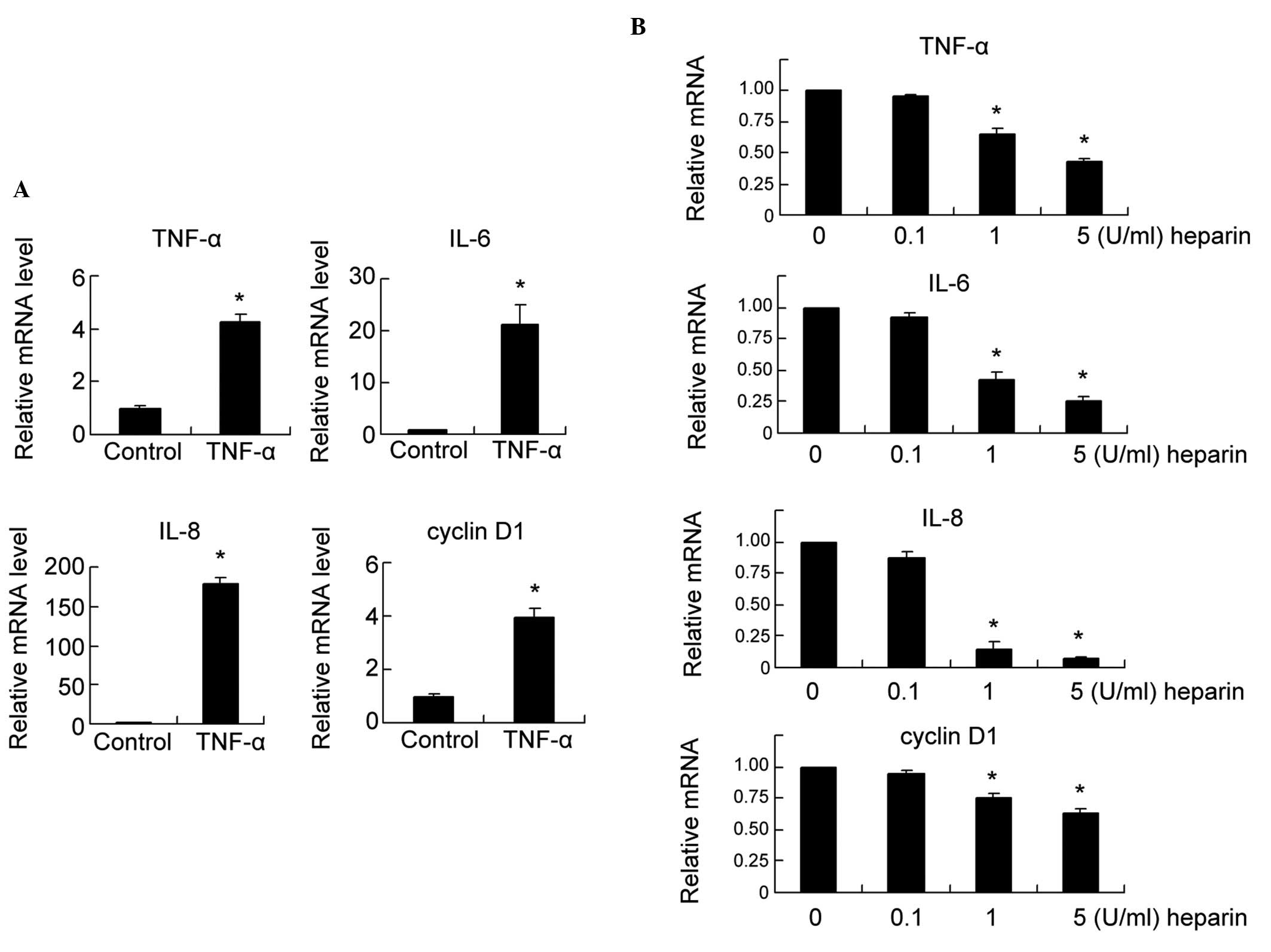
TNF-α is a downstream target of the NF-κB pathway,
which also activates NF-κB signaling. Previous studies have
reported a positive feedback loop between TNF-α and NF-κB signaling
(22–24). Therefore, the present study
examined the effect of TNF-α on the expression of NF-κB target
genes using RT-qPCR. As shown in Fig.
2A, 24 h of TNF-α treatment (10 ng/ml) significantly
upregulated the mRNA expression of the NF-κB target genes,
including TNF-α, IL-6, IL-8 and cyclin D1, compared with the
untreated control (P<0.05).
Subsequently, the role of heparin on the mRNA
expression levels of the TNF-α, IL-6, IL-8 and cyclin D1 NF-κB
target genes was investigated. The FLSs were treated with 10 ng/ml
of TNF-α for 24 h, following which the FLSs were exposed to
different concentrations of heparin (0, 0.1, 1 and 5 U/ml). As
shown in Fig. 2B, heparin
treatment decreased the mRNA levels of TNF-α, IL-6, IL-8 and cyclin
D1 in a dose-dependent manner (1 and 5 U/ml groups; P<0.05).
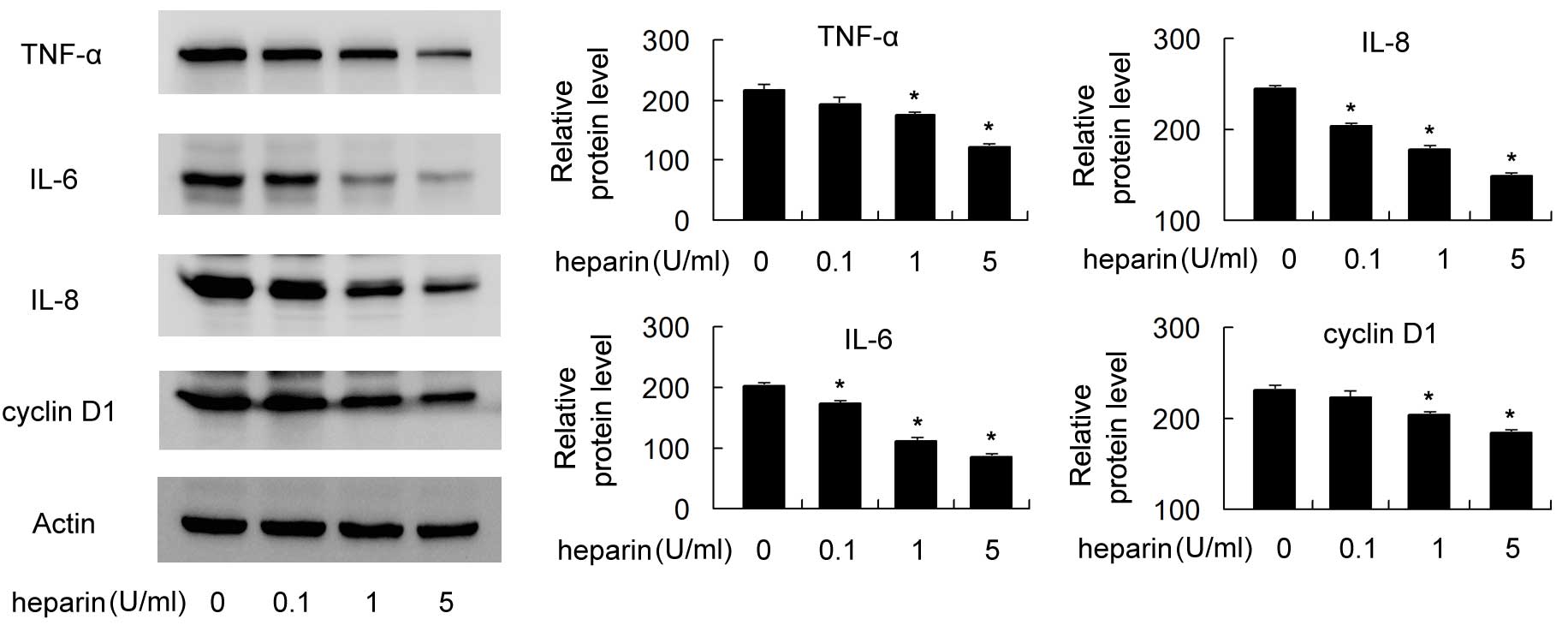
Effect of heparin on the protein
expression levels of TNF-α, IL-6, IL-8 and cyclin D1 in FLSs
Western blot analysis was used to examine
heparin-induced changes in the protein levels of TNF-α, IL-6, IL-8
and cyclin D1. The FLSs were treated with 10 ng/ml of TNF-α for 24
h, following which the FLSs were exposed to different
concentrations of heparin (0, 0.1, 1 and 5 U/ml) or another 24 h.
As shown in Fig. 3, heparin
treatment significantly decreased the protein levels of TNF-α,
IL-6, IL-8 and cyclin D1 in a dose-dependent manner (P<0.05). In
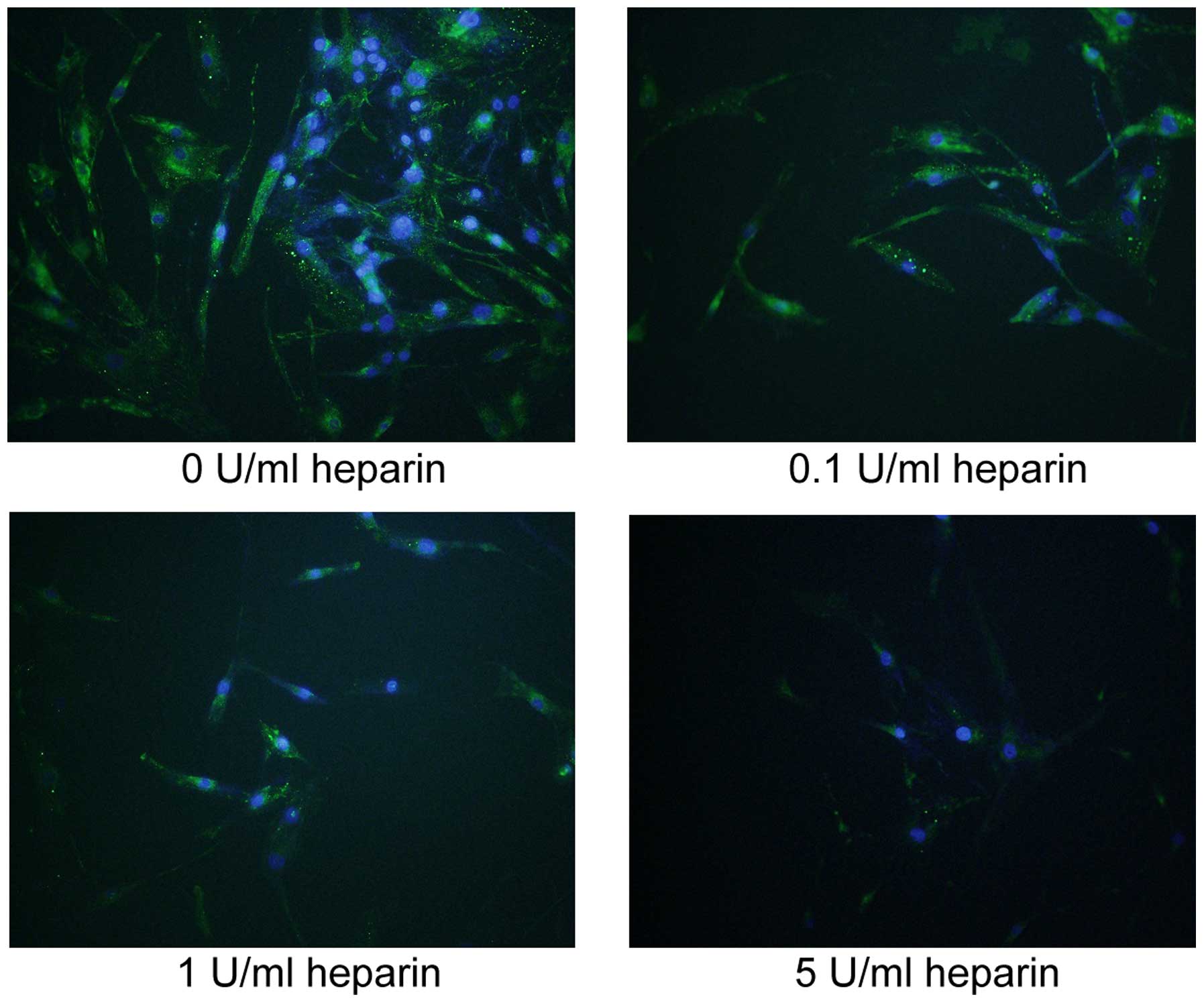
addition, immunofluorescence was used to examine protein expression
and localization of TNF-α in the heparin-treated FLS. As shown in
Fig. 4, TNF-α was located in the
cytoplasm of the FLSs, and heparin treatment dose-dependently
decreased the cellular protein expression of TNF-α.
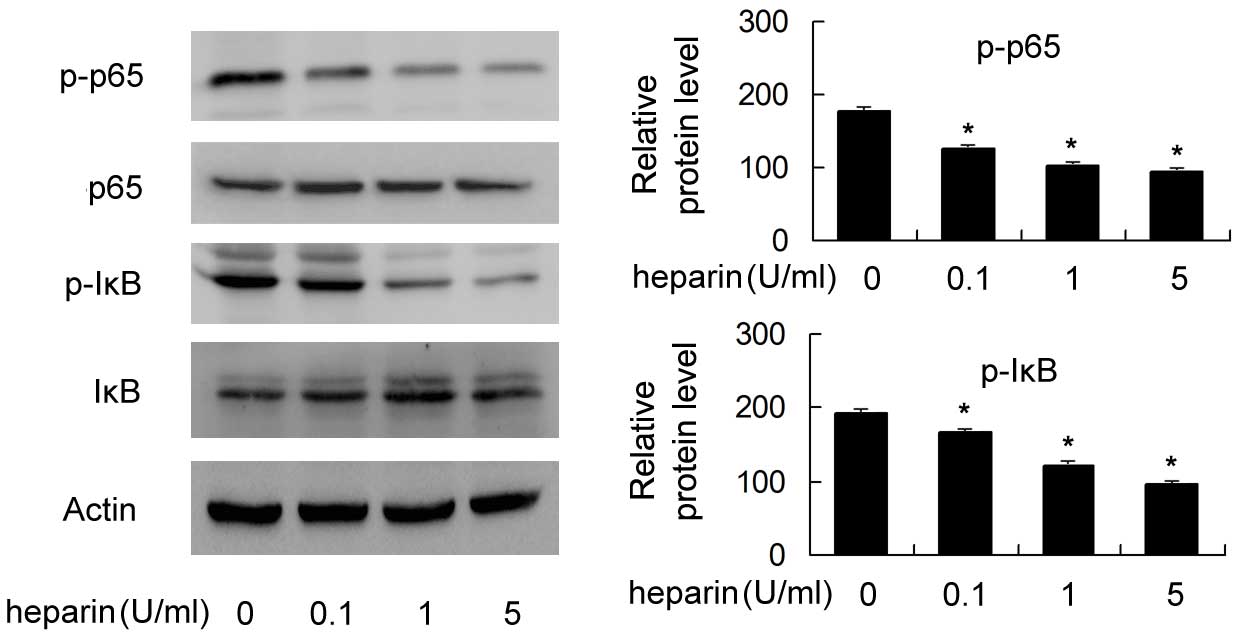
Effect of heparin on NF-κB activity
To investigate the mechanism by which heparin
inhibits proinflammatory cytokines, the present study examined the
effect of heparin on the phosphorylation of p65 and IκB, which are
key proteins of the NF-κB signaling pathway. As shown in Fig. 5, the levels of p-p65 and p-IκB were
significantly reduced following heparin treatment in a
dose-dependent manner, suggesting that heparin inhibited NF-κB
activity in the FLSs (P<0.05).
Discussion
In the present study, it was found that heparin (0,
0.1, 1 and 5 U/ml) dose-dependently inhibited TNF-α-induced NF-κB
activation and attenuated the TNF-α-induced production of IL-6,
IL-8, cyclin D1 and TNF-α in FLSs at mRNA and protein levels. In
addition, heparin reduced TNF-α-induced cell proliferation.
NF-κB signaling is important in the development of
inflammation-associated diseases, including RA. NF-κB signaling can
be induced by several stimuli, including cytokines, bacteria and
oxidative stress (25). Among
these stimuli, TNF-α is one of the most important cytokines
involved in the development of RA through the activation of several
signaling pathways, including NF-κB signaling (26). In addition, a previous study
reported the presence of a positive feedback loop between NF-κB and
TNF-α (12). Inhibition of the
function of TNF-α significantly suppresses the progression of
inflammation in RA and has become an important therapeutic approach
for the treatment of RA (27).
Heparin is a natural glycosaminoglycan, which is
widely used as an anticoagulant (19,28).
Aside from its anticoagulant function, it has other biological
functions, which remain to be fully elucidated. A previous study
suggested a role of heparin in the inhibition of NF-κB signaling
(19). However, the effects of
heparin on the process of inflammation in FLSs and the underlying
mechanisms have not been examined. In the present study, the role
of heparin in TNF-α-induced activation of NF-κB was investigated.
It was found that heparin attenuated TNF-α-induced NF-κB signaling
in a dose-dependent manner.
It has been reported that TNF-α can initiate joint
destruction and synovial inflammation during the development of RA
through the induction of inflammatory cytokines (2). The present study demonstrated that
the production of IL-6, IL-8 and cyclin D1 were significantly
enhanced in the FLSs treated with TNF-α. Heparin significantly
attenuated the TNF-α-induced production of TNF-α, IL-6, IL-8 and
cyclin D1 in the FLSs in a concentration-dependent manner,
suggesting its anti-inflammatory role in the RA process. This
TNF-α-induced production of cytokines was associated with NF-κB
activation in the FLSs. As heparin inhibited TNF-α-induced NF-κB
signaling, it decreased the expression of IL-6, IL-8, cyclin D1 and
TNF-α, possibly through the inhibition of NF-κB.
Heparin has been shown to attenuate the
proliferation of endothelial cells (29–31).
In the present study, it was found that heparin inhibited the
proliferation of the FLSs induced by TNF-α. In addition, heparin
inhibited the expression of cyclin D1, which is an NF-κB target and
acts as a cell cycle regulator involved in cell proliferation
(32). These results indicated
that cyclin D1 is critical in heparin-induced growth inhibition
through the inhibition of NF-κB. The molecular mechanism of
heparin-induced NF-κB inhibition remains to be elucidated. It was
previously reported that this effect may be associated with KLF-5
and growth factors, including EGF and VEGF (33,34).
The precise mechanism requires further investigation.
In conclusion, the present study demonstrated that,
by suppressing NF-κB activation, heparin inhibited TNF-α-induced
cell proliferation and the production of cytokines, including IL-6
and IL-8, in FLSs, suggesting that heparin may be beneficial in
preventing the progression of RA. Therefore, heparin may be a novel
therapeutic approach for the treatment of RA.
Acknowledgments
This study was supported by the Science and
Technology Project of Shenyang (grant no. F12-193-9-41).
References
|
1
|
Huber LC, Distler O, Tarner I, Gay RE, Gay
S and Pap T: Synovial fibroblasts: Key players in rheumatoid
arthritis. Rheumatology (Oxford). 45:669–675. 2006. View Article : Google Scholar
|
|
2
|
Noss EH and Brenner MB: The role and
therapeutic implications of fibroblast-like synoviocytes in
inflammation and cartilage erosion in rheumatoid arthritis. Immunol
Rev. 223:252–270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lafyatis R, Thompson NL, Remmers EF,
Flanders KC, Roche NS, Kim SJ, Case JP, Sporn MB, Roberts AB and
Wilder RL: Transforming growth factor-beta production by synovial
tissues from rheumatoid patients and streptococcal cell wall
arthritic rats. Studies on secretion by synovial fibroblast-like
cells and immuno-histologic localization. J Immunol. 143:1142–1148.
1989.PubMed/NCBI
|
|
4
|
Chu CQ, Field M, Feldmann M and Maini RN:
Localization of tumor necrosis factor alpha in synovial tissues and
at the cartilage-pannus junction in patients with rheumatoid
arthritis. Arthritis Rheum. 34:1125–1132. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chu CQ, Field M, Allard S, Abney E,
Feldmann M and Maini RN: Detection of cytokines at the
cartilage/pannus junction in patients with rheumatoid arthritis:
Implications for the role of cytokines in cartilage destruction and
repair. Br J Rheumatol. 31:653–661. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang HG, Wang Y, Xie JF, Liang X, Liu D,
Yang P, Hsu HC, Ray RB and Mountz JD: Regulation of tumor necrosis
factor alpha-mediated apoptosis of rheumatoid arthritis synovial
fibroblasts by the protein kinase Akt. Arthritis Rheum.
44:1555–1567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alonso-Ruiz A, Pijoan JI, Ansuategui E,
Urkaregi A, Calabozo M and Quintana A: Tumor necrosis factor alpha
drugs in rheumatoid arthritis: Systematic review and meta-analysis
of efficacy and safety. BMC Musculoskelet Disord. 9:522008.
View Article : Google Scholar
|
|
8
|
Li G, Zhang Y, Qian Y, Zhang H, Guo S,
Sunagawa M, Hisamitsu T and Liu Y: Interleukin-17A promotes
rheumatoid arthritis synoviocytes migration and invasion under
hypoxia by increasing MMP2 and MMP9 expression through NF-κB/HIF-1α
pathway. Mol Immunol. 53:227–236. 2013. View Article : Google Scholar
|
|
9
|
Criswell LA: Gene discovery in rheumatoid
arthritis highlights the CD40/NF-kappaB signaling pathway in
disease pathogenesis. Immunol Rev. 233:55–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Foxwell B, Browne K, Bondeson J, Clarke C,
de Martin R, Brennan F and Feldmann M: Efficient adenoviral
infection with IkappaB alpha reveals that macrophage tumor necrosis
factor alpha production in rheumatoid arthritis is NF-kappaB
dependent. Proc Natl Acad Sci USA. 95:8211–8215. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hwang SY, Kim JY, Kim KW, Park MK, Moon Y,
Kim WU and Kim HY: IL-17 induces production of IL-6 and IL-8 in
rheumatoid arthritis synovial fibroblasts via NF-kappaB- and
PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 6:R120–R128.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kagoya Y, Yoshimi A, Kataoka K, Nakagawa
M, Kumano K, Arai S, Kobayashi H, Saito T, Iwakura Y and Kurokawa
M: Positive feedback between NF-κB and TNF-α promotes
leukemia-initiating cell capacity. J Clin Invest. 124:528–542.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wendling D and Toussirot E:
TNF-alpha-targeted therapy in rheumatoid arthritis. Rev Rhum Engl
Ed. 66:187–191. 1999.PubMed/NCBI
|
|
14
|
Ding R, Li P, Song D, Zhang X and Bi L:
Predictors of response to TNF-α antagonist therapy in Chinese
rheumatoid arthritis. Clin Rheumatol. 34:1203–1210. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cavazzana I, Taraborelli M, Fredi M,
Tincani A and Franceschini F: Aseptic meningitis occurring during
anti-TNF-alpha therapy in rheumatoid arthritis and ankylosing
spondylitis. Clin Exp Rheumatol. 32:732–734. 2014.PubMed/NCBI
|
|
16
|
Sakthiswary R and Das S: The effects of
TNF α antagonist therapy on bone metabolism in rheumatoid
arthritis: A systematic review. Curr Drug Targets. 14:1552–1557.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oudemans-van Straaten HM, Kellum JA and
Bellomo R: Clinical review: Anticoagulation for continuous renal
replacement therapy-heparin or citrate? Crit Care. 15:2022011.
View Article : Google Scholar
|
|
18
|
Ramacciotti E, Clark M, Sadeghi N,
Hoppensteadt D, Thethi I, Gomes M and Fareed J: Review:
Contaminants in heparin: Review of the literature, molecular
profiling, and clinical implications. Clin Appl Thromb Hemost.
17:126–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Zheng Z, Li X and Ma X:
Unfractionated heparin inhibits lipopolysaccharide-induced
inflammatory response through blocking p38 MAPK and NF-κB
activation on endothelial cell. Cytokine. 60:114–121. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JH, Lee J, Seo GH, Kim CH and Ahn YS:
Heparin inhibits NF-kappaB activation and increases cell death in
cerebral endothelial cells after oxygen-glucose deprivation. J Mol
Neurosci. 32:145–154. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Crisostomo PR, Wang Y, Markel TA, Wang M,
Lahm T and Meldrum DR: Human mesenchymal stem cells stimulated by
TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B
- but not JNK-dependent mechanism. Am J Physiol Cell Physiol.
294:675–682. 2008. View Article : Google Scholar
|
|
23
|
Zheng Y, Ouaaz F, Bruzzo P, Singh V,
Gerondakis S and Beg AA: NF-kappa B RelA (p65) is essential for
TNF-alpha-induced fas expression but dispensable for both
TCR-induced expression and activation-induced cell death. J
Immunol. 166:4949–4957. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schütze S, Wiegmann K, Machleidt T and
Krönke M: TNF-induced activation of NF-kappa B. Immunobiology.
193:193–203. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McDonald PP, Bald A and Cassatella MA:
Activation of the NF-kappaB pathway by inflammatory stimuli in
human neutrophils. Blood. 89:3421–3433. 1997.PubMed/NCBI
|
|
26
|
Youn J, Kim HY, Park JH, Hwang SH, Lee SY,
Cho CS and Lee SK: Regulation of TNF-alpha-mediated hyperplasia
through TNF receptors, TRAFs, and NF-kappaB in synoviocytes
obtained from patients with rheumatoid arthritis. Immunol Lett.
83:85–93. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Emery P: Rheumatoid arthritis:
International disparities in access to anti-TNF therapy. Nat Rev
Rheumatol. 7:197–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ribic C, Lim W, Cook D and Crowther M:
Low-molecular-weight heparin thromboprophylaxis in medical-surgical
critically ill patients: A systematic review. J Crit Care.
24:197–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cao G, Wu JX and Wu QH: Low molecular
weight heparin suppresses lymphatic endothelial cell proliferation
induced by vascular endothelial growth factor C in vitro. Chin Med
J (Engl). 122:1570–1574. 2009.
|
|
30
|
Khorana AA, Sahni A, Altland OD and
Francis CW: Heparin inhibition of endothelial cell proliferation
and organization is dependent on molecular weight. Arterioscler
Thromb Vasc Biol. 23:2110–2115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cariou R, Harousseau JL and Tobelem G:
Inhibition of human endothelial cell proliferation by heparin and
steroids. Cell Biol Int Rep. 12:1037–1047. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS Jr: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ettelaie C, Fountain D, Collier ME, Elkeeb
AM, Xiao YP and Maraveyas A: Low molecular weight heparin
downregulates tissue factor expression and activity by modulating
growth factor receptor-mediated induction of nuclear factor-κB.
Biochim Biophys Acta. 1812:1591–1600. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Li X, Zheng Z, Liu Y and Ma X:
Unfractionated heparin suppresses lipopolysaccharide-induced
monocyte chemoattractant protein-1 expression in human
microvascular endothelial cells by blocking Krüppel-like factor 5
and nuclear factor-κB pathway. Immunobiology. 219:778–785. 2014.
View Article : Google Scholar : PubMed/NCBI
|