Introduction
Percutaneous angioplasty, including stent
implantation, is currently the most frequently performed medical
procedure for vascular diseases. However, long-term success is
limited by in-stent restenosis, which occurs in 20–30% of all
patients following intervention (1).
The primary underlying mechanism and pathology of
in-stent restenosis has only recently been elucidated. Various
events contribute to neointimal formation; of these, the migration
and proliferation of vascular smooth muscle cells (VSMCs) is
critical (2). Drug-eluting stents
(DES) markedly reduce the restenosis rate via the controlled
release of nonspecific antiproliferative agents, and are therefore
a promising therapeutic strategy. However, these nonspecific drugs
may cause endothelial toxicity, attenuating re-endothelialization
and resulting in late in-stent thrombosis (3–6).
Therefore, the identification of a novel therapeutic agent
specifically targeting VSMCs is essential for the development of
safer and more effective DES.
Platelet-derived growth factor (PDGF) is crucial for
the proliferation and migration of VSMCs that results in restenosis
(7,8). Activation of the PDGF receptor
(PDGFR) was observed a few days following the application of
mechanical force. In addition, the expression of PDGF ligands and
PDGFR has been detected in restenosis lesions (9,10).
Sunitinib malate (sunitinib) is a tyrosine kinase inhibitor of
PDGFR, which only weakly inhibits vascular endothelial growth
factor receptor. This agent exerts anticancer effects and has been
widely used for the treatment of metastatic renal cell carcinoma
and gastrointestinal stromal tumors (11,12).
Previous studies have administered sunitinib orally to prevent
restenosis in a balloon-injury carotid artery model (13). However, systemic administration of
a PDGFR inhibitor requires a relatively high dose, which may cause
adverse effects. Clinical trials of imatinib (a PDGFR-targeting
drug) detected no beneficial effects on in-stent restenosis when
the agent was administered orally (100 mg/day for 10 days)
(14). Therefore, the prevention
of restenosis via sunitinib targeting of PDGF may require a local
delivery system.
In the present study, it was hypothesized that a
sunitinib-eluting stent (STES) may be an effective therapeutic
strategy for the prevention of neointima formation in the rabbit
carotid model. In addition, the effects of sunitinib on VSMCs were
evaluated in vitro.
Materials and methods
Stent coating
Bare-metal stents (diameter, 2 mm; length, 20 mm)
were purchased from Dalian Yinyi Biomaterials Development Co., Ltd.
(Dalian, China). Sunitinib was obtained from CST Biological
Reagents Co., Ltd. (Shanghai, China) and sunitinib-eluting stents
were produced in our laboratory as previously described by Chen
et al (15). Sunitinib was
dissolved in 100 µl ethanol and mixed with lubricating jelly
(Jiangsu Province Jianerkang Medical Dressing Co., Ltd., Jintan,
China) to a final concentration of 2 mg/g of jelly. Lubricating
jelly is a commercially available product that consists of
propylene glycol, a viscous liquid commonly used as an additive in
food and medicine. Each balloon-mounted stent was individually
dipped in sunitinib jelly to produce a thin coating weighing
approximately 0.015 g; the amount of sunitinib eluted was
determined from the additional weight of the coating. The stents
were air-dried to completely evaporate the solvent. A coated stent
was crimped onto a shrunken catheter balloon, and a pressure of 10
atm was applied to make the stent expand fully. The amount of
sunitinib prior to and following expansion was analyzed.
Sunitinib release was measured using high
performance liquid chromatography (HPLC; Agilent 1100; Agilent
Technologies, Inc., Santa Clara, CA, USA), as previously described
(16). The STES was immersed in a
tube containing phosphate-buffered saline (PBS; pH=7.35–7.45). All
tubes were incubated at 37°C throughout the release study. Samples
were characterized at each predetermined time point.
Animal preparation and stent
implantation
All care and handling of the animals was in
accordance with guidelines for the care and use of laboratory of
animals, and the present study was approved by Nanjing University's
ethical research committee (Nanjing, China). A total of 24 male New
Zealand rabbits (2.5–3.5 kg) obtained from Jiangning Qinglongshan
Animal Center (Nanjing, China), were used in the present study.
Animals were maintained at 22°C and 65% humidity, and allowed ad
libitum access to water and a basal diet. The rabbits were
randomly assigned to two treatment groups: Bare-metal stent (BMS;
n=12) and sunitinib-coated stents (STES; n=12). Under anesthesia
with 5 mg droperidol and 100 mg ketamine (Fujian Fukang
Pharmaceutical Co., Ltd., Fuzhou, China) administered
intramuscularly, the carotid artery was accessed by dissection and
isolated from the surrounding tissue. The balloon-expandable stent
was inserted into the carotid artery through a small incision, and
the stent was deployed by inflation with 10 atm. for 30 sec.
Finally, the access site in the carotid artery was closed with 8–0
Prolene suture (Ethicon, Inc, Somerville, NJ, USA).
Postoperatively, all rabbits received 25 mg aspirin prior to
sacrifice, and a prophylactic antibiotic (300 mg cefuroxime) was
administered for 7 days. The implanted stents were monitored by a
Doppler handle probe (HP Sonos 4500; Philips Medical Systems, Inc.,
Bothell, WA, USA), and images of the blood flow were recorded every
month. Diagnostic angiography was performed to confirm the position
and patency rate of the stents.
Tissue harvest and pathological sample
preparation
At 1–3 months following stent implantation, rabbits
were euthanized by direct intracardiac injection of potassium
chloride and the stents and surrounding arteries were harvested.
The arteries containing stents were divided into three segments.
The proximal segment was opened longitudinally, flattened and fixed
in 1.6% glutaraldehyde. It was then dehydrated, dried with
CO2, coated with gold and visualized under a scanning
electron microscope (SEM; 3600N; Hitachi, Ltd., Tokyo, Japan). The
percentage of the re-endothelialized area compared with the total
luminal surface area was determined by examining SEM
photomicrographs of each specimen. Protein was extracted from the
ventral segment for western blot analysis. The distal segment was
fixed in 10% neutral-buffered formalin for 24 h, embedded in
methylmethacrylate, circumferentially cross-sectioned into 5-µm
thick sections on a microtome fitted with a D-profile tungsten
carbide knife (Delaware Diamond Knives, Inc., Wilmington, DE, USA)
and stained with hematoxylin and eosin (17) for the quantification of
histomorphological parameters. Tissue sections were visualized
using a light microscope (BX51; Olympus Corporation, Tokyo, Japan),
and the injury score (IS), average intimal thickness (AIT), areas
of neointima (AN), media area, fibrin score (FI) and inflammation
score (IMS) of each section was measured by digital morphometry
using Image-Pro Plus software version 6.2 (Media Cybernetics, Inc.,
Rockville, MD, USA). IS was graded using the standardized protocol
developed by Schwartz et al (18), which determined the degree of
injury of the vessel wall in a semi-quantitative manner. The
maximum score of 3 was applied when medial laceration extended
through the external elastic lamina, and a score of 0 corresponded
to an intact internal elastic lamina. Similarly, the extent of
cellular infiltration was scored from 0–3 using a system described
by Hong et al (19).
Cell proliferation analysis in
vitro
Rat smooth muscle cells (RSMCs) and rat endothelial
cells (RECs) were purchased from the Cell Bank of the National
Academy of Science (Shanghai, China). RSMCs were grown in
Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) containing 10% fetal bovine serum
(FBS; Sigma-Aldrich; Merck Millipore). RECs were cultured in
Endothelial Basal Medium 2 (Sigma-Aldrich; Merck Millipore). Rat
SMCs/ECs were chosen for the present preliminary study as rat
antibodies are more commonly available. RSMCs and RECs were seeded
in 96-well culture plates (7,500 cells/well). Sunitinib (0.1, 1 or
10 µM) or vehicle was added to the wells. Two days later, a
3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium (MTT) assay was
performed as described previously (20) to determine the cytotoxicity of
sunitinib.
To assess the direct effect of sunitinib on the
proliferation of RSMCs, a 3H-thymidine incorporation assay was
performed. (Methyl-3H)-thymidine (1 µCi/ml; PerkinElmer, Inc.,
Waltham, MA, USA) was added to wells 6 h prior to the termination
of each experiment. Radioactivity was measured using a liquid
scintillation counter.
Evaluation of apoptosis and necrosis
in vitro
To detect cell death, cells were stained with
annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) according to the manufacturer's instructions (BD Biosciences,
San Jose, CA, USA). RSMCs were cultured in a serum-free medium in
the presence or absence of sunitinib for 24 h and then stimulated
with 20 ng/ml PDGF-BB (CST Biological Reagents Co., Ltd.)
homodimers for a further 24 h. The cells were subsequently
trypsinized, collected and incubated with 5 µl annexin V-FITC and 5
µl PI (stock solution, 50 µg/ml) for 30 min at room temperature in
the dark. Apoptotic (annexin V+/PI−) and
necrotic (annexin V+/PI+) cells were detected
on a FACSCalibur (BD Biosciences).
RSMC attachment assay in vitro
RSMCs were cultured in 6-well plates with BMS and
STES for 12 h prior to examination. RSMCs attached to the stent
struts were stained with 4,6-diamidino-2-phenylindole (DAPI; a blue
fluorescent dye for cell nuclei) and observed under a fluorescent
microscope. The numbers of attached SMCs were counted in three
random high-power fields (magnification, ×200) per strut.
Scratch wound assay
RSMCs were seeded in 35-mm dishes in DMEM and
starved of FBS for 12 h, and cells were scratched with a 100-µl
pipette tip. Cells were pretreated with 0, 0.1, 1 or 10 µM
sunitinib and subsequently stimulated with 20 ng/ml PDGF-BB for 24
h. Cells were visualized under a light microscope and images were
captured. Image-Pro Plus software version 6.2 (Media Cybernetics,
Inc., Rockville, MD, USA) was used for quantification.
α-smooth muscle actin (α-SMA)
cytoskeleton staining
RSMCs were seeded in 6-well culture plates in DMEM
and starved of FBS for 24 h, and treated with sunitinib or vehicle
supplemented with 20 ng/ml PDGF-BB for a further 24 h. RSMCs were
fixed with 4% paraformaldehyde for 10 min and permeabilized in 0.2%
Triton X-100 for 10 min. The primary rabbit polyclonal anti-α-SMA
antibody (1:100; catalog no. 14968; CST Biological Reagents Co.,
Ltd.) was applied to cells. Following washing with PBS three times,
cells were incubated with the secondary antibody FITC-anti-rabbit
IgG (1:400; catalog no. ab6717; Abcam, Cambridge, MA, USA) for 2 h
at 37°C and observed under a fluorescent microscope.
Western blot analysis
For the in vitro experiment, protein was
extracted using radioimmunoprecipitation assay buffer
(Sigma-Aldrich; Merck Millipore) from RSMC cultured with various
concentrations of sunitinib and 20 ng/ml PDGF-BB. Protein (20 µg)
was loaded onto a 12% SDS-PAGE gel, separated by electrophoresis
and transferred onto a polyvinylidene difluoride membrane.
Membranes were blocked with 5% non-fat milk for 2 h, and incubated
overnight at 4°C with the following primary antibodies: Rabbit
phosphorylated (p)-PDGFR-β (1:1,000; catalog no. 3124), rabbit
p-extracellular signal-regulated kinase (ERK; 1:1,000; catalog no.
9101) and rabbit glyceraldehyde-3-phosphate dehydrogenase (GAPDH;
1:2,000; catalog no. 5174), all purchased from CST Biological
Reagents Co., Ltd. A horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (1:3,000; catalog no. ab6721; Abcam)
was then added to the membranes at room temperature for 2 h. The
signal was developed using Enhanced Chemiluminescence reagent (EMD
Millipore, Bedford, MA) and visualized with the FlourChem FC2
Imaging System (ProteinSimple, San Jose, CA, USA). The protein
bands were quantified using Image J software version 1.51b
(National Institutes of Health, Bethesda, MD, USA). For the in
vivo experiment, protein from neointimal tissue and media plus
adventitia from BMS and STES was extracted and the protein
expression levels of p-ERK and GAPDH were analyzed.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Unpaired Student's t-test was used to compare control and
sunitinib-treated groups. Statistical analyses were performed in
SPSS version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Stent coating
Incubation of bare-metal stents in the solution of
sunitinib jelly resulted in coating with this compound in the
present preliminary study; 32±5.4 µg sunitinib was bound to the
stent. The amount of sunitinib on the stent did not alter following
expansion. The results of the in vitro release of sunitinib
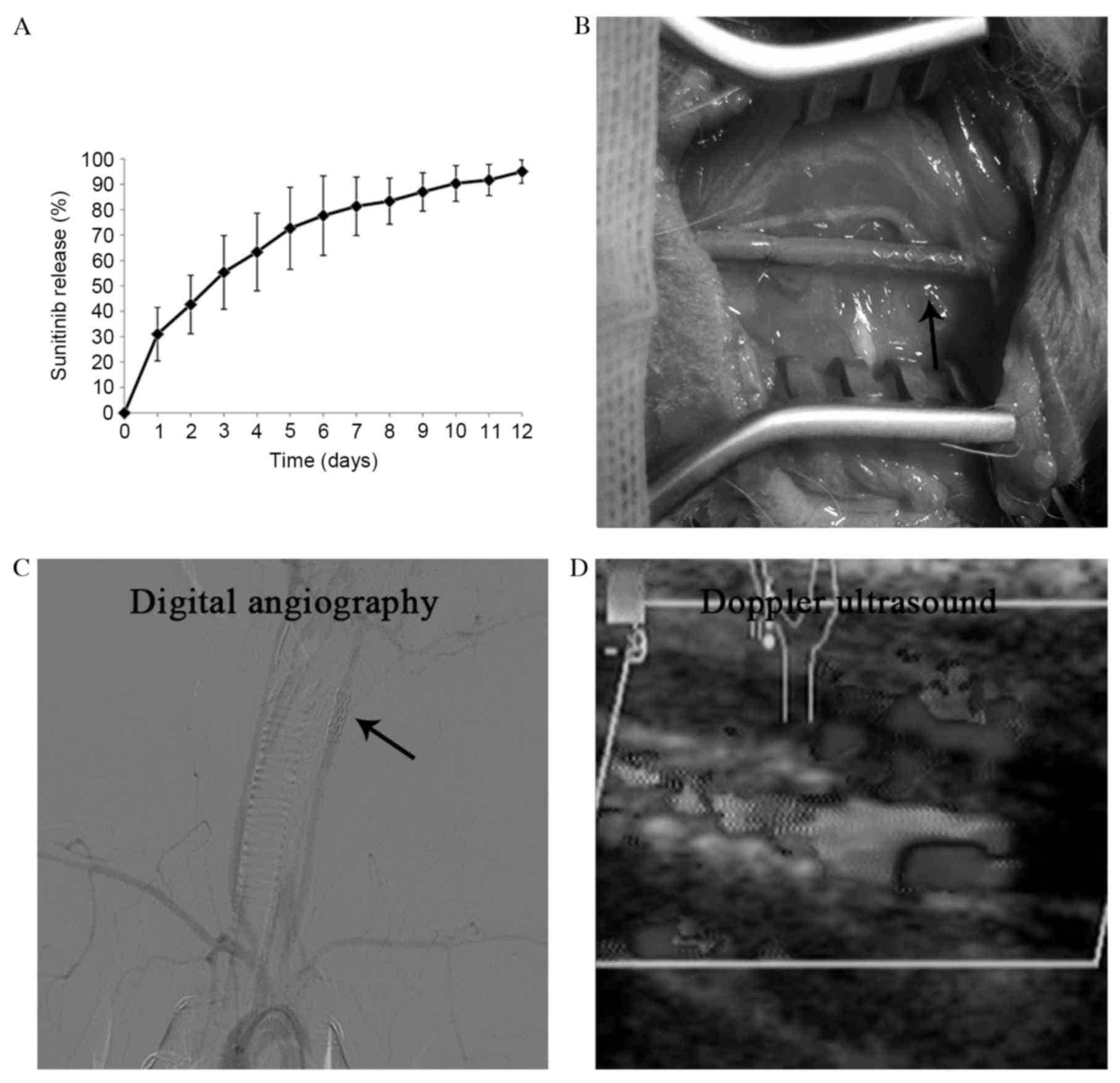
are presented in Fig. 1A.
STES reduces neointimal formation in
rabbit carotid arteries
Stents were successfully implanted into the carotid
arteries of 24 New Zealand rabbits (Fig. 1B), and the patency of the stent
grafts was demonstrated by diagnostic angiography and Doppler
ultrasound (Fig. 1C and D).
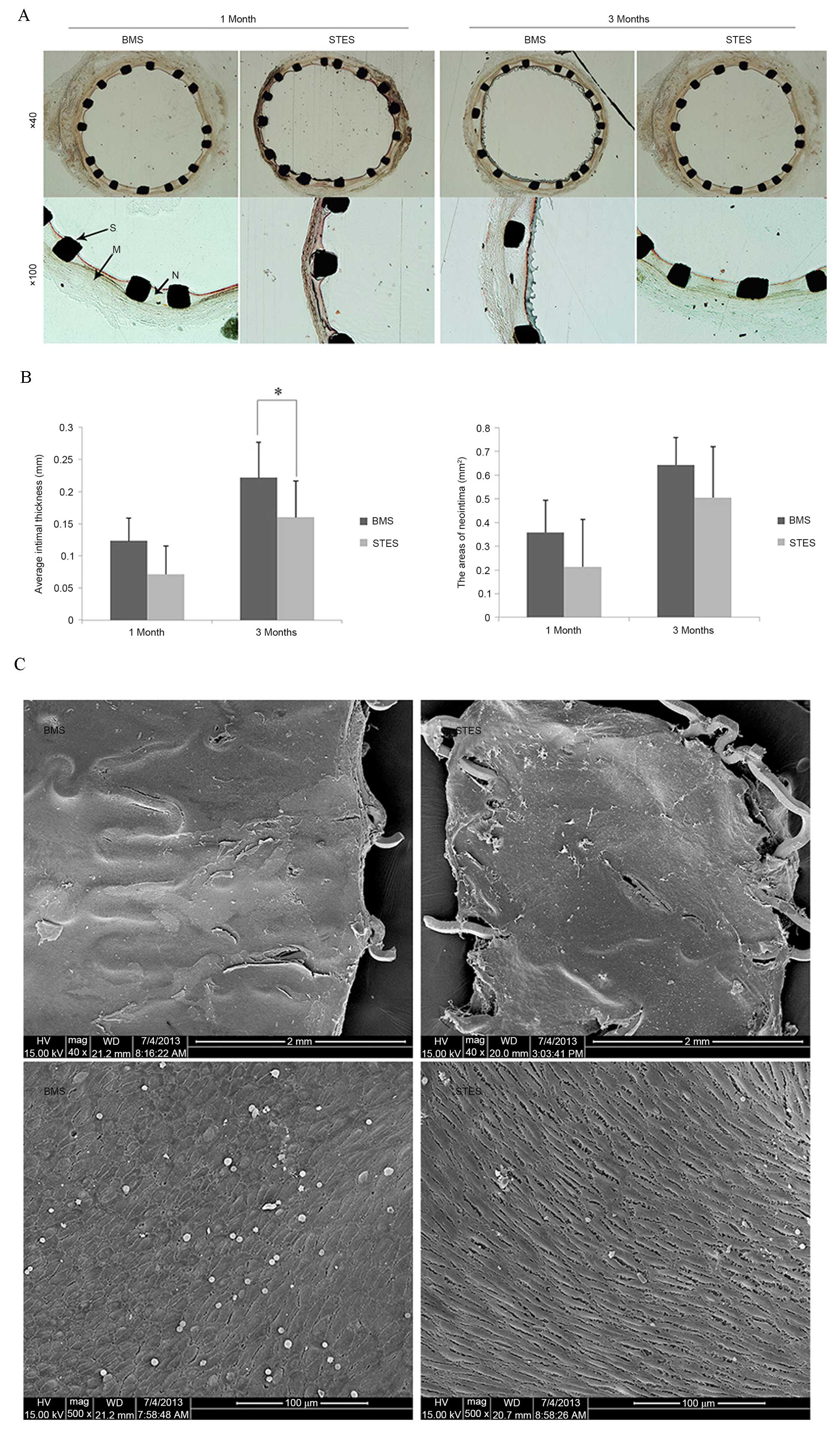
In-stent restenosis was examined by morphological
analysis, as presented in Fig. 2.
One month following implantation, the AIT was 0.12±0.03 mm in the
BMS group and 0.07±0.04 mm in the STES group (P=0.06). Three months
following implantation, clear neointimal formation was observed in
BMS arteries; however, AIT was markedly reduced in STES (0.16±0.06
vs. 0.22±0.05 mm; P=0.03). In addition, the AN was 0.35±0.14
mm2 in the BMS group and 0.21±0.12 mm2 in the
STES group (P=0.07) one month following implantation, and 0.64±0.20
mm2 in the BMS group and 0.50±0.21 mm2 in the
STES group (P=0.09) three months following implantation (Fig. 2A and B).
No differences were observed in the IS or IMS
between the two groups at one and three months following
implantation, indicating that the observed effects on neointima
formation did not result from structural differences or
inflammation. IS were 1.17±0.41 (BMS group) and 1.00±0.63 (STES
group; P=0.31), and 1.16±0.41 (BMS group) and 1.17±0.98 (STES
group; P=0.50), at one and three months post-implantation,
respectively. IMS were 0.33±0.52 (BMS group) and 0.17±0.41 (STES
group; P=0.18), and 0.33±0.52 (BMS group) and 0.50±0.55 (STES
group; P=0.31), at one and three months post-implantation,
respectively.
Regarding the cytotoxicity of sunitinib, media area
and FI were calculated. Media area was 0.42±0.03 in the BMS group
and 0.40±0.04 in the STES group (P=0.21), and 0.42±0.06 in the BMS
group and 0.39±0.04 in the STES group (P=0.25), at one and three
months post-implantation, respectively. The FI values were
1.83±0.41 (BMS) and 2.00±0 (STES; P=0.18), and 2.00±0 (BMS) and
1.67±0.52 (STES; P=0.09), at one and three months
post-implantation, respectively. These results suggest that local
administration of sunitinib inhibited neointimal formation without
damaging normal vascular media.
STES does not affect
re-endothelialization
Re-endothelialization was analyzed by SEM. The
re-endothelialized areas at one month following implantation were
91.7±3.7 and 89.5±6.8% in the BMS and STES groups, respectively
(P=0.24; Fig. 2C), indicating that
BMS and STES were efficient at inducing re-endothelialization.
Notably, BMS and STES were observed to almost fully
re-endothelialize areas, demonstrating that sunitinib did not delay
endothelialization in the rabbit carotid model.
Sunitinib inhibits proliferation and
induces necrosis in RSMCs
An MTT assay was performed in vitro with
RSMCs and RECs to evaluate the effect of sunitinib on
proliferation. Sunitinib treatment significantly suppressed the
proliferation of RSMCs in a dose-dependent manner (0.1 µM, P=0.040;
1 µM, P=0.001; 10 µM, P<0.001); these suppressive effects were
attenuated in RECs. No suppression was observed when RECs were
treated with 0.1 or 1 µM sunitinib. The proliferation of RECs was
slightly inhibited at 10 µM sunitinib (Fig. 3A). In addition, sunitinib treatment
significantly suppressed DNA synthesis in RSMCs in a dose-dependent
manner (0.1 µM, P=0.014; 1 µM, P=0.021; 10 µM, P=0.021). However,
sunitinib had no significant effects on DNA synthesis in RECs.
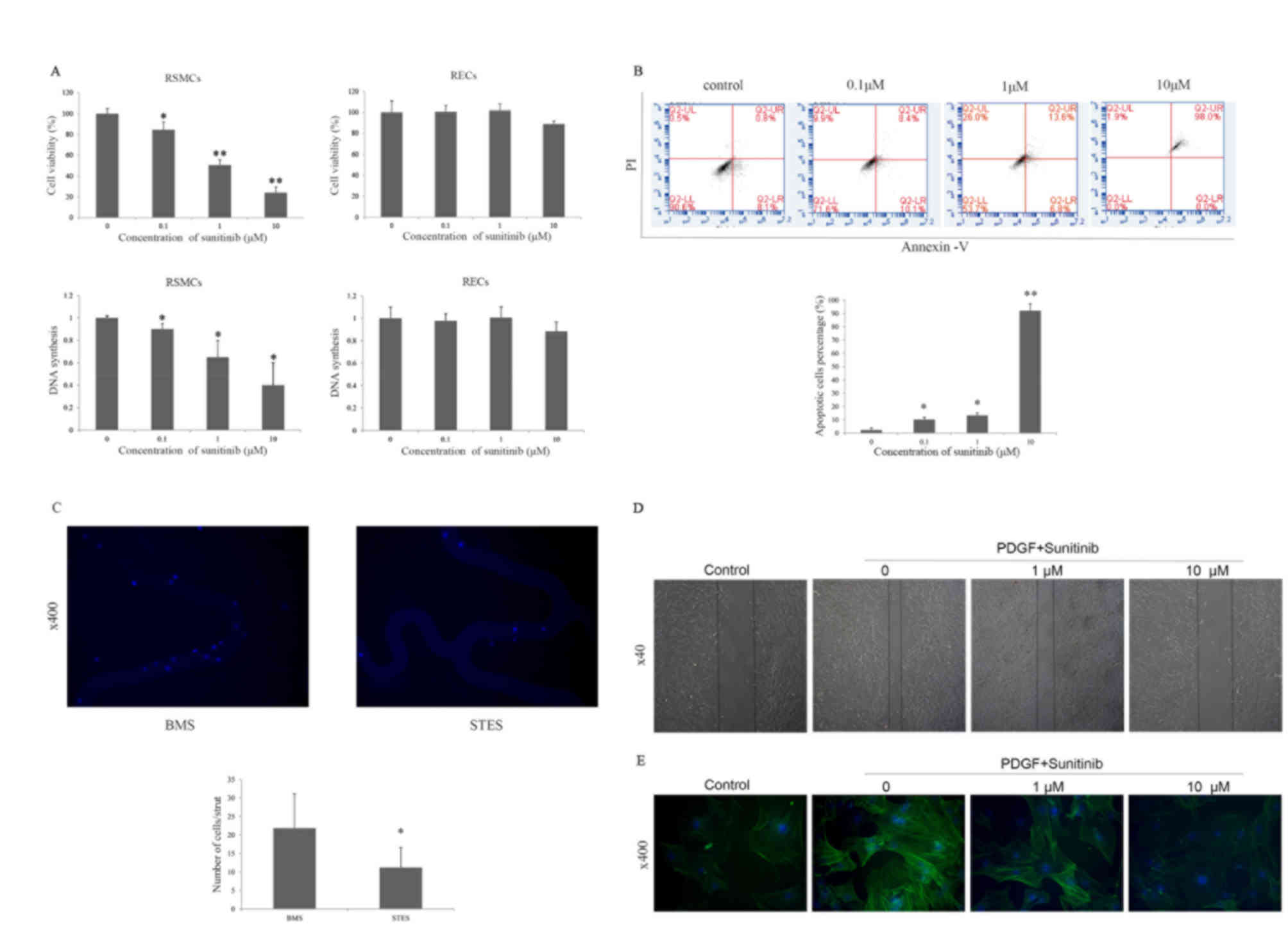 | Figure 3.(A) Effect of sunitinib on the
proliferation of RSMCs and RECs. Sunitinib inhibited proliferation
and DNA synthesis in RSMCs, but not in RECs. *P<0.05 and
**P<0.01 vs. vehicle. (B) Effect of sunitinib on apoptosis and
necrosis of PDGF-BB-treated RSMCs. Apoptotic (annexin
V+/PI−) or necrotic (annexin
V+/PI+) cells were identified by flow
cytometry. *P<0.05 and **P<0.01 vs. vehicle. (C)
Representative images reveal that greater numbers of cells attach
to the BMS compared with the STES. Cell nuclei were stained blue
with DAPI. Magnification, ×400. *P<0.05 vs. BMS. (D)
Representative images reveal that sunitinib attenuated PDGF-induced
migration of RSMCs in a scratch wound assay in a dose-dependent
manner. Solid lines represent the edges of cell migration.
Magnification, ×40. (E) Effect of sunitinib on α-SMA cytoskeleton
polymerization in RSMCs; α-SMA was stained with fluorescein
isothiocyanate (green), and nuclei were stained with DAPI (blue).
Magnification, ×400. RMSCs, rat smooth muscle cells; RECs, rat
endothelial cells; PDGF, platelet-derived growth factor; PI,
propidium iodide; BMS, bare-metal stent; STES, sunitinib-eluting
stent; DAPI, 4′,6-diamidino-2-phenylindole; α-SMA, α-smooth muscle
actin. |
As an antiproliferative effect is often accompanied
by apoptosis or necrosis, the apoptotic effects of sunitinib on
RSMCs were examined. Sunitinib significantly increased necrosis in
a dose-dependent manner (0.1 µM, P=0.010; 1 µM, P=0.015; 10 µM,
P=0.009), as assessed by PI staining, indicating that sunitinib may
block RSMC proliferation by inducing necrosis (Fig. 3B). The in vitro effects of
STES and BMS on RSMC adherence were then assessed. RSMC attachment
to STES was reduced compared with BMS (21.8±9.3 vs. 11.2±5.4/strut;
P=0.03; Fig. 3C).
Sunitinib-antagonizes PDGF-induced
migration of RSMCs
Migration of SMCs from media to intima occurs
following vascular injury, and PDGF is crucial for this migration.
A scratch wound assay and cytoskeleton staining was therefore
performed to investigate the effect of sunitinib on migration. The
scratch wound assay is a classic two-dimensional migration model.
As presented in Fig. 3D,
stimulation with PDGF-BB induced migration was attenuated by
sunitinib in a dose-dependent manner. Actin cytoskeleton
polymerization is critical for cell migration; therefore, α-SMA in
RSMCs was visualized with FITC. Actin polymerization was decreased
following sunitinib treatment (Fig.
3E), suggesting that downregulated expression levels of α-SMA
may represent one of the mechanisms underlying the inhibition of
RSMC migration by sunitinib.
Sunitinib blocks PDGF-induced ERK
phosphorylation
Various signaling pathways (particularly the ERK
signaling pathway) are involved in SMC migration and growth.
Therefore, ERK activation was analyzed by assessing
phosphorylation, to further study the mechanisms underlying the
inhibitory effects of sunitinib on proliferation and migration.
In the in vitro experiment, protein
expression levels of p-PDGFR were decreased by sunitinib treatment
in a dose-dependent manner, and PDGF-induced downstream p-ERK
protein expression levels were suppressed (0.1 µM, P=0.021; 1 µM,
P=0.024; 10 µM, P=0.031; Fig. 4A and
B). This suggested that sunitinib specifically inhibited
PDGF-induced ERK activation. In the in vivo experiment, the
media, adventitia and neointima were harvested. The protein
expression levels of p-ERK in the neointima were significantly
decreased in STES compared with BMS (P=0.027; Fig. 4C and D).
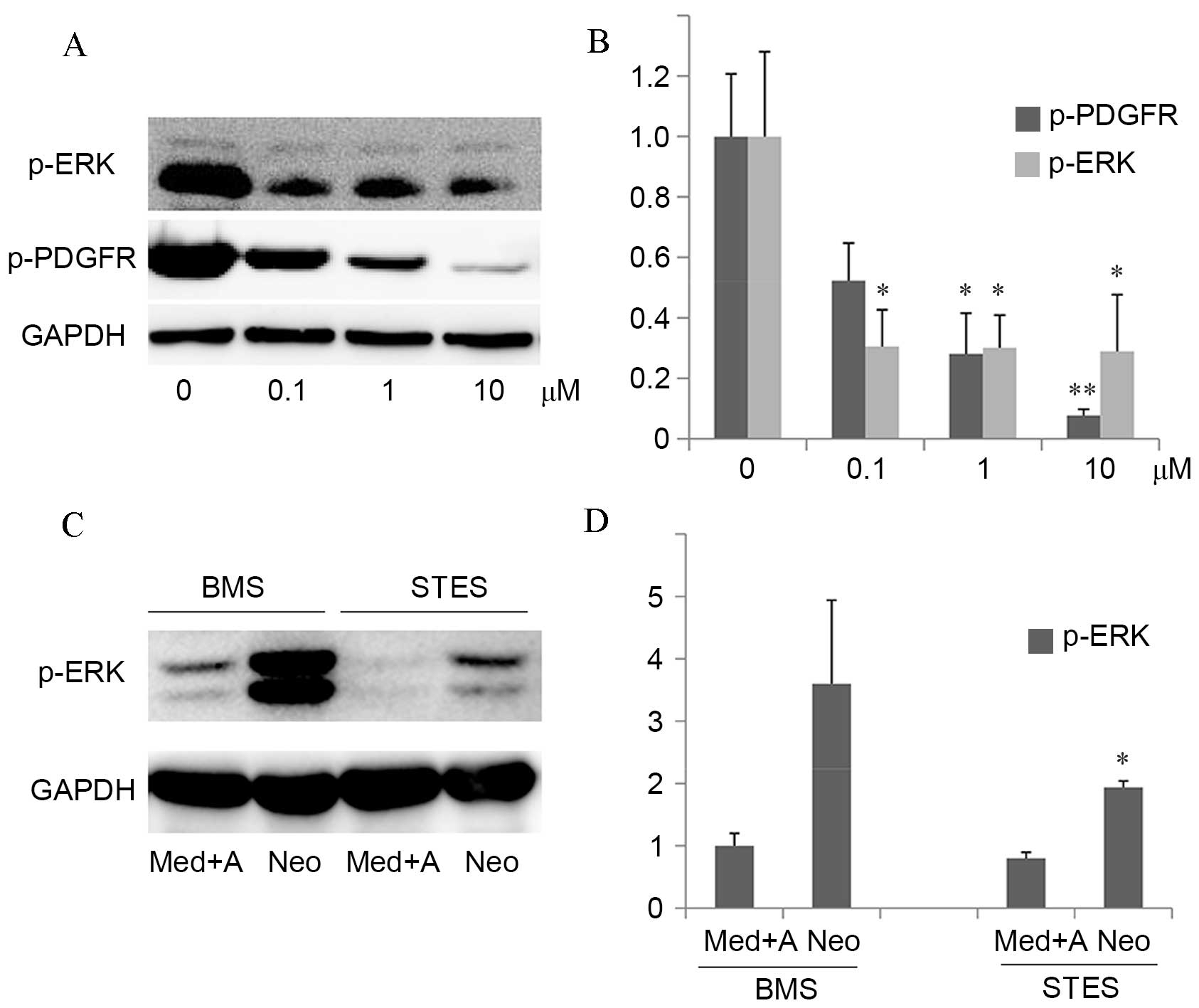 | Figure 4.The inhibitory effect of sunitinib on
PDGF-induced intracellular signaling in vitro and in
vivo. (A) Left panel: RSMCs were stimulated with 20 ng/ml
PDGF-BB and 0–10 µM sunitinib in serum-free medium for 24 h.
Western blot analysis of p-ERK, p-PDGFR and GAPDH, which served as
a loading control, was performed. (B) Quantification of western
blots demonstrated that protein expression levels of p-PDGFR were
decreased by sunitinib treatment in a dose-dependent manner, and
PDGF-induced downstream p-ERK protein expression levels were
suppressed. *P<0.05 and **P<0.01 vs. vehicle. (C) The media,
adventitia and neointima were harvested from rabbits implanted with
BMS or STES, and western blot analysis of p-ERK was performed, with
GAPDH serving as a loading control. (D) Quantification of western
blots demonstrated that protein expression levels of p-ERK in the
neointima were significantly decreased in STES compared with BMS.
*P<0.05 vs. BMS Neo. PDGF, platelet-derived growth factor;
RMSCs, rat smooth muscle cells; p-, phosphorylated; ERK,
extracellular signal-regulated kinase; PDGFR, PDGF receptor; BMS,
bare-metal stent; STES, sunitinib-eluting stent; Med, media; A,
adventitia; Neo, neointima; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase. |
Discussion
The importance of the PDGFR signaling pathway in the
pathophysiology of atherosclerosis and restenosis following
angioplasty has been highlighted by recent studies (10,13,14).
The potential to reverse in-stent restenosis through inhibiting
this signaling pathway has been investigated (7,8,21,22).
The majority of these studies administered PDGFR inhibitors orally.
This systemic application may cause unwanted side-effects,
including the development of cardiotoxicity. The present study
demonstrates that the local application of sunitinib through STES
may reduce in-stent restenosis without impairing endothelial
regeneration in the carotid model. The estimated dose of sunitinib
was 32±5.4 µg/stent, markedly reduced compared with the dose
required for oral administration. In addition, sunitinib attenuated
the proliferation and migration of SMCs in vitro,
potentially via the inhibition of the p-ERK signaling pathway.
These results indicate that STES may represent a potential DES for
the treatment of in-stent restenosis.
The mechanisms underlying the effect of STES on
in-stent restenosis require further investigation prior to its
clinical application. The abnormal proliferation of SMCs is a
prominent feature of neointimal formation (1). Sunitinib attenuated RSMC
proliferation in a dose-dependent manner. As an anti-proliferative
effect is often accompanied by apoptosis or necrosis, the apoptotic
effects of sunitinib in RSMCs were examined. Sunitinib increased
necrosis, indicating that it may inhibit RSMC proliferation via the
induction of necrosis. SMC migration from media to intima is
another mechanism underlying restenosis (23); the scratch wound assay and α-SMA
staining demonstrated the inhibitory effect of sunitinib on RSMC
migration. In addition, extracellular matrix production is critical
for the development of intimal hyperplasia; future studies in our
laboratory will therefore investigate the effect of sunitinib on
collagen synthesis.
ERK is an important downstream effector of
PDGF-induced SMC migration and proliferation (24). In the present study, sunitinib
inhibited PDGF-induced ERK phosphorylation in vitro and
in vivo. In addition, other downstream signaling molecules,
including protein kinase B and c-Jun N-terminal kinase, may
contribute to the inhibitory effect of sunitinib (25–27).
These molecules differentially regulate SMC growth and migration
via distinct signaling pathways.
Delayed healing and re-endothelialization following
implantation are features of DES, which may be the result of
adverse effects of the nonspecific antiproliferative agent used
(28–30). Theoretically, PDGFR inhibition
should not interfere with re-endothelialization, as endothelial
cells do not express PDGFR. SEM revealed no differences in
re-endothelialization between BMS and STES, suggesting that
sunitinib inhibited only RSMC proliferation and neointima
formation. Re-endothelialization was almost complete in the present
study one month following implantation. In humans, the timescale of
re-endothelialization remains to be fully elucidated. Stents
implanted in human arteries encounter long, heavily calcified or
chronically occluded lesions, and elderly patients have defective
endothelial progenitor cells (EPC). Therefore, rapid
re-endothelialization is difficult in humans. Certain pro-healing
strategies have been developed to promote endothelialization.
Stents coated with anti-cluster of differentiation 34 antibodies
have demonstrated promise in capturing EPC (31). The combination of the promotion of
re-endothelialization and the inhibition of neointima formation may
be a potential strategy for the development of novel DES.
Limitations of the present, preliminary, study
include the lack of pharmacokinetic data. The tissue distribution
and concentration of sunitinib following STES implantation remains
unknown. In addition, the division of implanted arteries into three
segments for separate analyses may introduce error, as the most
rapid re-endothelialization occurs in the proximal region of the
stent. Furthermore, the effect of STES was not assessed in a
hypercholesterolemic rabbit model. The use of rat instead of human
SMCs and ECs may also reduce the clinical relevance of the present
study. Further studies are required to evaluate the clinical
application of STES.
In conclusion, the results of the present study
demonstrate that the inhibition of PDGF signaling by sunitinib
attenuated the proliferation and migration of RSMCs but not RECs
in vitro, and that STES inhibited in-stent neointimal
formation in the rabbit carotid artery in vivo. The results
of the present study support the potential use of PDGFR inhibitors
in DES.
References
|
1
|
Costa MA and Simon DI: Molecular basis of
restenosis and drug-eluting stents. Circulation. 111:2257–2273.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lafont A and Libby P: The smooth muscle
cell: Sinner or saint in restenosis and the acute coronary
syndromes? J Am Coll Cardiol. 32:283–285. 1998.PubMed/NCBI
|
|
3
|
Spaulding C, Henry P, Teiger E, Beatt K,
Bramucci E, Carrié D, Slama MS, Merkely B, Erglis A, Margheri M, et
al: Sirolimus-eluting versus uncoated stents in acute myocardial
infarction. N Engl J Med. 355:1093–1104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stone GW, Lansky AJ, Pocock SJ, Gersh BJ,
Dangas G, Wong SC, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie
BR, et al: Paclitaxel-eluting stents versus bare-metal stents in
acute myocardial infarction. N Engl J Med. 360:1946–1959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Serruys PW, Silber S, Garg S, van Geuns
RJ, Richardt G, Buszman PE, Kelbaek H, van Boven AJ, Hofma SH,
Linke A, et al: Comparison of zotarolimus-eluting and
everolimus-eluting coronary stents. N Engl J Med. 363:136–146.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luscher TF, Steffel J, Eberli FR, Joner M,
Nakazawa G, Tanner FC and Virmani R: Drug-eluting stent and
coronary thrombosis: Biological mechanisms and clinical
implications. Circulation. 115:1051–1058. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Han Y, Lin C, Zhen Y, Song X, Teng
S, Chen C, Chen Y, Zhang Y and Hui R: PDGF-D contributes to
neointimal hyperplasia in rat model of vessel injury. Biochem
Biophys Res Commun. 329:976–983. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nurminskaya M, Beazley KE, Smith EP and
Belkin AM: Transglutaminase 2 promotes PDGF-mediated activation of
PDGFR/Akt1 and β-catenin signaling in vascular smooth muscle cells
and supports neointima formation. J Vasc Res. 51:418–428. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zimmermann O, Zwaka TP, Marx N, Torzewski
M, Bucher A, Guilliard P, Hannekum A, Hombach V and Torzewski J:
Serum starvation and growth factor receptor expression in vascular
smooth muscle cells. J Vasc Res. 43:157–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kappert K, Paulsson J, Sparwel J, Leppänen
O, Hellberg C, Ostman A and Micke P: Dynamic changes in the
expression of DEP-1 and other PDGF receptor-antagonizing PTPs
during onset and termination of neointima formation. FASEB J.
21:523–534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: A randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili
R, Bjarnason GA, et al: Overall survival and updated results for
sunitinib compared with interferon alfa in patients with metastatic
renal cell carcinoma. J Clin Oncol. 27:3584–3590. 2009. View Article : Google Scholar
|
|
13
|
Ishii S, Okamoto Y, Katsumata H, Egawa S,
Yamanaka D, Fukushima M and Minami S: Sunitinib, a small-molecule
receptor tyrosine kinase inhibitor, suppresses neointimal
hyperplasia in balloon-injured rat carotid artery. J Cardiovasc
Pharmacol Ther. 18:359–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zohlnhöfer D, Hausleiter J, Kastrati A,
Mehilli J, Goos C, Schühlen H, Pache J, Pogatsa-Murray G, Heemann
U, Dirschinger J and Schömig A: A randomized, double-blind,
placebo-controlled trial on restenosis prevention by the receptor
tyrosine kinase inhibitor imatinib. J Am Coll Cardiol.
46:1999–2003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen YX, Ma X, Whitman S and O'Brien ER:
Novel antiinflammatory vascular benefits of systemic and
stent-based delivery of ethylisopropylamiloride. Circulation.
110:3721–3726. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lankheet NA, Steeghs N, Rosing H,
Schellens JH, Beijnen JH and Huitema AD: Quantification of
sunitinib and N-desethyl sunitinib in human EDTA plasma by liquid
chromatography coupled with electrospray ionization tandem mass
spectrometry: Validation and application in routine therapeutic
drug monitoring. Ther Drug Monit. 35:168–176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Andrés-Manzano MJ, Andrés V and Dorado B:
Oil Red O and Hematoxylin and eosin staining for quantification of
atherosclerosis burden in mouse aorta and aortic root. Methods Mol
Biol. 1339:85–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schwartz RS, Edelman E, Virmani R, Carter
A, Granada JF, Kaluza GL, Chronos NA, Robinson KA, Waksman R,
Weinberger J, et al: Drug-eluting stents in preclinical studies:
Updated consensus recommendations for preclinical evaluation. Circ
Cardiovasc Interv. 1:143–153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hong MK, Kornowski R, Bramwell O, Ragheb
AO and Leon MB: Paclitaxel-coated Gianturco-Roubin II (GR II)
stents reduce neointimal hyperplasia in a porcine coronary in-stent
restenosis model. Coron Artery Dis. 12:513–515. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kok SH, Gambari R, Chui CH, Lau FY, Cheng
GY, Lai PB, Lam WS, Chan AS, Cheng CH, Teo IT, et al: Paradoxical
proliferative potential of iron (II) sulphate on cancer cells after
the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay. Int J Mol Med. 19:971–975. 2007.PubMed/NCBI
|
|
21
|
Jandt E, Mutschke O, Mahboobi S, Uecker A,
Platz R, Berndt A, Böhmer FD, Figulla HR and Werner GS: Stent-based
release of a selective PDGF-receptor blocker from the
bis-indolylmethanon class inhibits restenosis in the rabbit animal
model. Vascul Pharmacol. 52:55–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seo J, Lee HS, Ryoo S, Seo JH, Min BS and
Lee JH: Tangeretin, a citrus flavonoid, inhibits PGDF-BB-induced
proliferation and migration of aortic smooth muscle cells by
blocking AKT activation. Eur J Pharmacol. 673:56–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Raines EW: PDGF and cardiovascular
disease. Cytokine Growth Factor Rev. 15:237–254. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hua Y, Dolence J, Ramanan S, Ren J and
Nair S: Bisdemethoxycurcumin inhibits PDGF-induced vascular smooth
muscle cell motility and proliferation. Mol Nutr Food Res.
57:1611–1618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Murrell M, Khachigian L and Ward MR: The
role of c-jun in PDTC-sensitive flow-dependent restenosis after
angioplasty and stenting. Atherosclerosis. 194:364–371. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jia L, Wang R and Tang DD: Abl regulates
smooth muscle cell proliferation by modulating actin dynamics and
ERK1/2 activation. Am J Physiol Cell Physiol. 302:C1026–C1034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai Y, Knight WE, Guo S, Li JD, Knight PA
and Yan C: Vinpocetine suppresses pathological vascular remodeling
by inhibiting vascular smooth muscle cell proliferation and
migration. J Pharmacol Exp Ther. 343:479–288. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Joner M, Finn AV, Farb A, Mont EK,
Kolodgie FD, Ladich E, Kutys R, Skorija K, Gold HK and Virmani R:
Pathology of drug eluting stents in humans: Delayed healing and
late thrombotic risk. J Am Coll Cardiol. 48:193–202. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Finn AV, Nakazawa G, Joner M, Kolodgie FD,
Mont EK, Gold HK and Virmani R: Vascular responses to drug eluting
stents: Importance of delayed healing. Arterioscler Thromb Vasc
Biol. 27:1500–1510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakazawa G, Finn AV, Joner M, Ladich E,
Kutys R, Mont EK, Gold HK, Burke AP, Kolodgie FD and Virmani R:
Delayed arterial healing and increased late stent thrombosis at
culprit sites after drug-eluting stent placement for acute
myocardial infarction patients: An autopsy study. Circulation.
118:1138–1145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Houtgraaf JH and Duckers HJ: Endothelial
progenitor cell (EPC) capture to aid vascular repair following
coronary stenting: A new frontier in stent technology?
EuroIntervention. 4:C67–C71. 2008.PubMed/NCBI
|