Introduction
Parkinson's disease (PD) is a neurodegenerative
disorder characterized by the selective and progressive loss of
dopaminergic (DA) neurons in the substantia nigra. The majority of
cases are sporadic and of unknown etiology. Several lines of
evidence indicate that brain inflammation specifically activates
microglia, leading to the pathogenesis of PD (1,2). The
activated microglia secrete high levels of inflammatory mediators,
including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β),
eicosanoids, nitric oxide (NO) and reactive oxygen species
(2–4). These inflammatory mediators impair
neurons and further activate microglia, resulting in a vicious
cycle, which promotes further inflammation and neurodegeneration
(5–7). Microglial activation is an integral
aspect of inflammatory processes in the brain. The molecular
mechanism through which microglial activation occurs in patients
with PD remains to be fully elucidated.
The P2X7 receptor (P2X7R) is a purinergic,
ATP-binding receptor, which is expressed at high levels in cells of
monocyte/macrophage lineages. This receptor is important in the
innate immune system. In the central nervous system (CNS), the
extensive functional expression of P2X7R is detected in microglia,
which are resident macrophages of the brain (8). The leakage of ATP from damaged cells
signals the proliferation and activation of microglia by binding
P2X7R (9). As a unique member of
the purinergic receptor family, P2X7R is strictly associated with
the maturation and release of the IL-1β cytokine in microglia
(10). The activation of P2X7R in
microglia has been correlated with the production of
proinflammatory cytokines and chemokines, including TNF-α (11) and CC-chemokine ligand 3 (12). In microglial cells, activated P2X7R
also stimulates the production of superoxide and NO (13–15).
Therefore, P2X7R is considered to be a key in eliciting an
inflammatory response in microglia as it stimulates microglial
activation. Thus, it has the potential to lead to a deleterious
cycle of neuroinflammation and neurodegeneration (16,17).
The expression of P2X7R is enhanced in several types
of brain pathology, in which the presence of activated microglia is
a concurrent feature (18). The
expression of P2X7R is not only upregulated in the brains of
patients with PD (19), but also
in animal models of various neurodegenerative diseases, including
multiple sclerosis (MS), Alzheimer's disease (AD) and Huntington's
disease (HD) (20–22). Whether P2X7R has a beneficial or
detrimental role in the development of these diseases remains to be
elucidated, although a number of studies have demonstrated that the
inhibition or deficit of P2X7R has neuroprotective effects in
animal models of MS (20), HD
(22) and AD (21). Several lines of evidence have
confirmed that the P2X7R pathway causes neuronal injury, leading to
the progression of neurodegeneration (17,20).
Although reports on the role of P2X7R in PD are rare and
contradictory, previous studies have established that this receptor
is upregulated in an animal model of PD (23,24),
although a P2X7R antagonist was not associated with the reduction
of DA cell loss (24,25). However, a previous study provided
evidence supporting the suggestion that P2X7R antagonism attenuates
the neuronal dysfunction and damage in an animal model of PD
(23). This discrepancy in
previous reports may be attributed to the use of different animal
models. Animal models of PD, which are established through exposure
of the animals to toxins, including 6-hydroxydopamine,
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or rotenone, are based
on the destruction of DA neurons in the nigra. P2X7R may be
mediating the microglial inflammatory responses in PD. By
excessively activating microglia, P2X7R may be promoting DA neuron
damage. The present study hypothesized that inhibiting this
receptor may alleviate the progression of PD.
When P2X7Rs are stimulated, they activate
mitogen-activated protein kinases (MAPKs), including extracellular
signal-regulated kinases, c-Jun N-terminal kinases and p38 MAPKs
(26). p38 MAPKs have been
implicated in the release of immune-associated cytotoxic factors,
including NO and pro-inflammatory cytokines in neuroglia. Thus, p38
MAPK is pivotal in glial-mediated or inflammation-mediated
neurotoxicity (27). Inhibition of
p38 MAPK has been found to reduce the production of inflammatory
factors following exposure to the bacterial endotoxin,
lipopolysaccharide (LPS), in an animal model of PD (27,28).
Thus, the expression of p38 MAPK appears to be regulated following
activation of P2X7R in the substantia nigra. The intranigral LPS
model is suitable for examining the potential neuroprotective
effects of P2X7R antagonists on the neuroinflammatory processes
leading to cell death. LPS-induced inflammation causes the loss of
DA neurons and elicits a microglial inflammatory response in the PD
rat model (29,30).
In the present study, the immunoreactivity of P2X7R
in microglia present in the LPS-injured substantia nigra was
investigated. Furthermore, immunohistochemical analysis was
performed to determine whether the P2X7R antagonist, brilliant blue
G (BBG) (31) prevents LPS-induced
injury in nigrostriatal DA neurons (29,30).
The present study aimed to test the hypothesis that P2X7R causes
the loss of DA neurons in the substantia nigra by activating p38
MAPK in an LPS animal model of PD. It was found that selective
inhibition of the P2X7R affords marked protection of DA neurons
from LPS-induced cytotoxicity by suppressing p38 MAPK in the
microglia of the substantia nigra in rats.
Materials and methods
LPS model of PD
Male Sprague-Dawley rats (n=42; 250–300 g) used in
the present study were obtained from the Laboratory Animal Center
of the China Medical University (Shenyang, China). Rats were housed
in a room with the temperature maintained at 22±1°C. The relative
humidity of the room was 55%. The rats were subjected to a 12-h
light/dark cycle with free access to food and water. All animal
experiments were performed in accordance with the guidelines by the
Committee on Animal Research at the University of China Medical
University (Shenyang, China). The experimental protocol was
approved by the institutional ethics committee of China Medical
University (Shenyang, China).
The rats were anesthetized with chloral hydrate (400
mg/kg) and positioned in a stereotactic apparatus. LPS (5.0 µl; 2
µg/µl) was purchased from Sigma-Aldrich; Merck Millipore
(Darmstadt, Germany) and injected into the right substantia nigra
(30,32,33).
Injections were administered at the following locations: 5.5 mm
posterior to the bregma, 2 mm lateral to the midline, 8.0 mm
ventral to the dura. LPS was delivered over a period of 5 min, with
the needle remaining in situ for 5 min prior to removal (1
mm/min), thus reflux was prevented along the injection tract. A
total of 12 rats were treated only with intranigral injection of
LPS. BBG and SB203580 groups (n=12 for each group) were treated
with BBG or SB203580 following LPS administration. A total of six
rats were included in the control group and were administered with
0.5 µl phosphate-buffered saline (PBS) injections.
The preparation and administration of BBG, the P2X7R
antagonist, were performed according to previously described
procedures (21). The BBG
(Sigma-Aldrich; Merck Millipore) was dissolved in saline and
injected intraperitoneally at a dose of 50 mg/kg 1 h prior to
administering the LPS injection. The same dose was administered for
15 days (BBG injection administered 1 h prior to LPS). It has
previously been reported that this BBG treatment protocol is
effective at inhibiting LPS-induced inflammatory reactions in rat
brains (34).
In order to investigate the role of the p38 MAPK
signaling pathway in LPS-induced neuroprotection, SB203580
(Sigma-Aldrich; Merck Millipore), a selective p38 MAPK antagonist,
was injected intracerebroventricularly. Immediately following LPS
injection, a stainless steel guide cannula was lowered into the
right lateral cerebral ventricle using standard stereotaxic
procedures (1.0 mm posterior to the bregma, 1.5 mm lateral from the
midline, 3.5 mm ventral to dura). SB203580 solution (1 mg/ml) was
prepared in 3% dimethyl sulfoxide, and 10 µl of this solution was
injected directly into the right lateral ventricle of the
experimental rats via the stainless steel cannula connected with
polyethylene tubing (35,36). The same dose was injected daily for
15 days.
Immunohistochemistry
At 15 days post-LPS injection, the animals were
euthanized by chloral hydrate (600 mg/kg) and perfused with 100 ml
of 0.9% NaCl followed by 4% paraformaldehyde, and their brains were
processed into 30 µm slices via cryostat sectioning. Tyrosine
hydroxylase (TH) immunoreactivity was determined using an
avidin-biotin-peroxidase method. To inhibit endogenous peroxidase
activity, the sections were incubated for 30 min in 1%
H2O2 solution. These sections were then
blocked with 5% (v/v) normal goat serum in PBS for 1 h.
Subsequently; the sections were incubated overnight with rabbit
anti-TH polyclonal antibody (cat. no. 2792; 1:1,000; Cell Signaling
Technology, Beverly, MA, USA) at 4°C. The brain sections were then
incubated with biotinylated goat anti-rabbit secondary antibody
(cat. no. A0277; 1:500, Beyotime Institute of Biotechnology,
Haimen, China) for 2 h at room temperature. Subsequently, the brain
sections were incubated with avidin-conjugated horseradish
peroxidase for 1 h at 37°C. The sections were then incubated with
the peroxidase substrate, diaminobenzidine, to develop a stain of
desired intensity observed by a light microscope at ×100
magnification (BX60; Olympus Corporation, Tokyo, Japan).
The microglia in the substantia nigra were
identified by their immunoreactivity to the microglial marker
anti-ionized calcium binding adapter molecule 1 (Iba-1). Double
immunolabeling for P2X7R and Iba-1 through use of red and green
fluorescence labeling was performed with the aim of determining the
localization of P2X7R immunoreactivity on the microglia cells.
Similarly, to determine the localization of phosphorylated (p-)p38
MAPK immunoreactivity in microglia cells, double immunolabeling of
p-p38 MAPK and Iba-1 was performed. The sections were incubated
overnight at 4°C with the following primary antibodies: Rabbit
anti-P2X7R polyclonal antibody (cat. no. ab77413; 1:1,000; Abcam,
Cambridge, UK), goat anti-Iba-1 polyclonal antibody (cat. no.
ab5076; 1:500; Abcam), and rabbit anti-p-p38 MAPK monoclonal
antibody (cat. no. 4511; 1:1,000; Cell Signaling Technology). After
washing with 0.1 M PBS, the sections were incubated for 2 h at room
temperature in a solution containing appropriate donkey secondary
antibodies conjugated to Alexa Fluor 488 and 594 (cat. nos. A11055
and A21207; 1:500; Molecular Probes; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). For immunostaining controls, primary
antibody was omitted in all staining procedures. Digitized images
of the immunostained sections were captured using a Zeiss
Axioplan-2 light microscope (Zeiss GmbH, Jena, Germany), which was
equipped with a digital camera.
The number of TH-positive neurons in the substantia
nigra was estimated using an optical fractionator technique and
Stereo-Investigator™ software (MBF Bioscience, Inc., Williston, VT,
USA). Every sixth section in the entire substantia nigra was
selected for analysis (−4.56 to −6.48 mm posterior to the bregma).
A fractionator sampling scheme was used comprising counting frames
(80×40 mm), which were superimposed on sections at intervals of
x=150 mm and y=200 mm (37).
Measurements of the expression of P2X7R and p-p38
MAPK, and microgliosis (Iba-1 marker) were performed on
immunostained sections by quantifying the integrated optical
density (IOD) of immunoreactivity using Image-Pro Plus 6.0 software
(Media Cybernetics, Inc., Bethesda, MD, USA). In each stained
section, four non-overlapping fields within the substantia nigra
regions were selected. The nomenclature and boundaries of brain
structures were in agreement with those described in the rat brain
atlas of Paxinos and Watson (38).
A fixed setting was used throughout the entire process, and the
background reading was acquired from immune-negative regions of
each section.
Western blot analysis
The rats were sacrificed by decapitation, following
which the substantia nigra was isolated and immediately frozen in
liquid nitrogen. The nigral tissue was lysed with ultrasound for
<30 sec on ice and buffer containing the following reagents: 150
mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 1% sodium
deoxycholate, 1% Triton X-100, 0.1% SDS and 1 mM PMSF. The
homogenates were centrifuged at 12,000 × g for 20 min at 4°C and
the supernatants were carefully removed. Protein concentration was
determined using a bicinchoninic acid protein assay method with
bovine serum albumin (BSA) as the standard. The samples were boiled
in sodium dodecyl sulphate-polyacrylamide electrophoresis
(SDS-PAGE) loading buffer for 5 min. Protein samples (40 µg/lane)
were loaded and were separated by SDS-PAGE (12% polyacrylamide
gels). Subsequently, the samples were electrotransferred onto
nitrocellulose membranes. These membranes were blocked with 10%
non-fat dry milk in Tris-buffered saline for 1 h at room
temperature, and incubated overnight at 4°C with rabbit anti-p38
MAPK monoclonal antibody (cat. no. 8690; 1:1,000; Cell Signaling
Technology), p-p38 MAPK (cat. no. 4511; 1:1,000; Cell Signaling
Technology) and mouse monoclonal anti-tubulin (cat. no. AT819;
1:1,000; Beyotime Institute of Biotechnology) diluted in 2% BSA in
PBS. The immunoblots were processed with appropriate horseradish
peroxidase-conjugated secondary antibodies: Goat anti-mouse and
goat anti-rabbit (cat. no. A0216 and A0208; 1:1,000; Beyotime
Institute of Biotechnology) for 2 h at room temperature. The bands
were visualized with an ECL Plus chemiluminescence reagent kit
(Beyotime Institute of Biotechnology), and the density of
immunoreactive bands was quantified using NIH ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The results are expressed as the mean ± standard
error of the mean. Statistical analysis of the data was performed
using one-way analysis of variance, followed by the least
significant difference test and Student-Newman-Keuls test, which
were performed using the Statistical Package for Social Sciences
software (version 16.0; SPSS, Inc., Chicago, IL, USA). In all the
cases, P<0.05 was considered to indicate a statistically
significant difference.
Results
P2X7R antagonism and the inhibition of
p38 MAPK reduce the damage of DA neurons in the LPS model of PD in
rats
To determine whether P2X7R causes the loss of DA
neurons, the present study evaluated the effects of the P2X7R
antagonist, BBG, on the LPS-induced loss of DA neurons in the
substantia nigra of a rat model of PD. The loss of DA neurons,
which was caused by LPS injection, was assessed
immunohistochemically by counting TH-immunoreactive (TH-ir) neurons
in the substantia nigra (Fig. 1).
Compared with the sham control group, there was a significant
reduction (P<0.001) in the number of TH-ir nerve cells 15 days
following injury of DA neurons in the rats treated with LPS (PBS
group, 5,068±470; LPS group, 2,249±492). An intranigral injection
of LPS reduced TH-ir neurons in the substantia nigra by ~55.6%,
however, those in the adjacent ventral tegmental area were spared
(Fig. 1A). Treating the
LPS-treated animals with BBG partially reversed the reduction of
TH-ir neurons on the lesion side. The loss of TH-ir neurons in the
substantia nigra was initially 2,249±492 neurons following
induction of the LPS-induced lesion. This loss was significantly
reduced to 3,867±478 neurons in the LPS-injected rats, which were
treated with BBG for 15 days following LPS (Fig 1B; P=0.039). Compared with the
control animals treated with PBS, TH-ir cells were reduced by
approximately 23.7% in the LPS-treated animals treated with BBG for
15 days. To ascertain whether p38 MAPK was involved in the
LPS-induced loss of DA neurons, the p38 MAPK inhibitor, SB203580,
was applied following injection with LPS in the substantia nigra of
rats. The loss of TH-ir neurons was 2,249±492 when the rats were
treated with LPS; however, SB203580 led to a significant reduction
in the loss of TH-ir neurons to 3,353±424 neurons (Fig. 1B; P<0.001). Compared with the
control animals treated with PBS, TH-ir cells were reduced by 33.8%
in the LPS-treated animals exposed to SB203580, the p38 MAPK
inhibitor. These results indicated that BBG, the specific P2X7R
antagonist, and SB203580, the p38 MAPK inhibitor, offered
neuroprotection to the rats in the LPS model of PD.
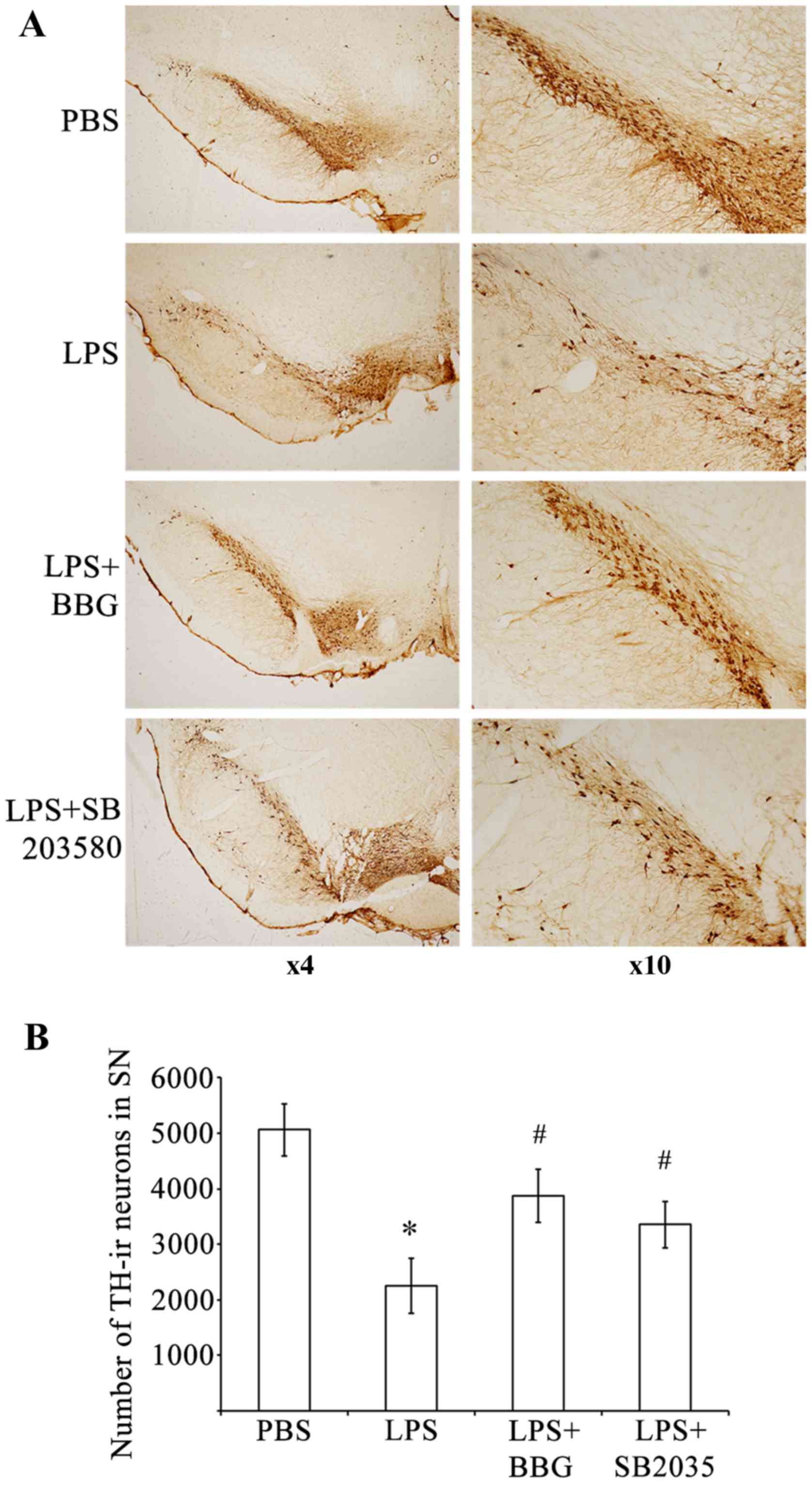 | Figure 1.BBG and SB203580 attenuate
LPS-induced death of DA neurons in the SNpc of rats. (A)
Representative examples of TH-stained sections of SNpc, adjacent
VTA and SNr in rats of the PBS control group, LPS group, LPS+BBG
group and LPS+SB203580 group. (B) Quantification of the number of
TH-ir neurons in the SNpc of rats in the PBS, LPS+BBG and
LPS+SB203580 groups. Data were obtained from six independent
animals (n=6). Compared with the SNpc in the PBS control group,
rats injected with LPS exhibited a significant reduction in the
number of TH-positive cells in the SNpc region. BBG and SB203580
significantly reduced the size of the SNpc lesion induced by LPS.
*P<0.001, compared with the PBS control; #P<0.001,
compared with the LPS group. LPS, lipopolysaccharide; PBS,
phosphate-buffered saline; DA, dopaminergic; BBG, brilliant blue G;
TH-ir, tyrosine hydroxylase immunoreactive; SNpc, substantia nigra
pars compacta. |
LPS-induced upregulation of the
expression of P2X7R and activation of microglia are attenuated by
P2X7R antagonism and p38 MAPK inhibition in the substantia nigra of
in the LPS model of PD
The present study performed immunohistochemical
analysis to determine whether P2X7R had an active role in
LPS-induced DA neuronal damage, which was mediated by microglia.
For this analysis, antibodies against P2X7R and the microglia
marker, Iba-1, were used. The analysis was performed in the
substantia nigra tissue of the LPS-treated rats. Whereas few
P2X7R-ir cells were present in the substantia nigra of the
PBS-injected control animals (Fig.
2), cells expressing a high level of P2X7R were identified in
the LPS-injured substantia nigra. Of note, the P2X7R antagonist,
BBG, was effective in reducing the number of P2X7R-ir cells. The
p38 MAPK inhibitor, SB203580, was also effective to a certain
extent in reducing P2X7R-ir.
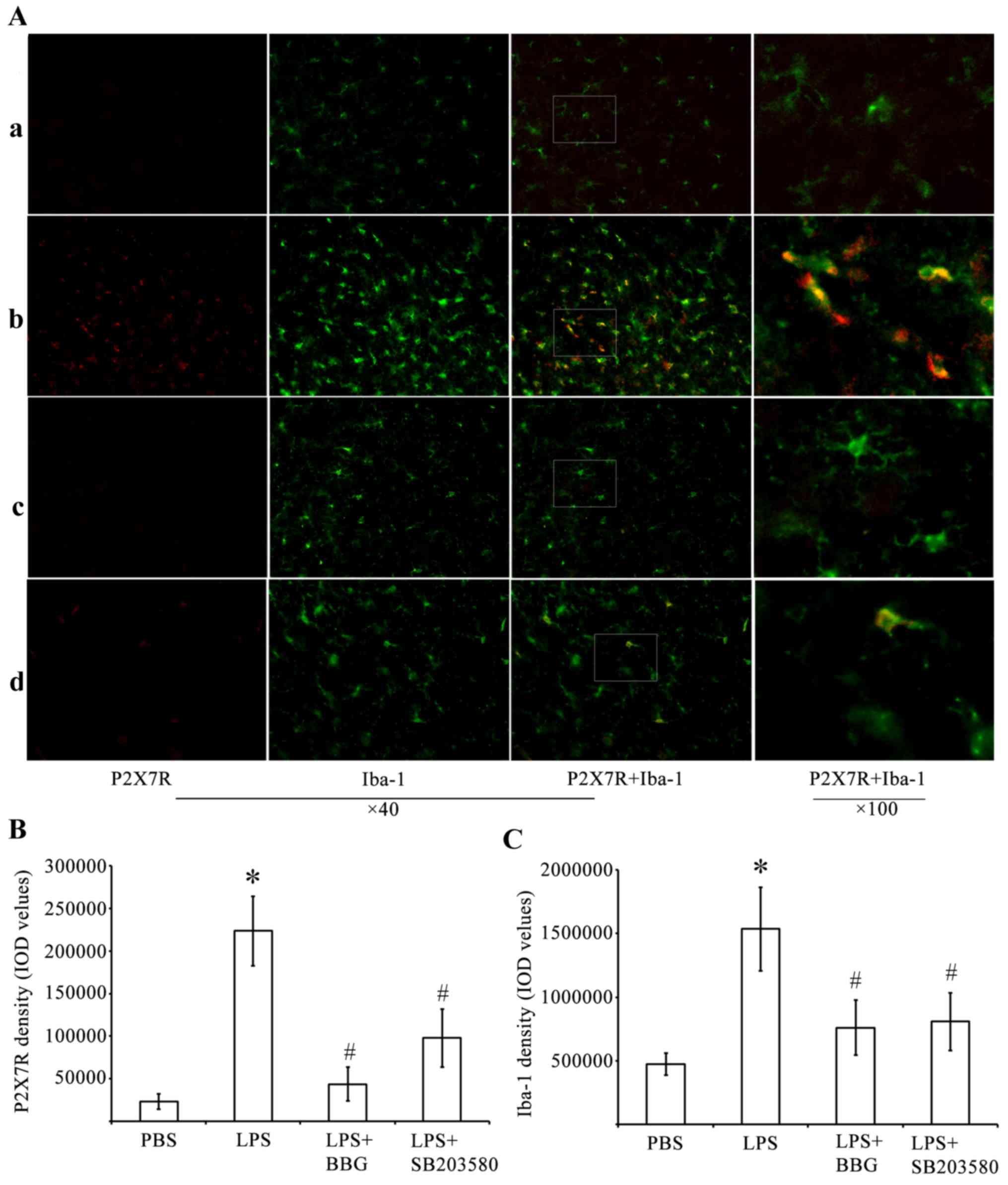 | Figure 2.Double immunofluorescence labeling of
P2X7R and Iba-1 in the SNpc of rats in the LPS model of Parkinson's
disease. (A) Immunofluorescence for P2X7R (red) and Iba-1, (green)
are shown for the (a) PBS, (b) LPS, (c) LPS+BBG and (d)
LPS+SB203580 groups. Co-immunolabeled P2X7R/Iba-1 microglia are
yellowish in color. Marked microglial activation was observed in
the SNpc of the LPS group. In the PBS group, microglia had an
inactive morphology with small soma and thin processes, whereas
microglia exposed to LPS had enlarged soma-lacking processes. BBG
significantly decreased microglia activation. Immunostaining for
P2X7R showed a marked increase in the LPS animals, compared with
PBS animals. BBG was effective in attenuating P2X7R-ir in
LPS-injected SNpc. Double-labeling immunohistochemistry identified
P2X7R-ir predominantly in activated, Iba-1-ir microglia, following
LPS treatment. SB203580 significantly inhibited the upregulation of
P2X7R and microglia activation in SNpc. Quantitative analysis of
IOD values of (B) P2X7R-ir and (C) Iba-1 in the groups (n=6) is
shown. *P<0.05, compared with the PBS control;
#P<0.05, compared with the LPS group. LPS,
lipopolysaccharide; PBS, phosphate-buffered saline; BBG, brilliant
blue G; SNpc, substantia nigra pars compacta; Iba-1, ionized
calcium binding adapter molecule 1; -ir, immunoreactive; IOD,
integrated optical density. |
Double-labeling experiments were performed using
antibodies against P2X7R and Iba-1. These experiments showed that
P2X7R-ir was distributed predominantly within activated
immunoreactive Iba-1 (Iba-1-ir) microglial cells of the LPS-treated
substantia nigra pars compacta (SNpc; Fig. 2A). A higher intensity of staining
of P2X7R in cells correlated with increased Iba-1-ir potency. LPS
induced microglial activation by increasing P2X7R-ir in the
substantia nigra. The activated microglial cells were characterized
by minimum ramification, hypertrophy and proliferation. The
activated microglia were positive for Iba-1, and were distributed
in the ventral tegmental area, SNpc and substantia nigra pars
reticulata (SNpr). In the substantia nigra of the PBS-injected
control animals, Iba-1-ir cells had elongated nuclei and ramifying
processes, which are typical of inactivated microglia.
By performing quantitative analysis of the IOD
values of P2X7R (Fig. 2B) and
Iba-1 (Fig. 2C), it was found that
the IOD of P2X7R-ir was significantly increased by 962% in the
substantia nigra of the LPS-treated rats. By contrast, the IOD
values of P2X7R-ir decreased by 80.5 and 56% in the substantia
nigra of BBG- and SB203580-treated LPS-injected rats, respectively
(Fig. 2B; P<0.05). At 15 days
post-LPS injection, the experimental rats exhibited a 3.25-fold
increase in the density of Iba-1 in the substantia nigra, compared
with the controls (Fig. 2C;
P<0.05). BBG and SB203580 were effective in reducing microglial
responses in the LPS-stimulated animals. The IOD of Iba-1 in the
substantia nigra decreased by 51 and 47% in the BBG- and
SB203580-treated rats, respectively, compared with the levels
observed in LPS-treated rats (Fig.
2C; P<0.05).
LPS-induced upregulation of p-p38 MAPK
is dependent on P2X7R
Immunohistochemistry and western blot analysis were
performed to determine whether LPS treatment activated p38 MAPK.
Compared with the control rats injected with PBS, the expression of
p-p38 MAPK was increased in the substantia nigra of the
experimental rats injected with LPS (Fig. 3). The immunoreactivity of p-p38
MAPK was decreased in the substantia nigra of BBG-treated and
SB203580-treated rats, which were also injected with LPS.
Double-labeling immunohistochemistry was performed using antibodies
against p-p38 MAPK (Fig. 3A; red)
and the microglial marker Iba-1 (Fig.
3A; green). The immunohistochemical analysis showed that,
compared with the PBS-injected control animals, the activated
microglia increased the expression levels of p-p38-MAPK in rats
administered with LPS. Therefore, quantitative analysis of IOD
values of p-p38 MAPK (Fig. 3B) was
performed. The analysis revealed that the IOD for p-p38-ir was
significantly increased by 845% in the substantia nigra of the
LPS-treated rats, whereas the same parameter decreased by 77.9 and
88.7% in the BBG- and SB203580-treated LPS-induced rats,
respectively (Fig. 3B; P<0.05).
Compared with the controls, the density of Iba-1 in the substantia
nigra of the experimental rats was increased by 3.62-fold 15 days
post LPS injection (Fig. 3C;
P<0.05). BBG and SB203580 were effective in reducing the
microglial responses of the LPS-treated animals. Compared with the
IOD of Iba-1 in the substantia nigra of the LPS-treated rats, the
IOD of Iba-1 decreased by 60.6 and 52.8% in the BBG- and
SB203580-treated LPS-injected rats, respectively (Fig. 3C; P<0.05).
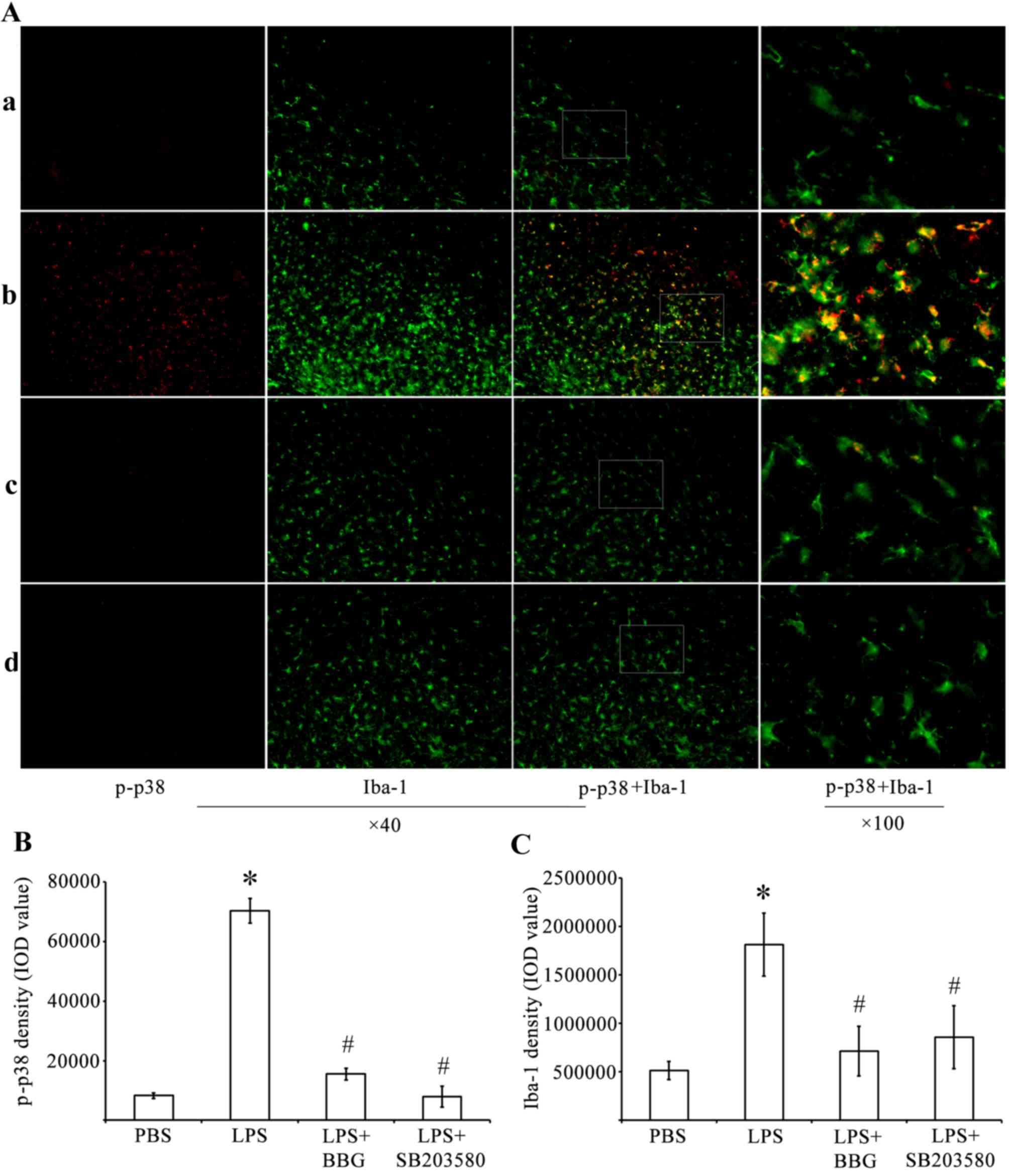 | Figure 3.Double immunofluorescence labeling of
p-p38 MAPK and Iba-1 in the SNpc of rats in the LPS model of
Parkinson's disease. (A) Immunofluorescence for p-p38 MAPK (red)
and Iba-1 (green) are shown for the (a) PBS, (b) LPS, (c) LPS+BBG
and (d) LPS+SB203580 groups. Co-immunolabeled p-p38/Iba-1 microglia
are a yellowish color. Compared with the PBS rats, LPS increased
the expression of p-p38 MAPK. Quantitative analysis of IOD values
of (B) p-p38 and (C) Iba-1 immunofluorescence in the SNpc of rats
(n=6). *P<0.05, compared with the PBS control;
#P<0.05, compared with the LPS group. MAPK,
mitogen-activated protein kinase; LPS, lipopolysaccharide; PBS,
phosphate-buffered saline; SNpc, substantia nigra pars compacta;
Iba-1, ionized calcium binding adapter molecule 1; p-,
phosphorylated; IOD, integrated optical density. |
Similar results were obtained when western blot
analysis was performed using antibodies against p-p38 MAPK
(Fig. 4A). Quantification of the
western blots revealed that, compared with the PBS-injected control
group, the LPS-injected group showed a significant increase in the
levels of p-p38 MAPK, but not p38 MAPK (Fig. 4B). In addition, BBG, the P2X7R
antagonist, and SB203580, the p38 MAPK inhibitor, significantly
reduced the activation of p38 MAPK in LPS-treated rats, as revealed
by the quantification of p38 MAPK bands. This indicated that BBG
and SB203580 offered protection against LPS-induced DA neuron death
in the substantia nigra of the experimental rats.
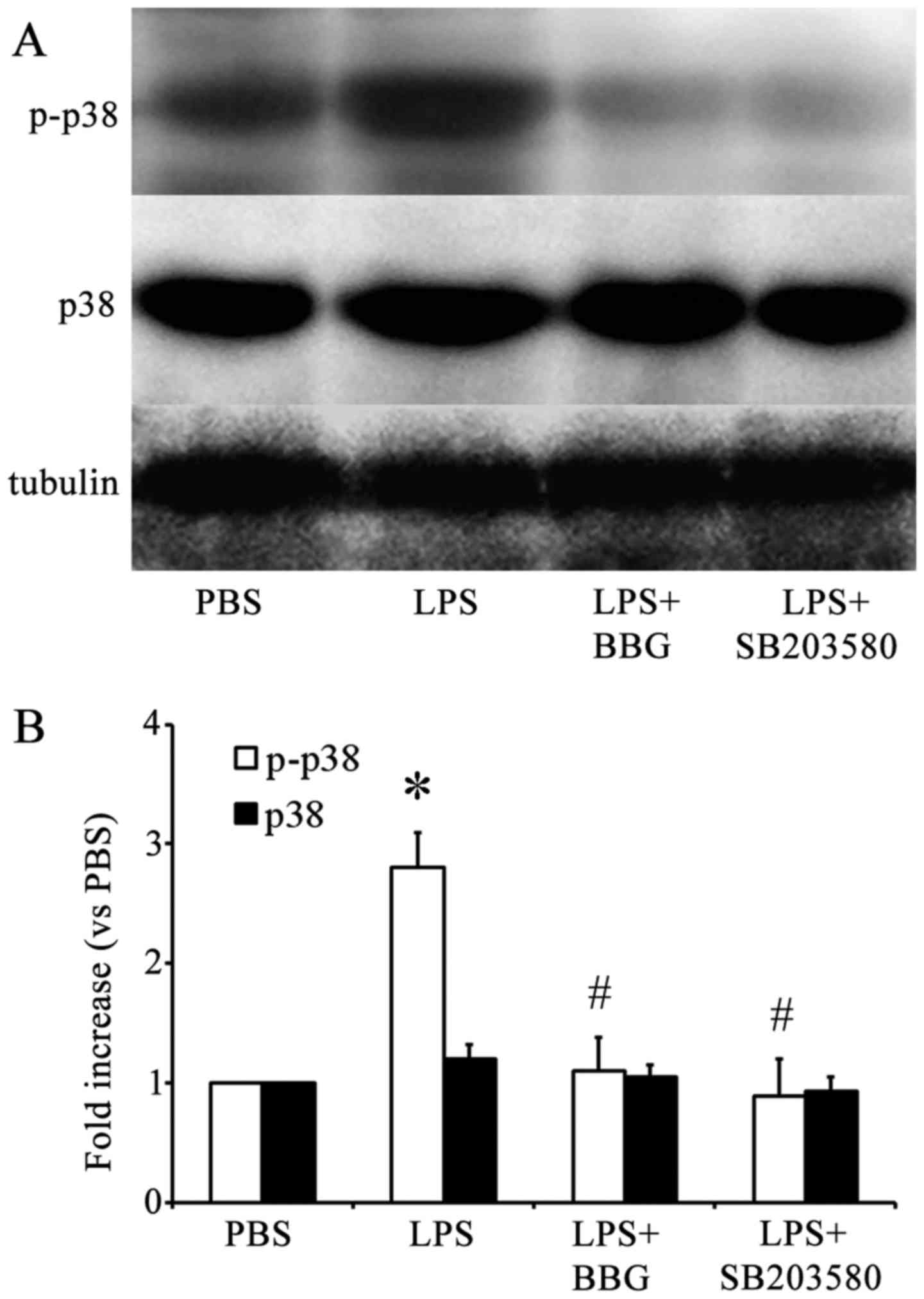 | Figure 4.Effect of BBG treatment on p38 MAPK
activation in substantia nigra in the LPS model of PD. (A)
Representative western blot analysis of the expression of p-p38 and
p38 MAPK in the substantia nigra of rats in the LPS model of PD.
(B) Quantitative analysis of western blots (n=6) of the expression
of p-p38 and p38 MAPK in the substantia nigra of rats in the PBS
control group, LPS group, LPS+BBG and LPS+SB203580 groups. Protein
bands were normalized to the expression levels of tubulin.
*P<0.05, compared with the PBS control; #P<0.05,
compared with the LPS group. PD Parkinson's disease; MAPK,
mitogen-activated protein kinase; LPS, lipopolysaccharide; PBS,
phosphate-buffered saline; BBG, brilliant blue G; SNpc, substantia
nigra pars compacta; Iba-1, ionized calcium binding adapter
molecule 1; p-, phosphorylated; IOD, integrated optical
density. |
Discussion
The chemical neuroanatomical analysis indicated that
the expression of P2X7R was upregulated in the substantia nigra
following LPS injection in experimental rats. When these
LPS-injected rats were treated with the P2X7R antagonist, BBG,
microglial activation was attenuated and a reduction in the loss of
TH-ir DA neurons was observed in the substantia nigra. In addition,
when the LPS-injected rats were treated with BBG, the activation of
p38 MAPK was reversed, microglial activation was attenuated and a
reduction in the loss of DA neurons was observed in the substantia
nigra. Similarly, SB203580, the p38 MAPK antagonist, attenuated the
activation of microglia, reduced the expression of P2X7R, and
protected DA neurons from LPS-induced neuronal damage in the
substantia nigra. These findings indicated that P2X7R activity,
mediated by the p38 MAPK signaling pathway, contributed to the
activation of microglia and the loss of DA neurons in LPS-injected
rats. These data are consistent with the hypothesis that P2X7R
contributes to the loss of DA neurons in the substantia nigra by
activating microglia via the p38 MAPK pathway.
Previously, it has been shown that the expression
and function of P2X7R are increased in patients with PD (19) and other neurodegenerative diseases
(39). The gene expression of
P2X7R has been found to be significantly upregulated in substantia
nigra samples obtained from patients, who were clinically and
neuropathologically diagnosed with idiopathic PD (19). In addition, genetic polymorphism of
P2X7R can affect the occurrence and development of sporadic PD
(40). Diseases, including
amyotrophic lateral sclerosis (ALS) and MS, are inflammatory
neurodegenerative disorders and, in tissue specimens of patients
with MS and ALS specimens, significantly higher densities of
P2X7R-ir microglial cells/macrophages have been found in the
affected regions of the brain (20). Another previous study provided
evidence that increased expression and function of P2X7R are
associated with the microglia of patients with AD, and indicates
that P2X7R is important in mediating microglial purinergic
inflammatory responses in AD brains (39). The levels of P2X7R have been
reported to be higher in the brains of HD mice, with P2X7R
antagonism attenuating neuronal apoptosis (22). In addition, P2X7R antagonists
ameliorated the motor performance of mice with experimental ALS
(20). In the present study, it
was found that P2X7R was upregulated in the substantia nigra
microglia of rats in the LPS model of PD. In addition, BBG, a P2X7R
antagonist, protected DA neurons from LPS-induced damage.
Therefore, the expression of P2X7R in microglia appeared to be a
critical factor in mediating microglial activity and stimulating
the loss of DA neurons in PD. These observations are consistent
with the suggestion that P2X7R is critical in neuroinflammation,
which is observed during the pathogenesis of a variety of
neurodegenerative diseases (14,17,18,20–23).
The mechanism through which P2X7R is upregulated in
neurodegenerative diseases remains to be fully elucidated, however,
it may be closely associated with the role of microglia in disease
progression. P2X7R triggers the maturation and release of the IL-1β
inflammatory cytokine from microglia (10). IL-1β is a crucial mediator in the
pathogenesis of inflammatory diseases of the CNS. Among the rats
included in the LPS model of PD, the LPS injection triggered
inflammatory mechanisms, which caused the degeneration of DA
neurons in the substantia nigra (29,33).
LPS is as a potent stimulator of glial cells, particularly
microglia, and has been a useful tool for modeling
inflammation-mediated neurodegeneration of DA in rats in an LPS
model of PD (30). At the
molecular level, LPS requires activated Toll-like receptor 4 (TLR4)
to induce its neurodegenerative effect, however, it also requires
activated microglia (41). A
previous study found that the production of microglial-derived
IL-1β occurs through the following mechanism: The activation of
multiple TLR isoforms (TLR2, TLR3 and TLR4) in the nervous system
elevates the levels of extracellular ATP and subsequently activates
P2X7R (42).
According to a previous in vivo study, when
LPS injection was administered into the striatum, it markedly
increased the expression of P2X7R in microglia, whereas the
inhibition of P2X7R increased neuronal survival in the striatum
(43). In addition, LPS stimulated
the cultured human microglia to enhance the cellular expression of
several proinflammatory factors, including cyclooxygenase-2, IL-1β,
IL-6, IL-12 and TNF-α, which are inhibited by P2X7R antagonists
(43). The double-labeling
experiments in the present study showed an upregulation of P2X7R in
activated IBA-1-ir microglial cells. BBG treatment provided
protection to DA neurons and reduced the activation of microglia.
This indicated that BBG exerted its neuroprotective effect by
suppressing the activation of microglia and inhibiting the
expression of P2X7R in activated microglia. These findings
suggested that P2X7R enhances its neuroinflammatory nigral
processes by activating microglia.
In the present study, it was also found that BBG
prevented the LPS-induced loss of DA neurons in the substantia
nigra. This finding supports an earlier study, which reported that
P2X7R antagonists significantly prevent 6-hydroxydopamine-induced
depletion of striatal DA stores (23,24).
However, other studies have reported that P2X7R deficiency or
inhibition is not effective against 1-methyl-4-phenylpyridinium or
rotenone-induced DA loss in chemical PD models (25). The discrepancies in different PD
models are attributed to the extent of the substantia nigra lesion
induced in different paradigms and/or the duration of treatment
with the P2X7R antagonist.
A previous study reported that P2X7R mediates the
phosphorylation of p38 MAPK during the progression of subarachnoid
hemorrhage (44). The significant
activation of p38 MAPK has also been observed in the substantia
nigra of other PD models (45),
and p38 MAPK inhibitors have provided significant neuroprotection
(28,45). Although LPS activates all the three
major MAPK pathways (46), the p38
MAPK pathway appears to the most closely associated with the
LPS-induced upregulation of inflammatory mediators (47). The p38 MAPK signaling pathway
inhibitor, SB203580, downregulates the expression of
pro-inflammatory mediators, including TNF-α and IL-1β (46). In glial cells, p38 MAPK induces NO
synthase to stimulate the production of NO, which underlies the
LPS-induced death of mesencephalic neurons (28). In the present study, it was found
that LPS induced an increase in the levels of p-p38 MAPK. In
addition, SB203580, the selective inhibitor of p38 MAPK, prevented
the LPS-induced loss of DA neurons in the substantia nigra of
experimental rats. BBG, a specific P2X7R antagonist, almost
completely inhibited p38 MAPK activation. Therefore, the inhibition
of P2X7R in the LPS-injected rats was neuroprotective as it reduced
p38 MAPK activation and the loss of DA neurons.
Several studies have suggested that P2X7R is present
in striatal DA terminals (23) and
astroglial cells (48), indicating
that P2X7R-mediated neurotoxicity is linked to microglial
activation. Other studies have reported that microglia are a
crucial contributing factor, which governs the ability of P2X7R in
controlling neurotoxicity (23,24,42,43).
In the present study, it was found that P2X7R was upregulated in
microglial cells following LPS-induced microgliosis; however, BBG
attenuated microgliosis. These findings support the hypothesis that
the localization of P2X7R on microglia is linked to its ability to
control the function of microglial cells.
In conclusion, the present study showed that the
increased expression of p38 MAPK and P2X7R in the substantia nigra
of rats caused the LPS-induced death of DA neurons. The results
provided evidence that the inhibition of P2X7R by BBG reduced
LPS-induced degeneration of DA neurons. Furthermore, there was a
reduction in the regional activation of microglia, which express
P2X7R protein. The interaction between P2X7R and the p38 MAPK
signaling pathway may have contributed to the loss of DA neurons in
the substantia nigra of the experimental PD rats. The results of
the present study suggested that substantia nigra DA neurons were
protected from neurodegeneration when P2X7R activity was inhibited
in activated microglial cells. These findings may be exploited for
developing neuroprotective therapies, which can be used in the
treatment of various neurodegenerative diseases.
Acknowledgements
This study was funded by the China National Nature
Science Fund (grant no. 81371421) and the Foundation of the
Liaoning Educational Committee (grant nos. L202013136 and
L2010560).
References
|
1
|
Hirsch EC, Vyas S and Hunot S:
Neuroinflammation in Parkinson's disease. Parkinsonism Relat
Disord. 18 Suppl 1:S210–S212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Appel SH: Inflammation in Parkinson's
disease: Cause or consequence? Mov Disord. 27:1075–1077. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qian L, Flood PM and Hong JS:
Neuroinflammation is a key player in Parkinson's disease and a
prime target for therapy. J Neural Transm (Vienna). 117:971–979.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ouchi Y, Yagi S, Yokokura M and Sakamoto
M: Neuroinflammation in the living brain of Parkinson's disease.
Parkinsonism Relat Disord. 15 Suppl 3:S200–S204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anderson KM, Olson KE, Estes KA, Flanagan
K, Gendelman HE and Mosley RL: Dual destructive and protective
roles of adaptive immunity in neurodegenerative disorders. Transl
Neurodegener. 3:252014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao HM, Jiang J, Wilson B, Zhang W, Hong
JS and Liu B: Microglial activation-mediated delayed and
progressive degeneration of rat nigral dopaminergic neurons:
Relevance to Parkinson's disease. J Neurochem. 81:1285–1297. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Politis M, Su P and Piccini P: Imaging of
microglia in patients with neurodegenerative disorders. Front
Pharmacol. 3:962012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaur C, Hao AJ, Wu CH and Ling EA: Origin
of microglia. Microsc Res Tech. 54:2–9. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Monif M, Reid CA, Powell KL, Smart ML and
Williams DA: The P2X7 receptor drives microglial activation and
proliferation: A trophic role for P2X7R pore. J Neurosci.
29:3781–3791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferrari D, Pizzirani C, Adinolfi E, Lemoli
RM, Curti A, Idzko M, Panther E and Di Virgilio F: The P2X7
receptor: A key player in IL-1 processing and release. J Immunol.
176:3877–3883. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki T, Hide I, Ido K, Kohsaka S, Inoue
K and Nakata Y: Production and release of neuroprotective tumor
necrosis factor by P2X7 receptor-activated microglia. J Neurosci.
24:1–7. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda
M and Inoue K: Activation of P2X7 receptors induces CCL3 production
in microglial cells through transcription factor NFAT. J Neurochem.
108:115–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gendron FP, Chalimoniuk M, Strosznajder J,
Shen S, González FA, Weisman GA and Sun GY: P2X7 nucleotide
receptor activation enhances IFN gamma-induced type II nitric oxide
synthase activity in BV-2 microglial cells. J Neurochem.
87:344–352. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Parvathenani LK, Tertyshnikova S, Greco
CR, Roberts SB, Robertson B and Posmantur R: P2X7 mediates
superoxide production in primary microglia and is up-regulated in a
transgenic mouse model of Alzheimer's disease. J Biol Chem.
278:13309–13317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartlett R, Yerbury JJ and Sluyter R: P2X7
receptor activation induces reactive oxygen species formation and
cell death in murine EOC13 microglia. Mediators Inflamm.
2013:2718132013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Monif M, Burnstock G and Williams DA:
Microglia: Proliferation and activation driven by the P2X7
receptor. Int J Biochem Cell Biol. 42:1753–1756. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Skaper SD, Facci L, Culbert AA, Evans NA,
Chessell I, Davis JB and Richardson JC: P2X(7) receptors on
microglial cells mediate injury to cortical neurons in vitro. Glia.
54:234–242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sperlágh B and Illes P: P2X7 receptor: An
emerging target in central nervous system diseases. Trends
Pharmacol Sci. 35:537–547. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Durrenberger PF, Grünblatt E, Fernando FS,
Monoranu CM, Evans J, Riederer P, Reynolds R and Dexter DT:
Inflammatory pathways in Parkinson's Disease; A BNE microarray
study. Parkinson's Dis. 2012:2147142012.
|
|
20
|
Yiangou Y, Facer P, Durrenberger P,
Chessell IP, Naylor A, Bountra C, Banati RR and Anand P: COX-2, CB2
and P2X7-immunoreactivities are increased in activated microglial
cells/macrophages of multiple sclerosis and amyotrophic lateral
sclerosis spinal cord. BMC Neurol. 6:122006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ryu JK and McLarnon JG: Block of
purinergic P2X(7) receptor is neuroprotective in an animal model of
Alzheimer's disease. Neuroreport. 19:1715–1719. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diaz-Hernández M, Díez-Zaera M,
Sánchez-Nogueiro J, Gómez-Villafuertes R, Canals JM, Alberch J,
Miras-Portugal MT and Lucas JJ: Altered P2X7-receptor level and
function in mouse models of Huntington's disease and therapeutic
efficacy of antagonist administration. FASEB J. 23:1893–1906. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carmo MR, Menezes AP, Nunes AC, Pliássova
A, Rolo AP, Palmeira CM, Cunha RA, Canas PM and Andrade GM: The
P2X7 receptor antagonist Brilliant Blue G attenuates contralateral
rotations in a rat model of Parkinsonism through a combined control
of synaptotoxicity, neurotoxicity and gliosis. Neuropharmacology.
81:142–152. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marcellino D, Suárez-Boomgaard D,
Sánchez-Reina MD, Aguirre JA, Yoshitake T, Yoshitake S, Hagman B,
Kehr J, Agnati LF, Fuxe K and Rivera A: On the role of P2X(7)
receptors in dopamine nerve cell degeneration in a rat model of
Parkinson's disease: Studies with the P2X(7) receptor antagonist
A-438079. J Neural Transm (Vienna). 117:681–687. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hracskó Z, Baranyi M, Csölle C, Gölöncsér
F, Madarász E, Kittel A and Sperlágh B: Lack of neuroprotection in
the absence of P2X7 receptors in toxin-induced animal models of
Parkinson's disease. Mol Neurodegener. 6:282011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lenertz LY, Gavala ML, Zhu Y and Bertics
PJ: Transcriptional control mechanisms associated with the
nucleotide receptor P2X7, a critical regulator of immunologic,
osteogenic, and neurologic functions. Immunol Res. 50:22–38. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhat NR, Zhang P, Lee JC and Hogan EL:
Extracellular signal-regulated kinase and p38 subgroups of
mitogen-activated protein kinases regulate inducible nitric oxide
synthase and tumor necrosis factor-alpha gene expression in
endotoxin-stimulated primary glial cultures. J Neurosci.
18:1633–1641. 1998.PubMed/NCBI
|
|
28
|
Jeohn GH, Cooper CL, Wilson B, Chang RC,
Jang KJ, Kim HC, Liu B and Hong JS: p38 MAP kinase is involved in
lipopolysaccharide-induced dopaminergic neuronal cell death in rat
mesencephalic neuron-glia cultures. Ann N Y Acad Sci. 962:332–346.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tufekci KU, Genc S and Genc K: The
endotoxin-induced neuroinflammation model of Parkinson's disease.
Parkinson's Dis. 2011:4874502011.
|
|
30
|
Dutta G, Zhang P and Liu B: The
lipopolysaccharide Parkinson's disease animal model: Mechanistic
studies and drug discovery. Fundam Clin Pharmacol. 22:453–464.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang LH, Mackenzie AB, North RA and
Surprenant A: Brilliant blue G selectively blocks ATP-gated rat
P2X(7) receptors. Mol Pharmacol. 58:82–88. 2000.PubMed/NCBI
|
|
32
|
Sui Y, Stanić D, Tomas D, Jarrott B and
Horne MK: Meloxicam reduces lipopolysaccharide-induced degeneration
of dopaminergic neurons in the rat substantia nigra pars compacta.
Neurosci Lett. 460:121–125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Herrera AJ, Castaño A, Venero JL, Cano J
and Machado A: The single intranigral injection of LPS as a new
model for studying the selective effects of inflammatory reactions
on dopaminergic system. Neurobiol Dis. 7:429–447. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gourine AV, Poputnikov DM, Zhernosek N,
Melenchuk EV, Gerstberger R, Spyer KM and Gourine VN: P2 receptor
blockade attenuates fever and cytokine responses induced by
lipopolysaccharide in rats. Br J Pharmacol. 146:139–145. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choe ES and McGinty JF:
N-Methyl-D-aspartate receptors and p38 mitogen-activated protein
kinase are required for cAMP-dependent cyclase response element
binding protein and Elk-1 phosphorylation in the striatum.
Neuroscience. 101:607–617. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu P, Zhan L, Zhu T, Liang D, Hu J, Sun
W, Hou Q, Zhou H, Wu B, Wang Y and Xu E: The roles of p38 MAPK/MSK1
signaling pathway in the neuroprotection of hypoxic
postconditioning against transient global cerebral ischemia in
adult rats. Mol Neurobiol. 49:1338–1349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stanic D, Finkelstein DI, Bourke DW, Drago
J and Horne MK: Timecourse of striatal re-innervation following
lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci.
18:1175–1188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paxinos G and Watson C: The Rat Brain: In
Stereotaxic Coordinates. Academic Press; Incorporated: 1998
|
|
39
|
McLarnon JG, Ryu JK, Walker DG and Choi
HB: Upregulated expression of purinergic P2X(7) receptor in
Alzheimer disease and amyloid-beta peptide-treated microglia and in
peptide-injected rat hippocampus. J Neuropathol Exp Neurol.
65:1090–1097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu H, Han X, Li Y, Zou H and Xie A:
Association of P2X7 receptor gene polymorphisms with sporadic
Parkinson's disease in a Han Chinese population. Neurosci Lett.
546:42–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lehnardt S, Massillon L, Follett P, Jensen
FE, Ratan R, Rosenberg PA, Volpe JJ and Vartanian T: Activation of
innate immunity in the CNS triggers neurodegeneration through a
Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA.
100:8514–8519. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Facci L, Barbierato M, Marinelli C,
Argentini C, Skaper SD and Giusti P: Toll-like receptors 2, −3 and
−4 prime microglia but not astrocytes across central nervous system
regions for ATP-dependent interleukin-1β release. Sci Rep.
4:68242014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Choi HB, Ryu JK, Kim SU and McLarnon JG:
Modulation of the purinergic P2X7 receptor attenuates
lipopolysaccharide-mediated microglial activation and neuronal
damage in inflamed brain. J Neurosci. 27:4957–4968. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen S, Ma Q, Krafft PR, Chen Y, Tang J,
Zhang J and Zhang JH: P2X7 receptor antagonism inhibits p38
mitogen-activated protein kinase activation and ameliorates
neuronal apoptosis after subarachnoid hemorrhage in rats. Crit Care
Med. 41:e466–e474. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu F, Wang Z, Gu JH, Ge JB, Liang ZQ and
Qin ZH: p38(MAPK)/p53-Mediated Bax induction contributes to neurons
degeneration in rotenone-induced cellular and rat models of
Parkinson's disease. Neurochem Int. 63:133–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pawate S, Shen Q, Fan F and Bhat NR: Redox
regulation of glial inflammatory response to lipopolysaccharide and
interferongamma. J Neurosci Res. 77:540–551. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krishna M and Narang H: The complexity of
mitogen-activated protein kinases (MAPKs) made simple. Cell Mol
Life Sci. 65:3525–3544. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Franke H, Verkhratsky A, Burnstock G and
Illes P: Pathophysiology of astroglial purinergic signalling.
Purinergic Signal. 8:629–657. 2012. View Article : Google Scholar : PubMed/NCBI
|














