Introduction
Beta-ketothiolase deficiency, also termed
mitochondrial acetoacetyl-CoA thiolase (T2) deficiency (Online
Mendelian Inheritance in Man nos. 607809, 203750), is an inherited
autosomal recessive disease caused by mutations in the acetyl-CoA
acetyltransferase 1 (ACAT1) gene (1–4). T2
deficiency affects ketone body metabolism and isoleucine
catabolism. The first description of T2 deficiency was in 1971
(2). Currently, >100 cases have
been identified worldwide, with no ethnic predisposition (5).
T2 deficiency typically presents between 6 and 18
months of age with intermittent ketoacidotic episodes; patients are
generally asymptomatic between episodes. Urinary organic acid
analysis typically reveals increased excretion of
2-methyl-3-hydroxybutyrate (2M3HB), 2-methylacetoacetate (2M-AcAc),
and tiglylglycine (TIG). However, certain cases with atypical
clinical and/or biochemical presentations have been identified
(6). This disorder often has a
favorable outcome, when damaging ketoacidotic episodes are avoided
(3).
The human ACAT1 gene (that encodes T2) is
located on chromosome 11q22.3–23.1, spans ~27 kb, and comprises 12
exons and 11 introns (7,8). The human T2 cDNA is ~1.5 kb long and
encodes a precursor protein of 427 amino acids, including a
33-amino-acid leader polypeptide (9). Numerous different mutations (>50)
have been identified in ACAT1, 20% of which cause aberrant
splicing (10). Although the
majority of mutations that cause aberrant splicing are located at
splice acceptor or donor sites (11–15),
certain exonic mutations have been identified to activate cryptic
splice sites within their exons or alter the consensus sequences of
exonic splice enhancer (ESE) sites (16,17).
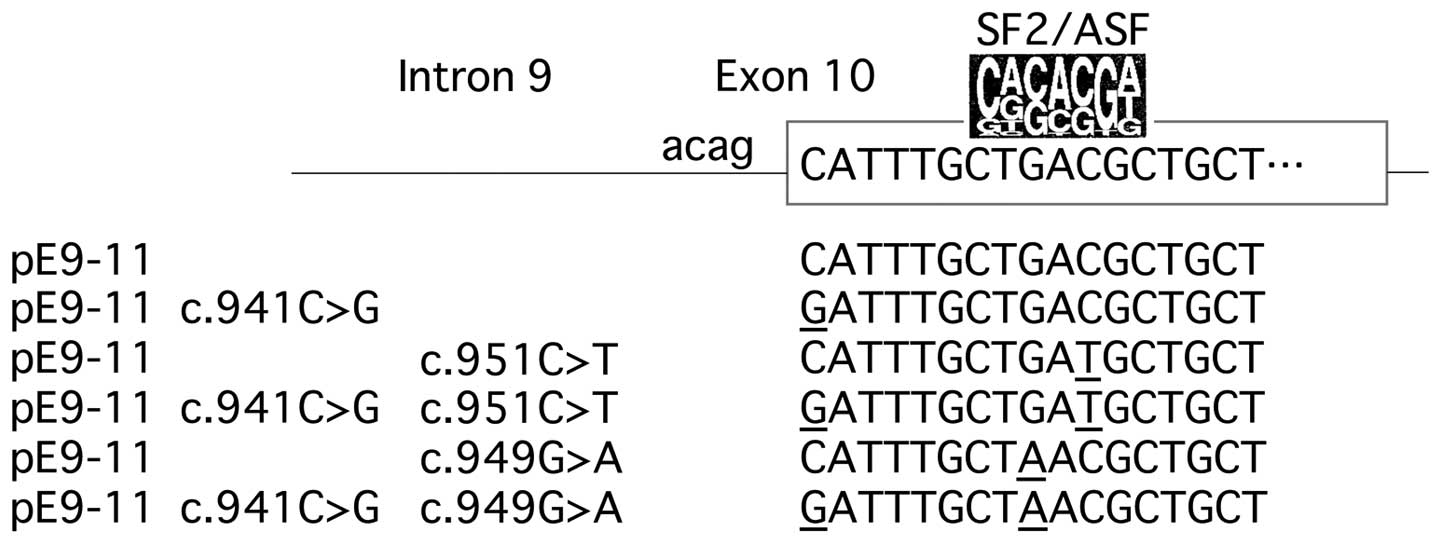
The present study reports a novel exonic mutation,
c.949G>A (nucleotide 9 in exon 10). A minigene splicing
experiment revealed that this mutation alters the sequence of an
ESE, serine/arginine-rich splicing factor 1 (SF2/ASF) binding site,
which results in exon 10 skipping.
Materials and methods
Patient clinical summary
The female patient (GK63) was born to
non-consanguineous German parents. She was in good health until 11
months of age when she was admitted to a hospital with vomiting,
acidotic breathing and somnolence. No hepatomegaly or cardiomegaly
was presented. Laboratory investigations revealed the following:
Blood pH 7.09; base excess, −22 mmol/l; blood glucose, 1.5 mmol/l;
lactate, 1.98 mmol/l; pyruvate, 0.1 mmol/l; and ketone bodies in
the urine. Urinary organic acid analysis at the time of a
ketoacidotic episode revealed excess excretion of 2M3HB, 2M-AcAc
and TIG. The patient was treated with intravenous glucose,
bicarbonate, fluids and electrolytes, and the condition improved
within 48 h. Repeated urinary organic acid analysis revealed excess
excretion of the metabolites listed above. The management plan
included avoiding prolonged fasting, protein restriction to 1.2–1.4
g/kg/day and 200 mg/kg/day L-carnitine supplementation. The patient
is currently 8 years old and has not experienced further episodes.
She has developed normally and has average intelligence
(IQ=99).
Mutation analysis
After parental consent was obtained, mutation
analysis was performed as part of the diagnostic work-up of the
patient using genomic DNA isolated from cultivated fibroblasts and
the SePaGene DNA extraction kit® (Sanko Junyaku Co.,
Ltd., Tokyo, Japan). Amplification of the 12 ACAT1 exons,
with their intron boundaries, was performed by polymerase chain
reaction (PCR) using primer pairs and conditions as previously
described (18). The 12 fragments
were sequenced using a BigDye® Terminator version 1.1
Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and an ABI PRISM® 3130xl genetic
analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
ESE identification
ESE finder version 3.0 (rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process)
was used to search for ESE sequences.
Minigene splicing experiment
A minigene construct, including a segment of
ACAT1 extending from the middle of exon 9 to the middle of
exon 11 (amplified by primers containing an EcoRI linker
sequence), was engineered using a pCAGGS eukaryote expression
vector as previously described (16,17,19,20).
This minigene construct produces a human T2-rabbit β-globin fusion
mRNA; therefore, reverse transcription-PCR amplification of this
specific mRNA was performed using a combination of a human T2 sense
primer and a rabbit β-globin antisense primer, as previously
described (16,17,19).
A KOD-Plus-Mutagenesis kit® (Toyobo Co., Ltd., Osaka,
Japan) was used to synthesize mutant constructs: c.949G>A
with/without c.941C>G (substitution of G for C at the first
nucleotide of exon 10); c.951C>T with/without c.941C>G; and
c. 941C>G (Fig. 1) (17).
Wild-type and mutant constructs were transfected
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) into 5×105 SV40-transformed
fibroblasts derived from a T2-deficient patient (GK03), which were
established by our group previously (9). A previous study demonstrated that, T2
mRNA was markedly decreased and T2 protein expression was virtually
undetectable in GK03 fibroblasts (9). RNA was extracted at 48 h
post-transfection using an ISOGEN kit® (Nippon Gene Co.,
Ltd., Tokyo, Japan), according to the manufacturer's instructions.
Transcription of the first-strand cDNA was performed using a rabbit
β-globin-specific antisense primer (β-glo2)
(5′-461AGCCACCACCTTCTGATA-3′), as described previously
(12). Amplification was performed
with the Ex9 (EcoRI) primer on T2 exon 9
(5′-cagctgcgaatt842CCAGTACACTGAATGATGGAGCAGCT873-3′,
lower case characters indicate linker sequence), and another
rabbit-specific antisense primer (β-glo3)
(5′-443GGCAGCCTGCACCTGAGGAGT-3′), as described
previously (12). Amplified
fragments were electrophoresed on a 5% polyacrylamide gel with
pUC13/HpaII DNA ladder marker, which was generated in our
lab using HpaII (Nippon Gene Co., Ltd.).
Transient expression analysis
Transient expression analysis of the D317N mutant
cDNA was performed using the pCAGGS eukaryotic expression vector
(Institute for Medical Genetics, Kumamoto University Medical
School, Kuhonji, Japan) (21) as
described previously (21,22). Following transfection, cells were
cultured at 37°C for 72 h, and then harvested and stored at −80°C
prior to use. The cells were freeze-thawed and sonicated in 50 mM
sodium phosphate (pH 8.0) containing 0.1% Triton X-100. Following
centrifugation at 10,000 × g for 10 min at 4°C, the
supernatant was used in an enzyme assay for acetoacetyl-CoA
thiolase activity, as previously described (22). The mean values and standard errors
of acetoacetyl-CoA thiolase activity, in the absence or presence of
potassium ions, of three independent experiments were
calculated.
Results
Mutation screening reveals a novel
mutation in the ACAT1 gene
Analysis demonstrated that patient GK63 was a
compound heterozygote, with a previously reported null mutation
(23), c.472A>G (N158D) and a
novel mutation, c.949G>A (D317N) in the ACAT1 gene,
located in exons 6 and 10, respectively. The latter mutation is
located at a potential SF2/ASF target ESE sequence, as is the
previously identified c.951C>T mutation (18). Genomic mutation screening
identified no further mutations.
c.949G>A results in exon 10
skipping
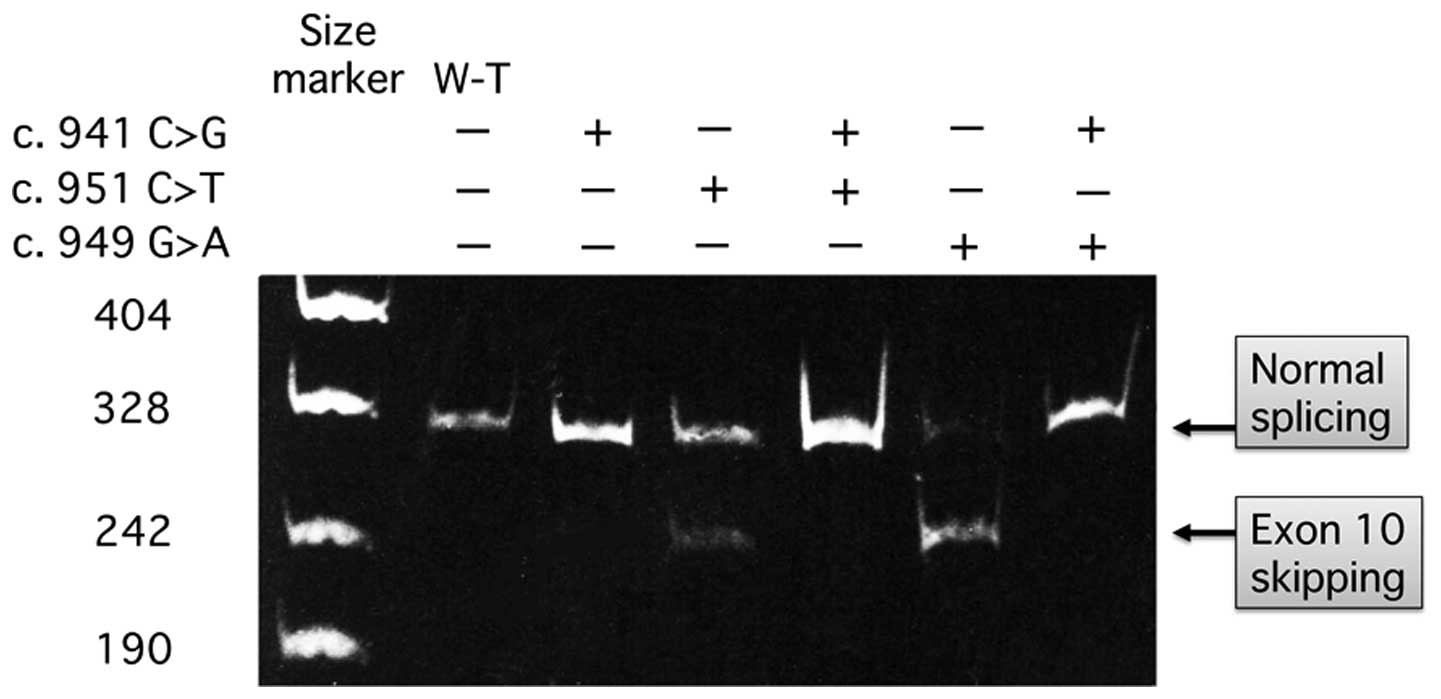
As presented in Fig.
2, in fibroblasts transfected with the minigenes, exon 10
skipping occurred in c.949G>A and c.951C>T mutant
transcripts. Normally spliced transcripts with the inclusion of
exon 10 were also produced from these mutant constructs. The
aberrant splicing was induced to a greater extent in c.949G>A
compared with c.951C>T mutant transcripts. The addition of the
c.941C>G mutation resulted in normal splicing from c.949G>A
and c.951C>T mutant constructs.
D317N mutant protein does not have
acetoacetyl-CoA thiolase activity
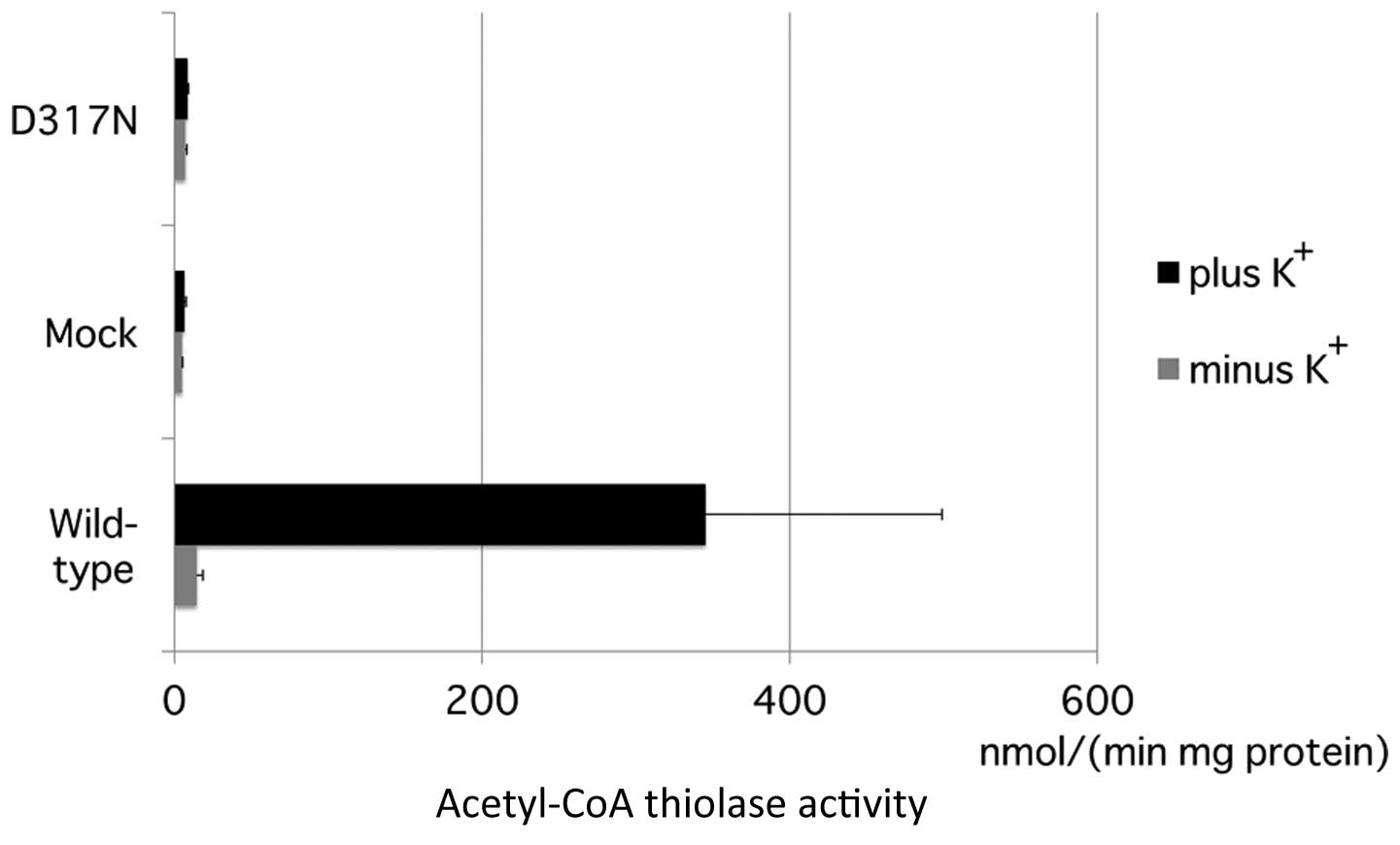
c.949G>A resulted in exon 10 skipping in the
majority of transcripts, however, normally spliced transcripts were
also detected in the minigene splicing experiment. Therefore, it
was investigated whether the D371N mutant protein retains residual
T2 activity via transient expression analysis of wild-type and
mutant cDNAs. Wild-type T2 protein produced high acetoacetyl-CoA
thiolase activity in the presence of potassium ions, which
represents T2 activity. D317N mutant protein did not retain any
potassium-ion-dependent acetoacetyl-CoA thiolase activity (Fig. 3). Based on the minigene splicing
experiment and transient expression analysis, c.949G>A was
determined to be a null mutation.
Discussion
The present study revealed that patient GK63 was a
compound heterozygote for a previously reported c.472A>G (N158D)
and a novel c.949G>A (D317N) mutation in the ACAT1 gene.
The latter mutation is a missense mutation and an ESE mutation,
which induces exon 10 skipping. This mutation is located at the
same codon as the previously reported c.951C>T mutation (D317D)
(17).
The accurate removal of introns from pre-mRNAs is
essential for functional gene expression. Splice sites, which
include the splice donor site, branch site and splice acceptor
site, do not contain all the information required for the precise
definition of exons (23–26); exonic sequences also contribute.
Regulatory elements in exons exist in the form of ESEs (23,24).
Exonic variants may inactivate an ESE, resulting in incorrect exon
inclusion. SF2/ASF is a prototypical serine- and arginine-rich (SR)
family protein and is an important protein for splicing and mRNA
metabolism. When bound to exonic sequences SR proteins mediate
recognition of the neighboring splice site (27).
Our previous study identified that the c.951C>T
mutation in ACAT1 caused exon 10 skipping (18). It was demonstrated that c.951C>T
is located within an ESE sequence for SF2/ASF (c.
947CTGACGC; from the nucleotide 7–13 of exon 10). A
minigene splicing experiment demonstrated that c.951C>T results
in aberrant splicing. Thus, c.951C>T, despite being a synonymous
substitution (D317D), was revealed to be a pathogenic mutation. In
addition, it was demonstrated that two additional nucleotide
substitutions located within the same ESE sequence, c.952G>A and
c.947C>T, caused exon 10 skipping in some transcripts (18). The novel mutation (c.949G>A) is
located within the same ESE sequence for SF2/ASF (c.
947CTGACGC); therefore, c.949G>A may affect splicing
in a similar manner to c.951C>T. In the absence of this
information, molecular analysis of DNA from patient GK63 may only
consider c.949G>A to be a missense mutation (D317N).
ESEs are more common in exons with weak splice sites
(28). G is the preferred first
nucleotide of an exon; however, ACAT1 exon 10 starts with C
(c.941C). Although the Shapiro and Senapathy score (26) of the splice acceptor site of intron
9 has a high score of 90.5, changing the first nucleotide of exon
10 from C to G (c.941C>G) further increases the score to 96.3
(26). It should be stressed that
the additional c.941C>G substitution abolished the aberrant
splicing and exon 10 skipping induced by all the ESE mutations in
the present study (c.949G>A and c.951C>T) and in our previous
study (c.951C>T, c.952G>A and c.947C>T) (18). This may indicate that the effect of
the ESE on splicing is weaker than the effect of G at position 941,
the preferable first nucleotide of exon 10.
A substitution in the ESE sequence for SF2/ASF in
exon 7 of the gene, survival of motor neuron 2 (SMN2) has
been well characterized to cause exon 7 skipping in about 90% of
transcripts (29). Although the
Shapiro and Senapathy score of the splice acceptor site of intron 6
is very high (99.7) and the first nucleotide of exon 7 is G,
substitution of only one nucleotide in the ESE causes exon 7
skipping in SMN2 (29,30).
This is in contrast with the findings of the present study.
Splicing is a complex process and numerous factors, including
ESEs/silencer and intronic splicing enhancer/silencer factors,
influence splicing efficiency together with splice acceptor/donor
sites (31).
In conclusion, the results of the present study
demonstrate that ACAT1 exonic mutations that affect ESE
sequences may result in aberrant splicing. This may affect the
activity of mitochondrial acetoacetyl-CoA thiolase. Ultimately,
minigene splicing experiments remain the most useful method to
detect the potential adverse effects of nucleotide substitution on
gene splicing.
Acknowledgements
The authors would like to thank Ms. Naomi Sakaguchi
(Department of pediatrics, Gifu University, Gifu, Japan) for
providing technical assistance. The present study was supported in
part by a Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan (grant
nos. 26114708, 24591505 and 16K09962), Health and Labour Science
Research Grants for Research on Intractable Diseases from the
Ministry of Health, Labour and Welfare of Japan, and the Practical
Research Project for Rare/Intractable Diseases from Japan Agency
for Medical Research and Development, AMED.
Glossary
Abbreviations
Abbreviations:
|
2M3HB
|
2-methyl-3-hydroxybutyrate
|
|
2M-AcAc
|
2-methylacetoacetate
|
|
ACAT1
|
acetyl-CoA acetyltransferase 1
|
|
ESE
|
exonic splice enhancer
|
|
SR
|
serine- and arginine-rich
|
|
T2
|
mitochondrial acetoacetyl-CoA
thiolase
|
|
TIG
|
tiglylglycine
|
References
|
1
|
Fukao T, Mitchell G, Sass JO, Hori T, Orii
K and Aoyama Y: Ketone body metabolism and its defects. J Inherit
Metab Dis. 37:541–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Daum RS, Lamm PH, Mamer OA and Scriver CR:
A ‘new’ disorder of isoleucine catabolism. Lancet. 2:1289–1290.
1971. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fukao T, Scriver CR and Kondo N: t2
Collaborative Working Group: The clinical phenotype and outcome of
mitochondrial acetoacetyl-CoA thiolase deficiency
(beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and
mutation-defined patients. Mol Genet Metab. 72:109–114. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sass JO: Inborn errors of ketogenesis and
ketone body utilization. J Inherit Metab Dis. 35:23–28. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hori T, Yamaguchi S, Shinkaku H, Horikawa
R, Shigematsu Y, Takayanagi M and Fukao T: Inborn errors of ketone
body utilization. Pediatr Int. 57:41–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abdelkreem E, Otsuka H, Sasai H, Aoyama Y,
Hori T, Abd El Aal M, Mahmoud S and Fukao T: Beta-ketothiolase
deficiency: Resolving challenges in diagnosis. Journal of Inborn
Errors of Metabolism & Screening. 4:2016. View Article : Google Scholar
|
|
7
|
Kano M, Fukao T, Yamaguchi S, Orii T,
Osumi T and Hashimoto T: Structure and expression of the human
mitochondrial acetoacetyl-CoA thiolase-encoding gene. Gene.
109:285–290. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Masuno M, Kano M, Fukao T, Yamaguchi S,
Osumi T, Hashimoto T, Takahashi E, Hori T and Orii T: Chromosome
mapping of the human mitochondrial acetoacetyl-coenzyme A thiolase
gene to 11q22.3-q23.1 by fluorescence in situ hybridization.
Cytogenet Cell Genet. 60:121–122. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fukao T, Yamaguchi S, Kano M, Orii T,
Fujiki Y, Osumi T and Hashimoto T: Molecular cloning and sequence
of the complementary DNA encoding human mitochondrial
acetoacetyl-coenzyme A thiolase and study of the variant enzymes in
cultured fibroblasts from patients with 3-ketothiolase deficiency.
J Clin Invest. 86:2086–2092. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fukao T, Maruyama S, Ohura T, Hasegawa Y,
Toyoshima M, Haapalainen AM, Kuwada N, Imamura M, Yuasa I, Wierenga
RK, et al: Three Japanese patients with beta-ketothiolase
deficiency who share a mutation, c.431A>C (H144P) in ACAT1:
Subtle abnormality in urinary organic acid analysis and blood
acylcarnitine analysis using tandem mass spectrometry. JIMD Rep.
3:107–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fukao T, Yamaguchi S, Orii T, Osumi T and
Hashimoto T: Molecular basis of 3-ketothiolase deficiency:
Identification of an AG to AC substitution at the splice acceptor
site of intron 10 causing exon 11 skipping. Biochim Biophys Acta.
1139:184–188. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fukao T, Yamaguchi S, Orii T, Schutgens
RB, Osumi T and Hashimoto T: Identification of three mutant alleles
of the gene for mitochondrial acetoacetyl-coenzyme A thiolase. A
complete analysis of two generations of a family with
3-ketothiolase deficiency. J Clin Invest. 89:474–479. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukao T, Song XQ, Yamaguchi S, Kondo N,
Orii T, Matthieu JM, Bachmann C and Hashimoto T: Identification of
three novel frameshift mutations (83delAT, 754insCT, and 435 + 1G
to A) of mitochondrial acetoacetyl-coenzyme A thiolase gene in two
Swiss patients with CRM-negative beta-ketothiolase deficiency. Hum
Mutat. 9:277–279. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thummler S, Dupont D, Acquaviva C, Fukao T
and de Ricaud D: Different clinical presentation in siblings with
mitochondrial acetoacetyl-CoA thiolase deficiency and
identification of two novel mutations. Tohoku J Exp Med. 220:27–31.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Law CY, Lam CW, Ching CK, Yau KC, Ho TW,
Lai CK and Mak CM: NMR-based urinalysis for beta-ketothiolase
deficiency. Clin Chim Acta. 438:222–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fukao T, Yamaguchi S, Wakazono A, Orii T,
Hoganson G and Hashimoto T: Identification of a novel exonic
mutation at −13 from 5′ splice site causing exon skipping in a girl
with mitochondrial acetoacetyl-coenzyme A thiolase deficiency. J
Clin Invest. 93:1035–1041. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukao T, Horikawa R, Naiki Y, Tanaka T,
Takayanagi M, Yamaguchi S and Kondo N: A novel mutation
(c.951C>T) in an exonic splicing enhancer results in exon 10
skipping in the human mitochondrial acetoacetyl-CoA thiolase gene.
Mol Genet Metab. 100:339–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fukao T, Nakamura H, Song XQ, Nakamura K,
Orii KE, Kohno Y, Kano M, Yamaguchi S, Hashimoto T, Orii T and
Kondo N: Characterization of N93S, I312T, and A333P missense
mutations in two Japanese families with mitochondrial
acetoacetyl-CoA thiolase deficiency. Hum Mutat. 12:245–254. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fukao T, Boneh A, Aoki Y and Kondo N: A
novel single-base substitution (c.1124A>G) that activates a
5-base upstream cryptic splice donor site within exon 11 in the
human mitochondrial acetoacetyl-CoA thiolase gene. Mol Genet Metab.
94:417–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Watanabe H, Orii KE, Fukao T, Song XQ,
Aoyama T, IJlst L, Ruiter J, Wanders RJ and Kondo N: Molecular
basis of very long chain acyl-CoA dehydrogenase deficiency in three
Israeli patients: Identification of a complex mutant allele with
P65L and K247Q mutations, the former being an exonic mutation
causing exon 3 skipping. Hum Mutat. 15:430–438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niwa H, Yamamura K and Miyazaki J:
Efficient selection for high-expression transfectants with a novel
eukaryotic vector. Gene. 108:193–199. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang GX, Fukao T, Rolland MO, Zabot MT,
Renom G, Touma E, Kondo M, Matsuo N and Kondo N: Mitochondrial
acetoacetyl-CoA thiolase (T2) deficiency: T2-deficient patients
with ‘mild’ mutation were previously misinterpreted as normal by
the coupled assay with tiglyl-CoA. Pediatr Res. 56:60–64. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goldstrohm AC, Greenleaf AL and
Garcia-Blanco MA: Co-transcriptional splicing of pre-messenger
RNAs: Considerations for the mechanism of alternative splicing.
Gene. 277:31–47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cooper TA and Mattox W: The regulation of
splice-site selection, and its role in human disease. Am J Hum
Genet. 61:259–266. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robberson BL, Cote GJ and Berget SM: Exon
definition may facilitate splice site selection in RNAs with
multiple exons. Mol Cell Biol. 10:84–94. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shapiro MB and Senapathy P: RNA splice
junctions of different classes of eukaryotes: Sequence statistics
and functional implications in gene expression. Nucleic Acids Res.
15:7155–7174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin S and Fu XD: SR proteins and related
factors in alternative splicing. Adv Exp Med Biol. 623:107–122.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caceres EF and Hurst LD: The evolution,
impact and properties of exonic splice enhancers. Genome Biol.
14:R1432013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cartegni L, Hastings ML, Calarco JA, de
Stanchina E and Krainer AR: Determinants of exon 7 splicing in the
spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet.
78:63–77. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh NN, Androphy EJ and Singh RN: An
extended inhibitory context causes skipping of exon 7 of SMN2 in
spinal muscular atrophy. Biochem Biophys Res Commun. 315:381–388.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakamura K, Fukao T, Perez-Cerda C, Luque
C, Song XQ, Naiki Y, Kohno Y, Ugarte M and Kondo N: A novel
single-base substitution (380C>T) that activates a 5-base
downstream cryptic splice-acceptor site within exon 5 in almost all
transcripts in the human mitochondrial acetoacetyl-CoA thiolase
gene. Mol Genet Metab. 72:115–121. 2001. View Article : Google Scholar : PubMed/NCBI
|