Introduction
Hepatitis B virus (HBV) infection is a major global
health issue, which has affected >2 billion people and results
in 0.5–1.2 million mortalities per year. There are ~350 million HBV
chronic carriers worldwide. Infection by HBV results in acute and
chronic liver diseases and may lead to chronic hepatitis,
cirrhosis, and hepatocellular carcinoma (1,2). Two
types of therapeutic agents are available for treatment of HBV
infection, interferon (IFN), and nucleoside and nucleotide
analogues (NA) (3). IFN exerts its
antiviral action by targeting the double-stranded RNA-activated
protein kinase R (PKR) (4). PKR
phosphorylates eukaryotic initiation factor-2α (eIF2α), a protein
synthesis initiation factor, and reduces the level of viral protein
synthesis (5). Theoretically, IFN
may be an ideal agent for treatment of HBV infection, however, the
response rate of interferon α is only 30–40% in HBV envelope
antigen-positive patients after 4–6 months IFN treatment (1). Other disadvantages of IFN include its
side effects and high costs (6).
Patients who use IFN may require symptomatic treatment, dose
modification or discontinuation of therapy (1). NAs, including lamivudine, tenofovir,
telbivudine and adefovir, have strong antiviral effects, however,
the development of drug resistance has limited their clinical
applications (7,8).
3-Hydroxy-3-methylglutaryl coenzyme A reductase
inhibitors, also known as statins, are widely used in the treatment
of hypercholesterolemia, however, they have also been reported to
inhibit hepatitis C virus (9) and
cytomegalovirus (10). HBV has
also been observed to be inhibited by statins, including
simvastatin (SIM) (11). However,
the mechanism underlying the inhibition of HBV by SIM remains to be
elucidated. Previously, it has been demonstrated that statins
inhibit vascular smooth muscle cell growth by downregulating
minichromosome maintenance (MCM) proteins (12). MCM proteins have ten conserved
factors functioning in gene replication (13). Of the ten conserved factors, MCM2-7
are connected to each other to form a complex. The MCM2-7 complex
acts as a replicative DNA helicase to regulate the initiation of
DNA synthesis (14,15). The complex is important in
restricting DNA replication to a single round per cell cycle
(16). During the G1 phase cell
division cycle (Cdc) 6 and DNA replication factor Cdt1 recruit the
MCM2-7 complex to form an origin recognition complex at the
replication origin. During S phase, the MCM2-7 complex is
phosphorylated by the Cdc7-Dbf4 kinase (17) and then changed conformation,
resulting in its association with Cdc45 at the replication origin.
The formation of Cdc45-MCM complex initiates the duplex DNA
unwinding and recruits various replication proteins to the unwound
DNA, initiating DNA synthesis (18–20).
MCM proteins interact with each other to form various complexes,
including MCM2-7, MCM4/6/7, MCM2/4/6/7, or MCM3/5 (21). Biochemical investigations into
these complexes have indicated that only the dimeric complex of the
MCM4/6/7 heterotrimer has DNA helicase, single-stranded DNA
binding, and DNA-dependent ATPase activities (22).
The MCM complex is an important host replication
factor that participates in the genome replication of viruses in
host cells, such as the influenza virus (23). It has been demonstrated that
nuclear MCM7 is correlated with hepatitis B virus infection
(P=0.020) (24). Our preliminary
experiments also indicated that the expression of exogenous green
fluorescent protein (GFP), which was transfected by adenoviral
vectors, was decreased when MCM7 was silenced by small interfering
RNA (siRNA) in murine normal fibroblast NIH3T3 cells. It indicated
that MCM7 silencing may contribute to the inhibition of adenoviral
vectors, a DNA virus. Thus, the present study hypothesized that SIM
attenuated the expression of HBV DNA via an MCM-dependent
mechanism.
The current study demonstrated for the first time,
to the best of our knowledge, that SIM suppressed HBV expression
levels by reducing the expression of MCM7 protein at the
translational level. The results of the present study also
demonstrated that the translational inhibition of MCM7 induced by
SIM may be associated with the increasing phosphorylation of eIF2α.
In addition, the LKB1-AMPK signaling pathway may be involved in the
phosphorylation of eIF2α as a result of SIM. Overall, the findings
of the current study demonstrated that decreasing MCM7 expression
by SIM at the translational level may contribute to inhibition of
HBV.
Materials and methods
Reagents
SIM was purchased from Merck Millipore (Darmstadt,
Germany). SIM (4 mg) was dissolved in 100 µl ethanol and 150 µl 0.1
M NaOH, incubated at 50°C for 2 h, and adjusted to pH 7.0 with HCl.
The final volume was corrected to 1 ml by adding absolute ethyl
alcohol. Antibodies against MCM7 (sc-9966; 1:500), GFP (sc-8334;
1:1,000) phosphorylated (p)-retinoblastoma (Rb; Ser567) (sc-32824;
1:500), Rb (sc-50; 1:1,000), cyclin D1 (sc-4074; 1:1,000), tumor
protein P53 (p53; sc-126; 1:1,000), LKB1 (sc-32245; 1:500), β-actin
(sc-47778; 1:1,000) and horse-radish peroxidase (HRP)-conjugated
anti-rabbit (sc-2370; 1:5,000) or anti-mouse (sc-2383; 1:5,000) IgG
were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Antibodies against cyclin-dependent kinase inhibitor 1B (p21;
10355-1-AP; 1:1,000), cyclin-dependent kinase inhibitor 1B (p27;
25614-1-AP; 1:1,000) and PERK (20582-1-AP; 1:1,000) were purchased
from Wuhan Sanying Biotechnology (Wuhan, China). p-eIF2α (Ser51;
#9721; 1:1,000) and p-AMPKα (Thr172; #2531; 1:500) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) was used for the siRNA transfection. The Cell
Cycle Detection kit (KGA511-KGA512) was purchased from Nanjing
KeyGen Biotech, Co., Ltd. (Nanjing, China). MG132 (M8699) and
lamivudine (Y0000426) was purchased from Sigma-Aldrich (Merck
Millipore).
Cell culture
The HepG2.2.15 human HBV-transfected liver cells
line used in the present study was kindly provided by School of
Basic Medical Sciences, Fourth Military Medical University (Xi'an,
China). HepG2.2.15 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM; Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA) supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a water-saturated
atmosphere of 5% CO2. SIM was added into the medium at different
concentrations and the medium was changed every two days. Cells
were divided into three groups according to the concentrations of
SIM added into the medium: i) Control group (not treated with SIM);
ii) low concentration group (treated with 5 µM SIM); and iii) high
concentration group (treated with 40 µM SIM).
Cell proliferation assay
Cell proliferation was evaluated using a cell growth
curve and a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium
bromide (MTT) assay. Exponentially growing cells
(3.0×104 cells/well) were seeded into 96-well plates
(Corning Incorporated, Corning, NY, USA) and cultured in DMEM
supplemented with 10% FBS. After 24 h, cells were added to the
medium containing different concentrations of SIM (control, 5, or
40 µM) and cultured for 7 days at 37°C. To create cell growth
curves, cell numbers were counted at indicated times. The MTT assay
was performed by adding 20 µl MTT solution (5 mg/ml) into each
well. Following incubation for an additional 4 h at 37°C, the
medium was removed and 150 µl dimethyl sulfoxide (Sigma-Aldrich;
Merck Millipore) was added to each well to dissolve the resultant
formazan crystals. The absorbance was measured at 570 nm using a
microplate reader (PerkinElmer, Inc., Waltham, MA, USA).
Cell cycle analysis
Cells were seeded in six-well plates and treated
with 5 or 40 µM of SIM for 1 and 4 days at 37°C in a humidified
atmosphere of 5% CO2. Cells were subsequently collected by
trypsinization. Samples of at least 1×106 cells were
stored in ice-cold 70% ethanol for at least 2 h at 4°C, washed
twice with phosphate-buffered saline (PBS) and stained with a
solution containing 50 mg/ml propidium iodide (PI) and 50 mg/ml
RNase at room temperature for 30 min in the dark. Cell cycle
analysis was performed by using a FACSCalibur flow cytometer (BD
Biosciences, San Jose, CA, USA) and the CellQuest Pro software,
version 6.0 (BD Biosciences).
Protein extraction and western blot
analysis
Following treatment with 5 or 40 µM SIM in 100 mm
diameter cell culture dishes for 1 and 4 days, cells were washed
twice with ice-cold PBS, and lysed in radioimmunoprecipitation
lysis buffer with protease inhibitor and phosphatase inhibitor
cocktails (Roche Diagnostics GmbH, Mannheim, Germany). Cell lysates
were centrifuged at 12,000 × g for 20 min at 4°C and the
supernatant was harvested. The protein concentration was determined
using a Pierce BCA Protein assay kit (Thermo Fisher Scientific,
Inc.). Protein samples (100 µg) were separated electrophoretic ally
using a 10% sodium dodecyl sulfate polyacrylamide gel
electrophoresis gel and transferred to a polyvinylidene fluoride
membrane (EMD Millipore, Billerica, MA, USA). The membrane was
blocked with 5% (w/v) non-fat dry milk in 1 M Tris buffer saline
(pH 7.4), 5 M NaCl and 0.1% Tween-20 (TBST) for 1 h at 37°C.
Subsequently, the membrane was incubated at 4°C overnight in 5%
non-fat dry milk in TBST containing primary antibodies. Subsequent
to washing with TBST three times for 10 min each time, the membrane
was incubated with a HRP-conjugated anti-rabbit or anti-mouse IgG
secondary antibodies for 1 h at room temperature. Signals were
detected using the Immobilon™ Western Chemiluminescent HRP
Substrate reagent (EMD Millipore).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following exposure of the cells to 5 or 40 µM SIM
for 1 and 4 days, total RNA was extracted from cells using
E.Z.N.A®. Total RNA Kit I (Omega Bio-Tek, Inc.,
Norcross, GA, USA) according to the manufacturer's protocols. Total
RNA (2 µg) was reverse transcribed using the PrimeScript™ RT Master
Mix kit (RR036a; Takara Biotechnology, Co., Ltd., Dalian, China)
according to the manufacturer's protocols. qPCR was performed using
an iQ5 system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Reactions were performed using GoTaq qPCR Master mix (Promega
Corporation, Madison, WI, USA). The thermocycling parameters were
as follows: One cycle of 50°C for 2 min; one cycle of 95°C for 10
min; and 40 amplification cycles of 95°C for 15 sec and 60°C for 1
min. The following primers were used: MCF7, F
5′-GCTGATTGCCGTACAAGAG-3′ and R 5′-AGCAGGGTACTGGTTCTG-3′; GAPDH, F
5′-CTCCTCCACCTTTGACGCTG-3′ and R 5′-TCCTCTTGTGCTCTTGCTGG-3′. MCM7
gene expression were defined as MCM7 mRNA expression normalized to
GAPDH mRNA expression. The 2−∆∆Cq method was used to
calculate relative expression levels (25).
Silencing of MCM7 by small interfering
RNAs
siRNA (100 pmol) and 5 µl Lipofectamine™ 2000 were
diluted in 245 µl of opti-MEM medium (Gibco; Thermo Fisher
Scientific, Inc.), respectively. Diluted siRNA and Lipofectamine™
2000 reagents were mixed and incubated at room temperature for 20
min. The siRNA-lipid complex was added to cells which had been
grown to 70–80% confluence in six-well plates and the cells were
incubated at 37°C in a 5% CO2 incubator. The sequences of sense and
antisense primers (obtained from Thermo Fisher Scientific, Inc.)
were as follows: Sense, 5′-AUCGGAUUGUGAAGAUGAATT-3′ and antisense,
5′-UUCAUCUUCACAAUCCGAUTT-3′ for MCM7 siRNA; and sense,
5′-UAGCGACUAAACACAUCAATT-3′ and antisense,
5′-UUGAUGUGUUUAGUCGCUATT-3′ for the negative control siRNA.
Detection of HBV DNA in HepG2.2.15
cells and cell culture supernatants
Expression levels of HBV DNA in HepG2.2.15 cells and
cell culture supernatants were collected by centrifugation at 3,220
× g for 5 min at room temperature (22–25°C) and were quantified by
fluorescence qPCR (FQ-PCR) using the HBV PCR kit purchased from
DAAN Gene Co., Ltd. (Guangzhou, China) according to the
manufacturer's protocols. The thermocycling parameters were as
follows: One cycle of 45°C for 10 min; one cycle of 95°C for 15
min; and 40 amplification cycles of 95°C for 15 sec and 58°C for 1
min.
Statistical analysis
Statistical analyses were performed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA). At least three replicate
experiments were conducted for each group and results were
presented as the mean ± standard deviation. Statistical differences
between groups were determined by using Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
MCM7 silencing inhibited HBV DNA
replication
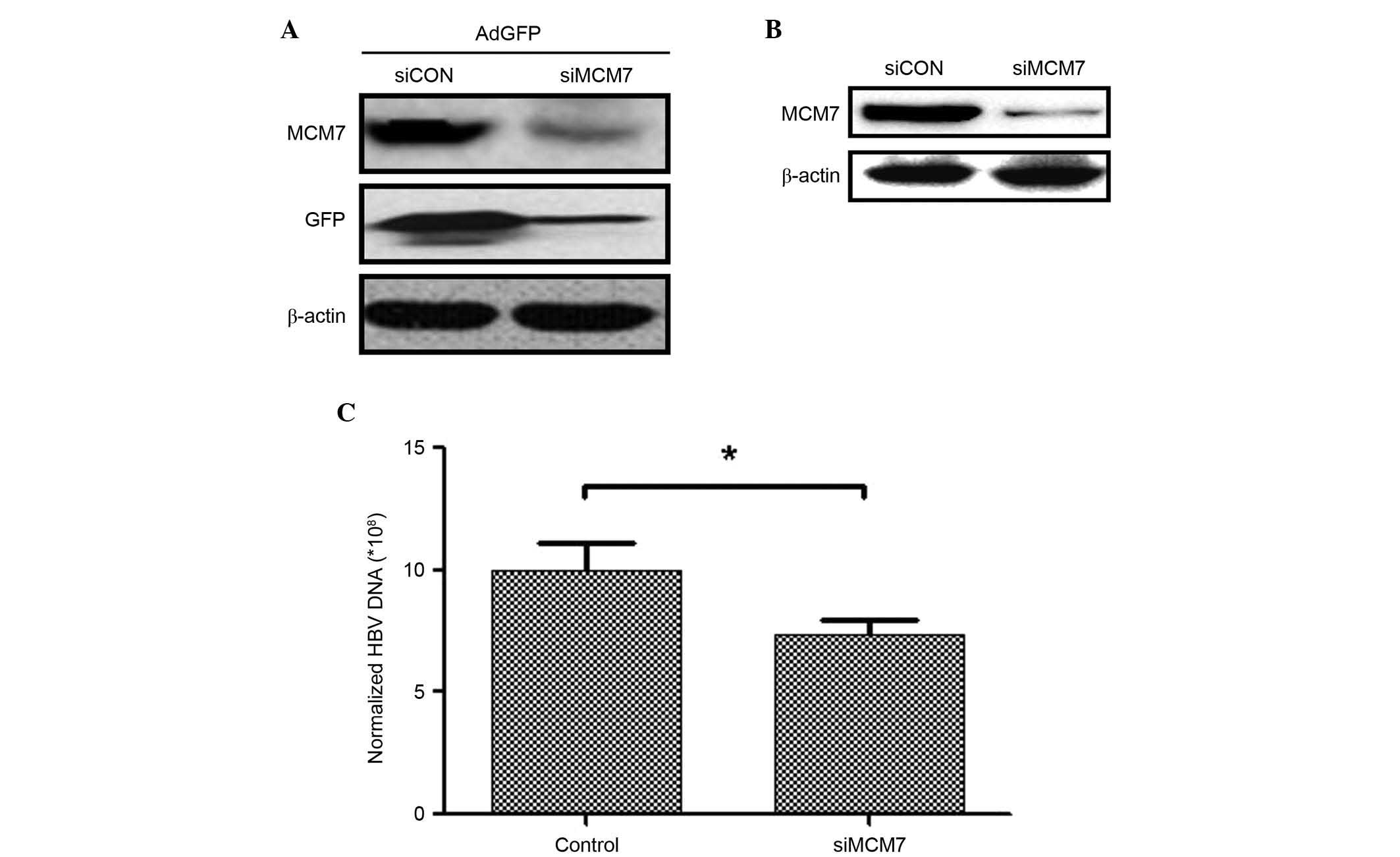
Our preliminary experiments indicated that the
expression of GFP was decreased when MCM7 was silenced by siRNA in
NIH3T3 cells transfected with AdGFP (Fig. 1A). In the present study, MCM7 was
silenced by transfecting siRNA into HepG2.2.15 cells to investigate
the association between MCM7 and HBV DNA replication. The MCM7
protein expression level was downregulated following siMCM7
transfection (Fig. 1B). In
addition, protein concentrations of HepG2.2.15 and
siMCM7-transfected HepG2.2.15 cells were determined to normalize
the expression level of HBV DNA. The results indicated that the HBV
DNA expression level was significantly decreased 7 days after
downregulating MCM7 expression (P<0.05; Fig. 1C). These results indicated that
MCM7 was associated with increased HBV DNA replication and that
silencing of MCM7 resulted in downregulation of HBV DNA
expression.
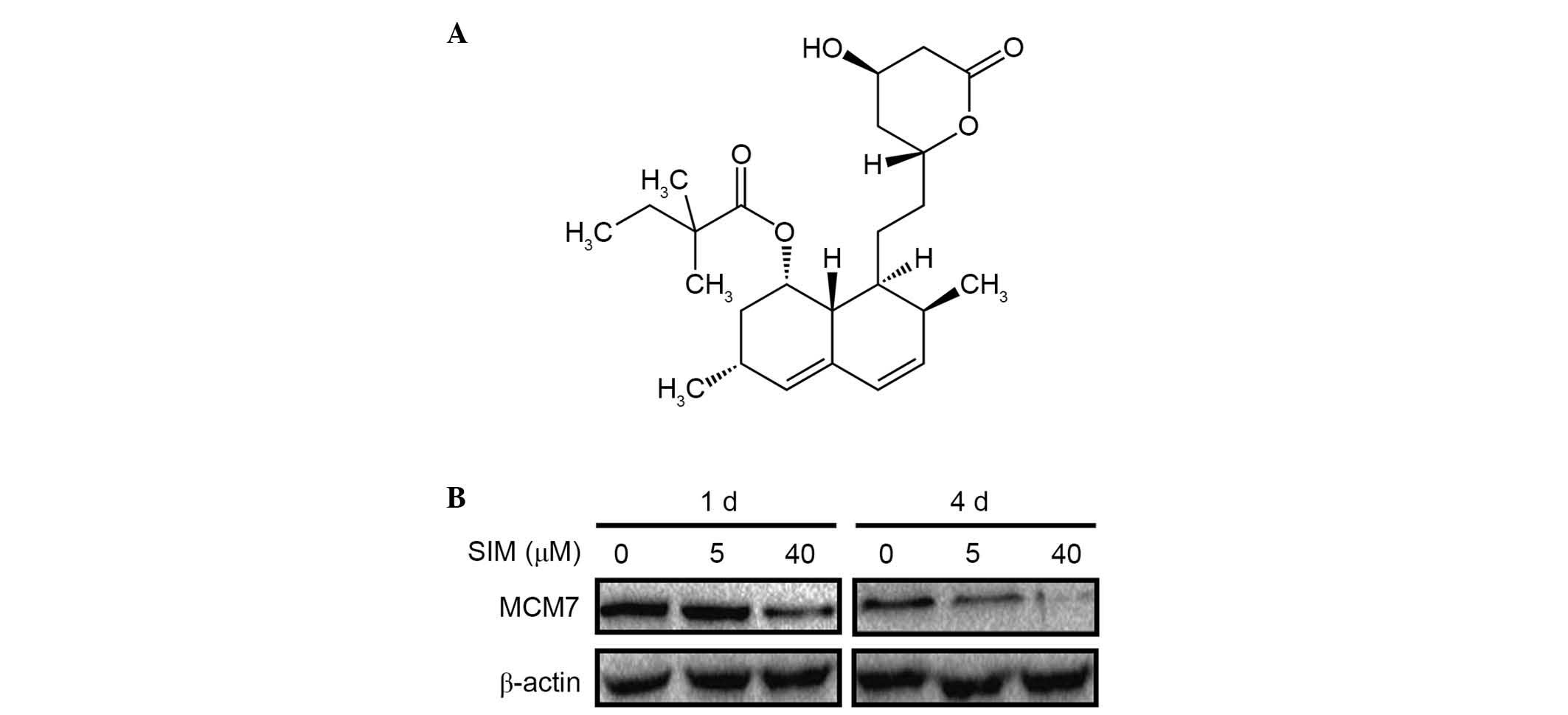
SIM treatment inhibited MCM7 protein
expression
HepG2.2.15 cells were treated with 5 or 40 µM SIM
(C25H38O5; molecular weight, 418.57 Da; Fig. 2A). Protein expression levels of
MCM7 protein were determined by western blotting at 1 and 4 days
after treatment. Notably, on day 1 of the treatment the expression
levels of the MCM7 protein was slightly downregulated only in the
high (40 µM) dose SIM treatment group, while, on day 4, the protein
expression levels of MCM7 decreased significantly in low (5 µM) and
high (40 µM) dose SIM treatment groups (Fig. 2B). Proteasome inhibitor MG132 was
used to detect whether MCM7 protein was degraded or not by SIM and
no difference was observed following the use of MG132 (data not
shown).
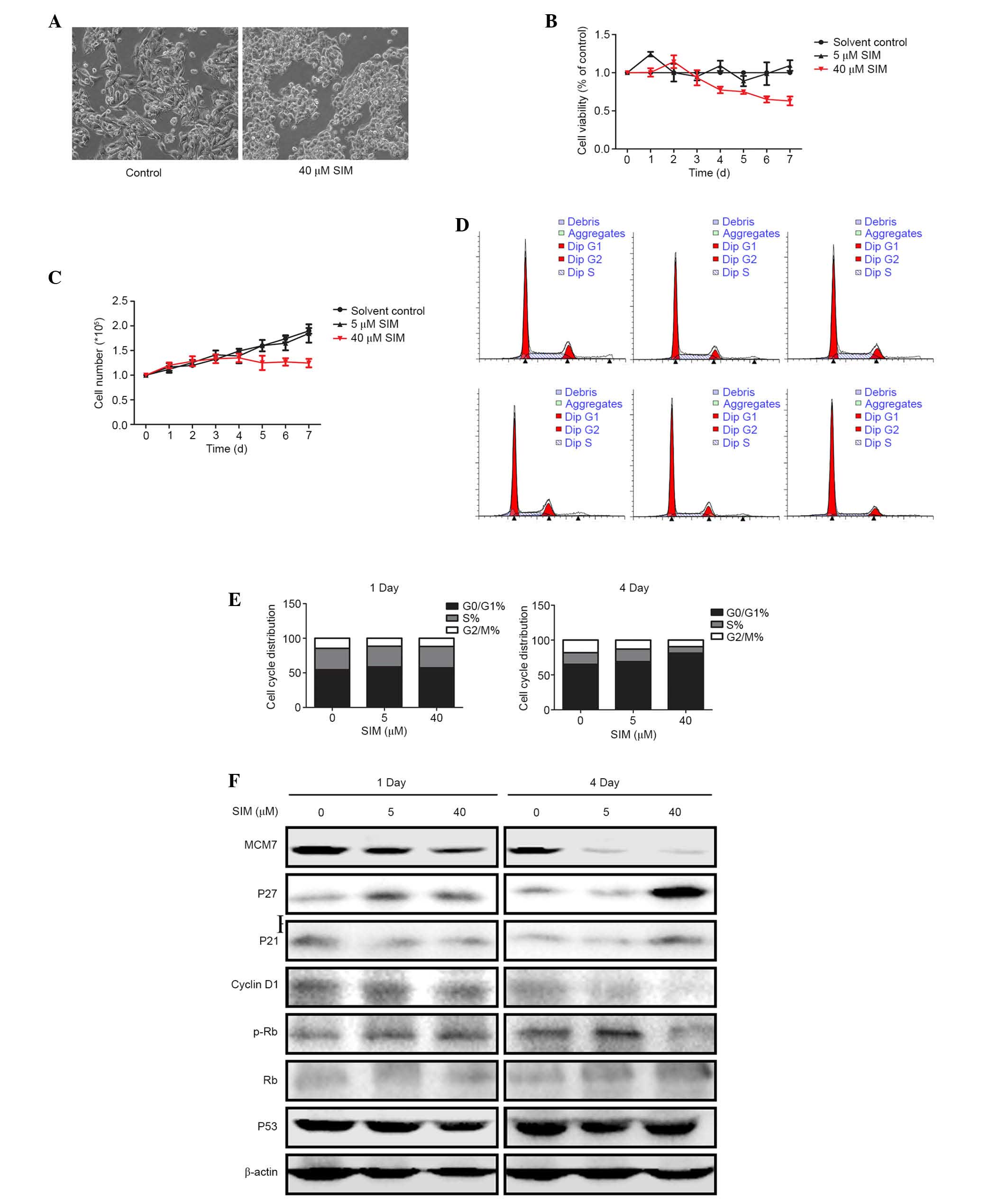
SIM suppressed HepG2.2.15 cell
proliferation
To investigate the effect of SIM on HepG2.2.15 cell
proliferation, HepG2.2.15 cells were treated with 5 or 40 µM SIM
for 1 and 4 days. Cell morphology changed after four days treatment
with 40 µM SIM. Cells became small and rounded and ceased
proliferation, compared with the cells in the control group
(Fig. 3A). MTT assay and cell
growth curve indicated that 40 µM SIM suppressed HepG2.2.15 cell
proliferation, particularly 4 days after SIM treatment (Fig. 3B and C).
To further investigate the effects of
SIM on HepG2.2.15 cell proliferation, the cell cycle was analyzed
by PI
Flow cytometry demonstrated that after 4 days SIM
treatment, the G1/S cell cycle arrest was more notable
in the 40 µM group compared with those in the control and low (5
µM) dose groups (Fig. 3D and E),
while on day 1 there was no significant G1/S cell cycle
arrest observed. This indicated that SIM generated G1/S
cell cycle arrest in a dose- and time-dependent manner. To further
investigate how expression of cell cycle proteins were changed in
SIM-treated HepG2.2.15 cells, western blotting was used to examine
the expression levels of proteins involved in G1/S
transition. The results demonstrated that MCM7 was notably
downregulated by SIM in the cells treated with 5 or 40 µM, while
cyclin D1 and p-Rb were downregulated, and p27 and p21 were
upregulated, only in cells treated with high dose (40 µM) SIM after
4 days. However, expression levels of Rb and p53 were not notably
different in either group (Fig.
3F).
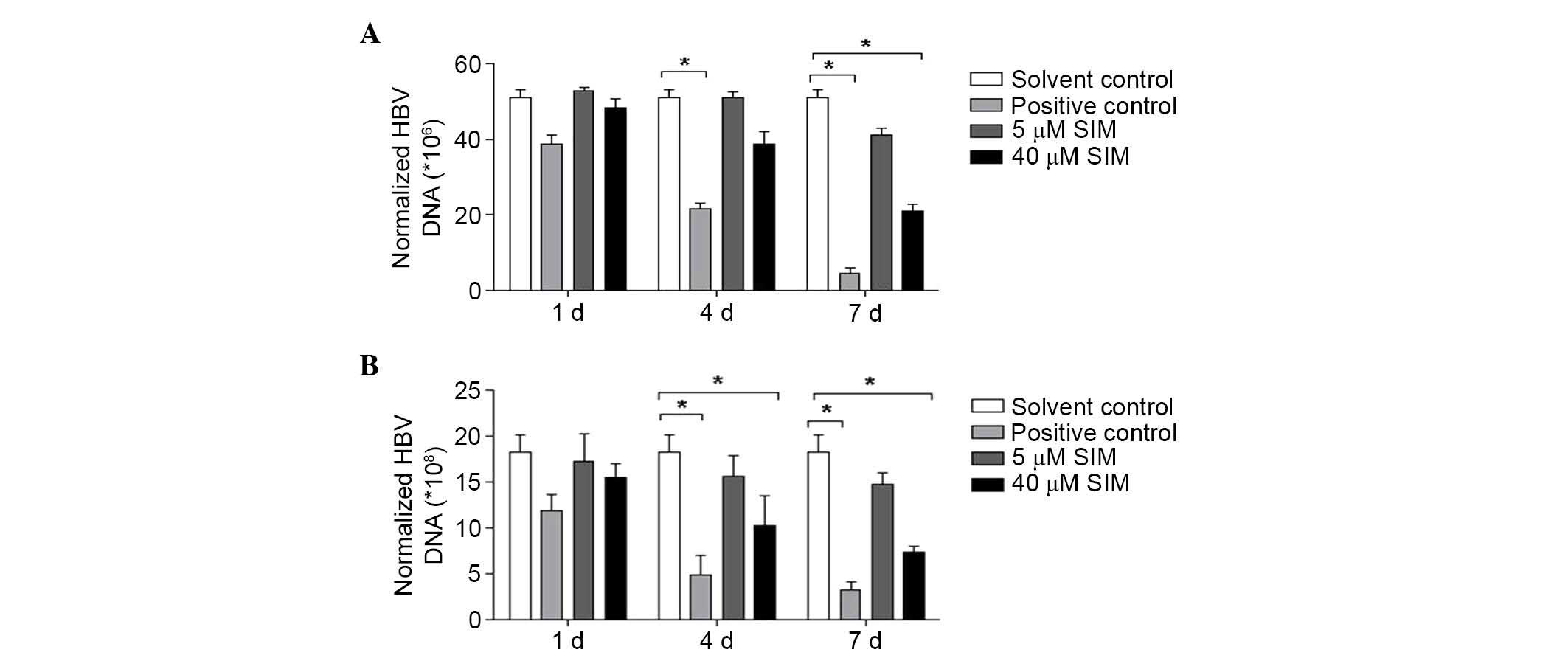
SIM repressed HBV DNA replication in
HepG2.2.15 cells and their culture supernatants
HepG2.2.15 cells were divided into four groups: i)
Solvent control group (treated with solvent); ii) positive control
group (treated with 5 µM lamivudine); iii) low dose (5 µM) SIM
group; and iv) high dose (40 µM) SIM group. FQ-PCR was conducted to
assess the level of HBV DNA replication in culture supernatants and
cells at 1, 4 and 7 days after treatment with SIM. After 4 days SIM
treatment, the expression of HBV DNA in HepG2.2.15 cell culture
supernatant were downregulated (without statistical significance),
while after 7 days SIM treatments, the HBV DNA expression was
significantly downregulated in the 40 µM SIM group (P<0.05;
Fig. 4A). Similar results were
observed for the cells, at 4 and 7 days after SIM treatment, HBV
DNA expression was downregulated, with a statistically significant
difference identified in the 40 µM SIM group (P<0.05; Fig. 4B).
LKB1-AMPK signaling pathway may be
involved in the anti-HBV effect of SIM
To investigate the underlying mechanism of the
effect of SIM on MCM7, MCM7 mRNA expression levels were assessed by
RT-qPCR at 1 and 4 days after HepG2.2.15 cells were treated with
SIM. Notably, the results were contrary to results obtained for
MCM7 protein expression levels. The expression level of MCM7 mRNA
was increased by SIM in a dose- and time-dependent manner (Fig. 5A). Thus, the present study
investigated whether SIM affects the translation of MCM7. As
initiation factor eIF2α is commonly involved in the translation,
eIF2α expression following SIM treatment was investigated. Western
blots indicated that after 4 days of treatment with high dose (40
µM) SIM, HepG2.2.15 cells indicated increased expression of protein
kinase RNA-like endoplasmic reticulum (ER) kinase (PERK) upstream
of p-eIF2α and eIF2α, which can phosphorylate eIF2α directly. As
PERK is downstream of the LKB1-AMPK signaling pathway, further
investigations were conducted into the activation of LKB1 and AMPK
by western blotting. The results demonstrated that the expression
levels of LKB1 and p-AMPK were increased after 4 days treatment
with SIM (40 µM) in HepG2.2.15 cells, however, expression of these
factors exhibited no notable changes after 1 day or in low dose (5
µM) treatment groups (Fig.
5B).
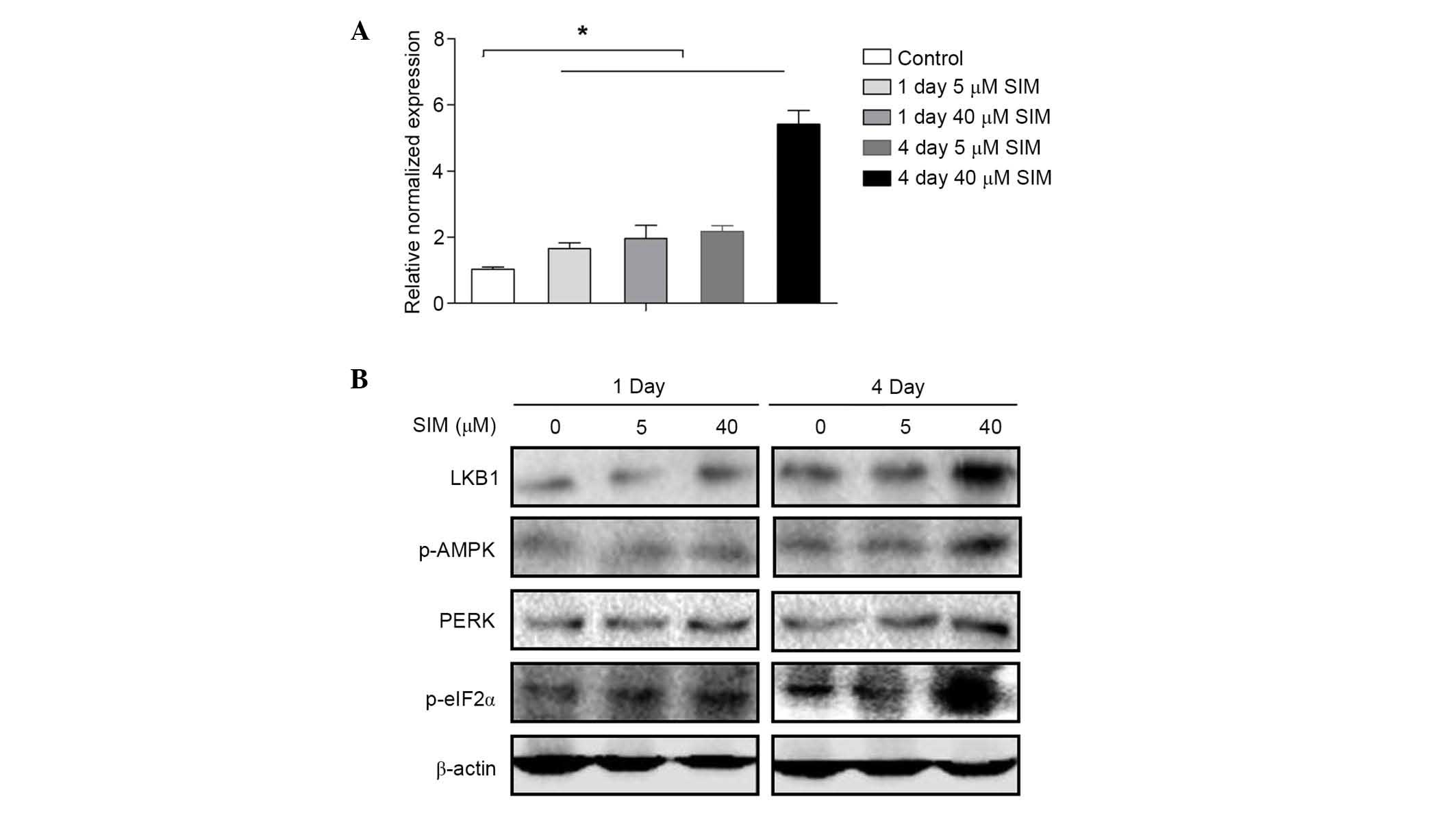 | Figure 5.LKB1-AMPK pathway may be involved in
the anti-HBV effect of SIM. (A) HepG2.2.15 cells were treated with
SIM (0, 5 and 40 µM) for 1 and 4 days and MCM7 mRNA levels were
determined by reverse transcription-quantitative polymerase chain
reaction. (B) HepG2.2.15 cells were treated with SIM (0, 5 and 40
µM) for 1 and 4 days. Total proteins were extracted and the
expression levels of LKB1, p-AMPK, PERK and p-eIF2α were examined
by western blot analysis. Equal protein loading was evaluated by
β-actin. Data were presented as the mean ± standard deviation from
three independent experiments. Significance was analyzed using
two-tailed Student's t-test. *P<0.05 vs. the untreated control
group. LKB1, liver kinase B1; AMPK, AMP-activated protein kinase;
HBV, hepatitis B virus; SIM, simvastatin; PERK, protein kinase
RNA-like endoplasmic reticulum kinase; eIF2α, eukaryotic initiation
factor-2α; p, phosphorylated. |
Discussion
HBV infection remains a major health problem,
particularly in developing countries. Due to varying response
rates, drug resistance and side effects, to investigate novel
effective therapeutic agents with fewer side effects is required.
SIM is a widely used therapeutic agent in the treatment of
hypercholesterolemia, and has been reported to inhibit the level of
HBV DNA (11), however, the
underlying mechanism remains unknown. Elucidation of a mechanism
underlying the effect of SIM on inhibiting HBV DNA replication may
enable development of novel therapies to treat HBV infections. In
the present study, it was determined that SIM induced HBV
downregulation involved a MCM-dependent mechanism, and SIM may
inhibit MCM7 expression via increasing eIF2α phosphorylation, which
is mediated by the LKB1-AMPK signaling pathway.
Numerous studies have reported that statins have
activities that prevent the replication of viruses, including
influenza virus and HBV (9,10,23).
SIM has been demonstrated to have a function to inhibit HBV
(11). Consistently, the present
study detected that the expression levels of HBV were downregulated
in HepG2.2.15 cells and their supernatants following treatment with
SIM. As SIM itself has an effect on inhibiting cell growth, the
total protein concentration of cells in each group was examined as
normalization. HBV copy number per unit protein concentration was
calculated to infer the treatment effects on HBV DNA
replication.
As HBV genomes are considerably small, they cannot
encode large numbers of genes alone. Thus, once they infect host
cells, viruses use a number of proteins from host cells termed
‘host factors’ to aid their replication. The MCM complex, which
controls DNA helicase in host cells, has previously been reported
as an important ‘host factor’ (23). Kawaguchi and Nagata (23) demonstrated that the MCM complex is
important in the replication of HBV in host cells. These effects
suggest that MCM complex may be an important antiviral target.
Notably, a previous study demonstrated that at orvastatin, a type
of statin, inhibits the expression of MCM proteins in vascular
smooth muscle cells (12). Thus,
it was hypothesized that SIM may inhibit the replication of HBV DNA
via downregulation of MCM7 protein. The results of the present
study demonstrated that SIM downregulated MCM7 protein expression
in HepG2.2.15 cells, and HBV DNA expression was decreased in cells
following MCM7 silencing.
Furthermore, the present study aimed to investigate
how SIM regulates MCM7. It was demonstrated that the mRNA
expression levels were increased while MCM7 protein expression was
decreased following SIM treatment. This indicates that certain
variations may have occurred during the process of translation or
protein degradation. Proteasome inhibitor MG132 was used to detect
whether MCM7 protein was degraded or not by SIM and no difference
was observed following the use of MG132 (data not shown). In
addition, the results demonstrated that SIM promoted
phosphorylation of eIF-2α and expression of PERK. Phosphorylation
of eIF-2α was one of the most well-studied mechanisms that regulate
translation (26,27). eIF2 contains three subunits, α,
βand γ (28) and it moves
Met-tRNAi to the ribosome to form the ternary complex
eIF2-GTP-Met-tRNAi (29). Due to
GTP hydrolysis, eIF2-GDP complex is released from the ternary
complex. The eIF2-GDP complex remains in an inactive state until
the GTP exchange factor, eIF-2B, catalyzes GDP to GTP; then the
eIF2-GTP complex is regenerated and a new round of transport is
started. Phosphorylation of eIF-2α at residue Ser51 inhibits eIF-2β
activity, resulting in the suppression of translation initiation
(27). When cells suffer from
various stress conditions, including anoxia and medication,
phosphorylation of eIF2α is initiated and translation initiation is
repressed. eIF2α is also associated with IFN, which has been
identified to protect cells from viral infection (5,30).
IFN can activate certain key biological functions of PKR, a
double-stranded RNA-dependent protein kinase. PKR phosphorylates
eIF-2α and the phosphorylated eIF-2α is key in the antiviral
mechanism of the host (31). PKR
is one of the four mammalian kinases that can phosphorylate eIF-2α,
the other three kinases are general control nonderepressible 2,
PERK (27), and heme-regulated
inhibitor kinase (32,33). It was assumed that SIM may inhibit
HBV DNA in a similar manner as IFN exerts its antiviral functions.
PERK is commonly activated via ER stress (34). In eukaryotic cells, ER is
understood to be important in folding and maturing most secreted
and transmembrane proteins (35).
Whether unfolded proteins can enter the ER or not depends on cell
differentiation programs, environmental conditions and the
physiological state of the cell. Unfolded proteins accumulate in
the ER and induce a coordinated adaptive program termed the
unfolded protein response. When cells are under stress conditions,
the ER cannot lead to efficient folding of proteins, thus, unfolded
proteins accumulate in the lumen of ER, ER stress is initiated,
then PERK consequently is activated, and eIF2α is phosphorylated,
attenuating protein synthesis (35).
A previous study has reported that the LKB1-AMPK
pathway is able to regulate the activation of PERK (36). The results of the present study
demonstrated increased PERK and p-eIF2α expression levels, coupled
with activation of LKB1-AMPK signaling pathway. LKB1, also known as
serine/threonine-protein kinase 11, phosphorylates and activates
AMPK proteins and is implicated as a central regulator of cell
polarity and energy metabolism in a variety of systems (37,38).
Perhaps certain associations exist between ER stress and the
LKB1-AMPK signaling pathway. Further research is required to
elucidate whether these exist or not.
In conclusion, this is the first study, to the best
of our knowledge, to indicate that the anti-HBV activity of SIM may
be, at least in part, mediated by inhibition of MCM7 expression in
HepG2.2.15 cells. Phosphorylation of eIF2α and activation of PERK
and the LKB1-AMPK pathway may be important in SIM-mediated
inhibition of MCM7 expression. Future research may determine
whether or not SIM can inhibit other viruses via the MCM-dependent
mechanism. MCMs may be a novel target for antiviral therapy in the
future.
Acknowledgements
The present study was financially supported by a
grant from the National Natural Science Foundation of China (grant
no. 81071876).
References
|
1
|
Liaw YF and Chu CM: Hepatitis B virus
infection. Lancet. 373:582–592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen L, Zhao H, Yang X, Gao JY and Cheng
J: HBsAg-negative hepatitis B virus infection and hepatocellular
carcinoma. Discov Med. 18:189–193. 2014.PubMed/NCBI
|
|
3
|
Tujios SR and Lee WM: Update in the
management of chronic hepatitis B. Curr Opin Gastroenterol.
29:250–256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gale MJ and Katze MG: Molecular mechanisms
of interferon resistance mediated by viral-directed inhibition of
PKR, the interferon-induced protein kinase. Pharmacol Ther.
78:29–46. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garcia MA, Gil J, Ventoso I, Guerra S,
Domingo E, Rivas C and Esteban M: Impact of protein kinase PKR in
cell biology: From antiviral to antiproliferative action. Microbiol
Mol Biol Rev. 70:1032–1060. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niederau C, Heintges T, Lange S, Goldmann
G, Niederau CM, Mohr L and Häussinger D: Long-term follow-up of
HBeAg-positive patients treated with interferon alfa for chronic
hepatitis B. N Engl J Med. 334:1422–1427. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zoulim F and Locarnini S: Management of
treatment failure in chronic hepatitis B. J Hepatol. 56:(Suppl 1).
S112–S122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song ZL, Cui YJ, Zheng WP, Teng DH and
Zheng H: Diagnostic and therapeutic progress of multi-drug
resistance with anti-HBV nucleos(t)ide analogues. World J
Gastroenterol. 18:7149–7157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ikeda M, Abe K, Yamada M, Dansako H, Naka
K and Kato N: Different anti-HCV profiles of statins and their
potential for combination therapy with interferon. Hepatology.
44:117–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Potena L, Frascaroli G, Grigioni F,
Lazzarotto T, Magnani G, Tomasi L, Coccolo F, Gabrielli L, Magelli
C, Landini MP and Branzi A: Hydroxymethyl-glutaryl coenzyme a
reductase inhibition limits cytomegalovirus infection in human
endothelial cells. Circulation. 109:532–536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bader T and Korba B: Simvastatin
potentiates the anti-hepatitis B virus activity of FDA-approved
nucleoside analogue inhibitors in vitro. Antiviral Res. 86:241–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bruemmer D, Yin F, Liu J, Kiyono T, Fleck
E, Van Herle A, Graf K and Law RE: Atorvastatin inhibits expression
of minichromosome maintenance proteins in vascular smooth muscle
cells. Eur J Pharmacol. 462:15–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maiorano D, Lutzmann M and Méchali M: MCM
proteins and DNA replication. Curr Opin Cell Biol. 18:130–136.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Namdar M and Kearsey SE: Analysis of
Mcm2-7 chromatin binding during anaphase and in the transition to
quiescence in fission yeast. Exp Cell Res. 312:3360–3369. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shechter D, Ying CY and Gautier J: DNA
unwinding is an Mcm complex-dependent and ATP hydrolysis-dependent
process. J Biol Chem. 279:45586–45593. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gonzalez MA, Tachibana KE, Laskey RA and
Coleman N: Control of DNA replication and its potential clinical
exploitation. Nat Rev Cancer. 5:135–141. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishitani H and Lygerou Z: Control of DNA
replication licensing in a cell cycle. Genes Cells. 7:523–534.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masuda T, Mimura S and Takisawa H: CDK-
and Cdc45-dependent priming of the MCM complex on chromatin during
S-phase in Xenopus egg extracts: Possible activation of MCM
helicase by association with Cdc45. Genes Cells. 8:145–161. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aladjem MI: Replication in context:
Dynamic regulation of DNA replication patterns in metazoans. Nat
Rev Genet. 8:588–600. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simon NE and Schwacha A: The Mcm2-7
replicative helicase: A promising chemotherapeutic target. Biomed
Res Int. 2014:5497192014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JK and Hurwitz J: Isolation and
characterization of various complexes of the minichromosome
maintenance proteins of Schizosaccharomyces pombe. J Biol Chem.
275:18871–18878. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JK and Hurwitz J: Processive DNA
helicase activity of the minichromosome maintenance proteins 4, 6,
and 7 complex requires forked DNA structures. Proc Natl Acad Sci
USA. 98:54–59. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawaguchi A and Nagata K: De novo
replication of the influenza virus RNA genome is regulated by DNA
replicative helicase, MCM. EMBO J. 26:4566–4575. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou YM, Zhang XF, Cao L, Li B, Sui CJ, Li
YM and Yin ZF: MCM7 expression predicts post-operative prognosis
for hepatocellular carcinoma. Liver Int. 32:1505–1509. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sonenberg N and Dever TE: Eukaryotic
translation initiation factors and regulators. Curr Opin Struct
Biol. 13:56–63. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi Y, Vattem KM, Sood R, An J, Liang J,
Stramm L and Wek RC: Identification and characterization of
pancreatic eukaryotic initiation factor 2 alpha-subunit kinase,
PEK, involved in translational control. Mol Cell Biol.
18:7499–7509. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmitt E, Naveau M and Mechulam Y:
Eukaryotic and archaeal translation initiation factor 2: A
heterotrimeric tRNA carrier. FEBS Lett. 584:405–412. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim JH, Park SM, Park JH, Keum SJ and Jang
SK: eIF2A mediates translation of hepatitis C viral mRNA under
stress conditions. EMBO J. 30:2454–2464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Isaacs A and Lindenmann J: Virus
interference. I. The interferon. Proc R Soc Lond B Biol Sci.
147:258–267. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang S, Sun Y, Chen H, Dai Y, Zhan Y, Yu
S, Qiu X, Tan L, Song C and Ding C: Activation of the PKR/eIF2α
signaling cascade inhibits replication of Newcastle disease virus.
Virol J. 11:622014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trinh MA, Ma T, Kaphzan H, Bhattacharya A,
Antion MD, Cavener DR, Hoeffer CA and Klann E: The eIF2α kinase
PERK limits the expression of hippocampal metabotropic glutamate
receptor-dependent long-term depression. Learn Mem. 21:298–304.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: Coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Avivar-Valderas A, Bobrovnikova-Marjon E,
Diehl J Alan, Bardeesy N, Debnath J and Aguirre-Ghiso JA:
Regulation of autophagy during ECM detachment is linked to a
selective inhibition of mTORC1 by PERK. Oncogene. 32:4932–4940.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shaw RJ: LKB1 and AMP-activated protein
kinase control of mTOR signalling and growth. Acta Physiol (Oxf).
196:65–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|