Introduction
Ventricular remodeling, which is characterized by
progressive dilation and deterioration in cardiac performance, is a
critical process underlying the progression to heart failure and
subsequent mortality following myocardial infarction (MI). As a
result, the attenuation of adverse myocardial remodeling is one of
the most important aspects for improving prognosis following MI
(1). However, despite previous
advances in reperfusion therapy and optimized pharmacological
treatments, left ventricular (LV) remodeling is still observed in a
substantial proportion of patients, and progression to heart
failure can still occur in up to one-third of patients who have had
a MI (2,3). These data suggest that the current
clinical treatments targeting myocardial remodeling post-MI remain
inadequate. The promotion of therapeutic angiogenesis has been
demonstrated to be a promising approach to ameliorate adverse
myocardial remodeling (4).
Intermedin (IMD), also known as adrenomedullin 2, is
a novel member of the calcitonin/calcitonin gene-related peptide
(CGRP) family. The human IMD gene encodes a prepropeptide of 148
amino acids (preproIMD); proteolytic cleavage of preproIMD yields a
series of biologically active C-terminal fragments. Previous
studies have revealed that an endogenous degraded fragment termed
IMD1–53, which contains cleavage sites located between
two basic amino acids at Arg93-Arg94, possessed potent
cardioprotective activities in multiple physiopathological
processes, including ischemia/reperfusion injury (5,6),
cardiac fibrosis (7) and cardiac
hypertrophy (8,9). However, the mechanisms underlying the
cardioprotective effects of IMD require further elucidation.
Notably, IMD was also reported to serve a role in the regulation of
angiogenesis in vitro and in vivo (10,11).
However, whether IMD1–53 may promote therapeutic
angiogenesis within the infarcted myocardium, and therefore
attenuate adverse myocardial remodeling post-MI, has not been
investigated. To test this hypothesis, the present study
investigated the effects of recombinant IMD1–53 on
angiogenesis in primary cultured myocardial microvascular
endothelial cells (MMVECs) and in a rat model of MI. The results
suggested that IMD1–53 attenuated adverse ventricular
remodeling post-MI by promoting therapeutic angiogenesis, possibly
through the activation of AMP-activated protein kinase (AMPK)
signaling.
Materials and methods
Materials
IMD1–53 peptide was purchased from
Phoenix Pharmaceuticals, Inc. (Burlingame, CA, USA). Dulbecco's
modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA
USA). Phosphorylated (p)-AMPKαThr-172 (cat. no. 2535),
AMPKα (cat. no. 2532), p-AktSer-473 (cat. no. 4060), and
p-endothelial nitric oxide synthase (p-eNOS)Ser-1179
(cat. no. 9570) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Anti-Akt (cat. no. sc-8312),
anti-eNOS (cat. no. sc-654), anti-GAPDH (cat. no. sc-25778) and
anti-VEGF (cat. no. sc-152), anti-CD31 (cat. no. sc-1505) anti-CD34
(cat. no. sc-7045) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Compound C (Comp C) was
purchased from Toronto Research Chemicals Inc. (North York, ON,
Canada). Goat anti-rabbit (cat. no. sc-2030) and goat anti-mouse
(cat. no. sc-3791) secondary antibodies were purchased from Santa
Cruz Biotechnology, Inc.
For all experiments concerning IMD1–53,
the same volume of PBS was used as a vehicle. For experiments
concerning Comp C, the same volume of DMSO was used as a
vehicle.
Isolation and identification of
MMVECs
Male Wistar rats (n=16; 4–6 weeks; 80–100 g) were
used for the isolation of primary MMVECs. The rats were housed at
room temperature with a 12/12 h light/dark cycle and free access to
food and water. The animal protocol was approved by the Animal Care
Committee of Shanghai Jiao Tong University (Shanghai, China). Rats
were sacrificed with an overdose of sodium pentobarbital (180
mg/kg) and heparinized by intraperitoneal injection of sodium
heparin (500 units/0.1 kg). Following thoracotomy, the heart was
rapidly dislodged and washed in PBS. The atria, right ventricle,
epicardial tissue and visible connective tissue were carefully
removed, and the remaining myocardial tissue was washed in PBS
prior to cutting into 1 mm3 sections without visible
vessels. Myocardial tissues were seeded onto culture plates that
were pre-coated with rat tail tendon gelatin and subsequently
incubated at 37°C in a humidified atmosphere containing 5%
CO2 for 30 min. Tissues were cultured in DMEM containing
4,500 mg/l D-glucose and supplemented with 20% FBS, 50 U/ml
heparin, 100 U/ml penicillin and 100 µg/ml streptomycin. Tissue
sections were discarded after the cells began to grow, and the
medium was replaced at 72 h intervals. MMVECs were identified by
typical ‘cobblestone’ appearance and positive CD31 and CD34
immunostaining. MMVECs at the second passage were used for
experiments. The cells were grown to 80–90% confluence and were
used in subsequent experimental analyses.
Immunostaining
MMVECs were plated on a 12×12 mm plate, fixed by
methanol for 15 min at −20°C, blocked with 10% normal goat serum
(cat. no. 31872; Gibco; Thermo Fisher Scientific, Inc.) for 30 min
at room temperature, incubated with CD31 (cat. no. sc-1505; Santa
Cruz Biotechnology, Inc.) and CD34 (cat. no. sc-7045; Santa Cruz
Biotechnology, Inc.) antibodies at 1:200 overnight at 4°C, and then
incubated with goat anti-mouse secondary antibody (cat. no.
sc-3791; Santa Cruz Biotechnology, Inc.) at 1:1,500 for 1 h at room
temperature. Finally, DAB working solution diluted by
H2O2 was added and the MMVECs were observed
under a light microscope (Olympus IX71; Olympus Corporation, Tokyo,
Japan).
Cell proliferation assay
The proliferation rate of MMVECs was assessed using
the MTT assay (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany).
The cells were seeded into 96-well plates at a density of
2×103 cells/well and incubated in DMEM containing 10%
FBS at 37°C with 5% CO2 for 24 h. IMD1–53,
Comp C or IMD1–53+Comp C was added to the medium at
different concentrations (0, 10, 20, 40, 80, 160 nm of IMD and/or
20 µmol compound C) at 0 h. Following 0, 8, 16, 24 and 48 h
incubation, 10 µl of MTT was added to each well and the cells were
incubated at 37°C for 4 h. Following incubation, the supernatant
fluid was removed and 100 µl dimethyl sulfoxide was added to each
well. The cells were incubated for 10 min and the absorbance at 490
nm was measured with an Epoch Microplate Spectrophotometer (BioTek
Instruments, Inc., Winooski, VT, USA).
Migration assay
MMVEC migration rate was assessed using the Boyden
Chemotaxis Chamber assay (Neuro Probe, Inc., Gaithersburg, MD,
USA). The cells were trypsinized and resuspended in 10% DMEM
containing 10% FBS. IMD1–53 (80 nmol) and/or Comp C (20
µmol) were added to the wells in the lower chamber, and
1.5×104 cells (200 µl/well) were added into the upper
chamber. Cells migrating through the filter were fixed in 4%
paraformaldehyde for 10 min and subsequently stained with 0.1%
crystal violet stain solution (Merck Millipore) for 30 min. Five
random microscopic fields per well were quantified using an Olympus
IX71 fluorescence microscope (Olympus Corporation). Each experiment
was repeated three times.
Tube formation assay
Tube formation of MMVECs was assessed using a
Matrigel assay (BD Biosciences, Franklin Lakes, NJ, USA). Matrigel
was chilled at 4°C overnight, melted prior to use and then quickly
added (70 µl/well) to 96-well plates using a pre-chilled pipette.
The plates were incubated at 37°C in 5% CO2 for 1 h. The
cells were seeded into the plates at a density of 1×104
cells/well and incubated for 12 h in the presence or absence of
IMD1–53 (80 nmol) and/or Comp C (20 µmol) at 37°C. Tube
formation was observed and images were captured using an Olympus
1X71 microscope (Olympus Corporation). Images were processed using
Image-Pro Plus software 6.0 (Media Cybernetics, Inc., Rockville,
MD, USA) to calculate the degree of tube formation by measuring the
length of tubes from five randomly selected fields (magnification,
×200) from each well. Each experiment was repeated three times.
Western blot analysis
Cell or tissue samples were homogenized as follows:
Freshly frozen myocardial tissue samples were ground into small
pieces (~1×1×1 mm) in liquid nitrogen with a mortar and a pestle.
The samples were transferred to microcentrifuges containing
radioimmunoprecipitation lysis buffer (~150 µl per 10 mg tissue;
Beyotime Institute of Biotechnology, Haimen, China) and 1 mM
phenylmethylsulfonyl fluoride. The samples were then kept on ice
for 1 h, vortexing every 10 min. The lysates were then centrifuged
at 10,000 × g for 30 min at 4°C, and supernatants were removed for
immediate western blot analysis. Protein samples (5 mg/ml) were
loaded onto a 10% Bis-Tris gel; following electrophoresis,
separated proteins were transferred to a polyvinylidene fluoride
membrane for 1 h. The membranes were then incubated with blocking
solution (5% skim milk) for 1–2 h at room temperature, followed by
three washes with TBS-Tween-20 (TBST; 0.1% Tween; 10 min each).
Membranes were incubated with one of the following primary
antibodies: Anti-AMPK (1:800); anti-p-AMPKThr172
(1:1,000); anti-Akt (1:1,500); anti-p-Aktser473
(1:1,000); anti-eNOS (1:500); anti-p-eNOSser1117
(1:500); anti-vascular endothelial growth factor (VEGF; 1:1,000);
and anti-GAPDH (1:8,000) at 4°C overnight. Primary antibodies were
recovered and the membranes were washed three times with TBST (10
min each), followed by incubation with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; cat.
no. sc-2030; 1:2,000) secondary antibody at room temperature for 1
h. Subsequently, the membranes were washed five times with TBST (5
min/wash), and the bands were detected using Western Blotting
Luminol Reagent (Santa Cruz Biotechnology, Inc.) and quantified by
densitometric analysis of digitized autoradiograms using Quantity
One (version 4.6.2) software (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Each immunoblotting experiment was repeated three times,
and the averages of the results were calculated.
Animal model
A total of 70 male Sprague-Dawley rats (age, 8
weeks; weight, 250–280 g; Experimental Animal Center, Fudan
University, Shanghai, China) were used in the present study, and
were maintained at room temperature with a 12/12 h light/dark cycle
and free access to food and water. The animal research study
protocol was in compliance with the Guide for the Care and Use of
Laboratory Animals published by the National Institutes of Health
(NIH Publication no. 85–23, revised 1996) and approved by the
Animal Care Committee of Shanghai Sixth Hospital, Shanghai Jiao
Tong University. All rats were acclimated for 2 weeks prior to
experimentation. All rats were anesthetized by intraperitoneal
injection of pentobarbital sodium (60 mg/kg) and ventilated using a
DW-200 animal respirator (Shanghai Alcott Biotech Co., Ltd.,
Shanghai, China) with room air following endotracheal intubation.
Electrocardiogram (ECG) lead II was continuously monitored during
the experiment. Heart exposure was performed by a thoracotomy at
the left fourth intercostal space. The left anterior descending
(LAD) coronary artery was ligated using a size 6–0 Prolene
polypropylene suture, 1–2 mm below the tip of the left atrial
appendage. LAD ligation was confirmed by a color change of the
myocardium and an elevation of the ST segment on the ECG. A total
of 4 weeks after LAD ligation, all the surviving rats were
sacrificed by injection of 180 mg/kg pentobarbital sodium
overdose.
Animal protocols
A total of 70 Sprague-Dawley rats were randomly
divided and assigned to five treatment groups: i) Sham group
(n=10), which underwent thoracotomy and incision of the pericardial
sac without LAD ligation; ii) MI group (n=15), which underwent LAD
ligation without any treatment; iii) IMD group (n=15), which
contained MI rats that received subsequent intraperitoneal
injections of IMD1–53 (10 nmol/kg/day) starting 3 days
after LAD ligation; iv) IMD+Comp C group (n=15), which contained MI
rats that received subsequent intraperitoneal injections of
IMD1–53 (10 nmol/kg/day) and Comp C (5 mg/kg/day)
starting 3 days after LAD ligation; and v) Comp C group (n=15),
which contained MI rats that received subsequent intraperitoneal
injections of Comp C (5 mg/kg) starting 3 days after LAD
ligation.
Echocardiography
An echocardiographic examination was performed on
day 28 following the induction of MI using a standard Acuson
Sequoia 512 ultrasound system equipped with a 15 MHz probe (Siemens
AG, Munich, Germany). Two-dimensional and motion-mode
echocardiography were performed. The LV end-diastolic diameter
(LVEDD) and the LV end-systolic diameter (LVESD) were measured. In
addition, the LV ejection fraction (LVEF) and fractional shortening
(FS) were calculated as previously described (12). All measurements from five
consecutive cardiac cycles were averaged and analyzed by a single
observer that was blinded to the treatment protocol.
Histological analysis
Under deep anesthesia with pentobarbital sodium
(intraperitoneal injection at 100 mg/kg), the heart was excised and
cut into 2-mm-thick transverse slices, which were then fixed in 10%
formalin solution at room temperature for 24 h. The samples were
subsequently embedded in paraffin and sectioned (4 µm).
Immunohistochemical analysis was performed as previously described
(13). Tissue sections were then
deparaffinized using xylene and graded ethanol and were blocked in
10% goat serum (Gibco, Thermo Fisher Scientific, Inc.) for 30 min
at 37°C. Samples were incubated at 4°C overnight with primary
rabbit polyclonal antibodies against CD31 (ab28364) and VEGF
(ab46154; Abcam, Cambridge). Sections were subsequently treated
with secondary goat polyclonal secondary antibody against rabbit
IgG (ab97047; Abcam). All sections were counterstained with
hematoxylin. Immunoreactivity for CD31 and VEGF was measured using
a fluorescence microscope and the Image-Pro Plus 4.0 analysis
system (Media Cybernetics, Inc.). Capillaries were identified by
positive staining for CD31. Results were expressed as the average
amount of capillaries per ×10 field. Masson's trichrome stain was
performed in the border area to evaluate collagen deposition.
Statistical analysis
Statistical analysis of the data was conducted using
the statistical software SPSS version 13.0 (SPSS, Inc., Chicago,
IL, USA). Experimental data were expressed as the mean ± standard
deviation and analyzed using one-way ANOVA followed by a
least-significant difference corrected multiple comparison test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
IMD1–53 increases
proliferation of MMVECs in a dose-dependent manner
The effects of IMD1–53 on the
proliferation of MMVECs were examined using the MTT assay. The
results indicated that IMD1–53 was able to promote the
proliferation of MMVECs in a dose-dependent manner with the maximum
effect observed at 80 nmol (P<0.05 vs. 0 nmol control, Fig. 1A). In addition, co-administration
of Comp C, a well-known inhibitor of AMPK, significantly attenuated
the effects of IMD1–53 on the proliferation of MMVECs
(P<0.05 vs. IMD at 8, 16, 24 and 48 h; Fig. 1B), whereas treatment with Comp C
alone had no significant influence on the proliferation of MMVECs
(P>0.05, Fig. 1B).
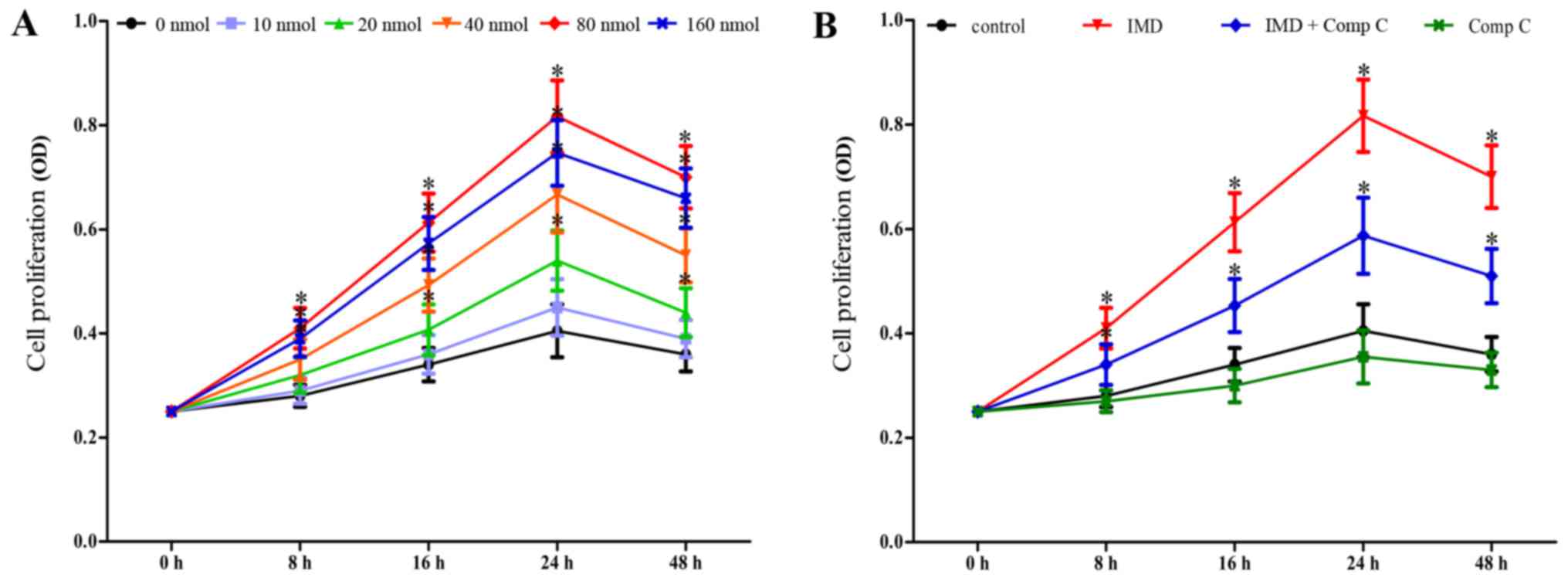 | Figure 1.IMD1–53 promotes the
proliferation of MMVECs. Proliferation was assessed using the MTT
assay. (A) MMVECs were seeded in 96-well dishes in the presence of
different concentrations (0–160 nmol) of IMD1–53 for 0,
8, 16, 24 and 48 h. (B) MMVECs were seeded in 96-well dishes in the
presence of IMD1–53 (80 nmol), Comp C (20 µmol), or both
for 0, 8, 16, 24 and 48 h. All data are expressed as the mean ±
standard deviation from three independent experiments. *P<0.05
vs. control group. Comp C, compound C; IMD, intermedin; MMVECs,
myocardial microvascular endothelial cells. |
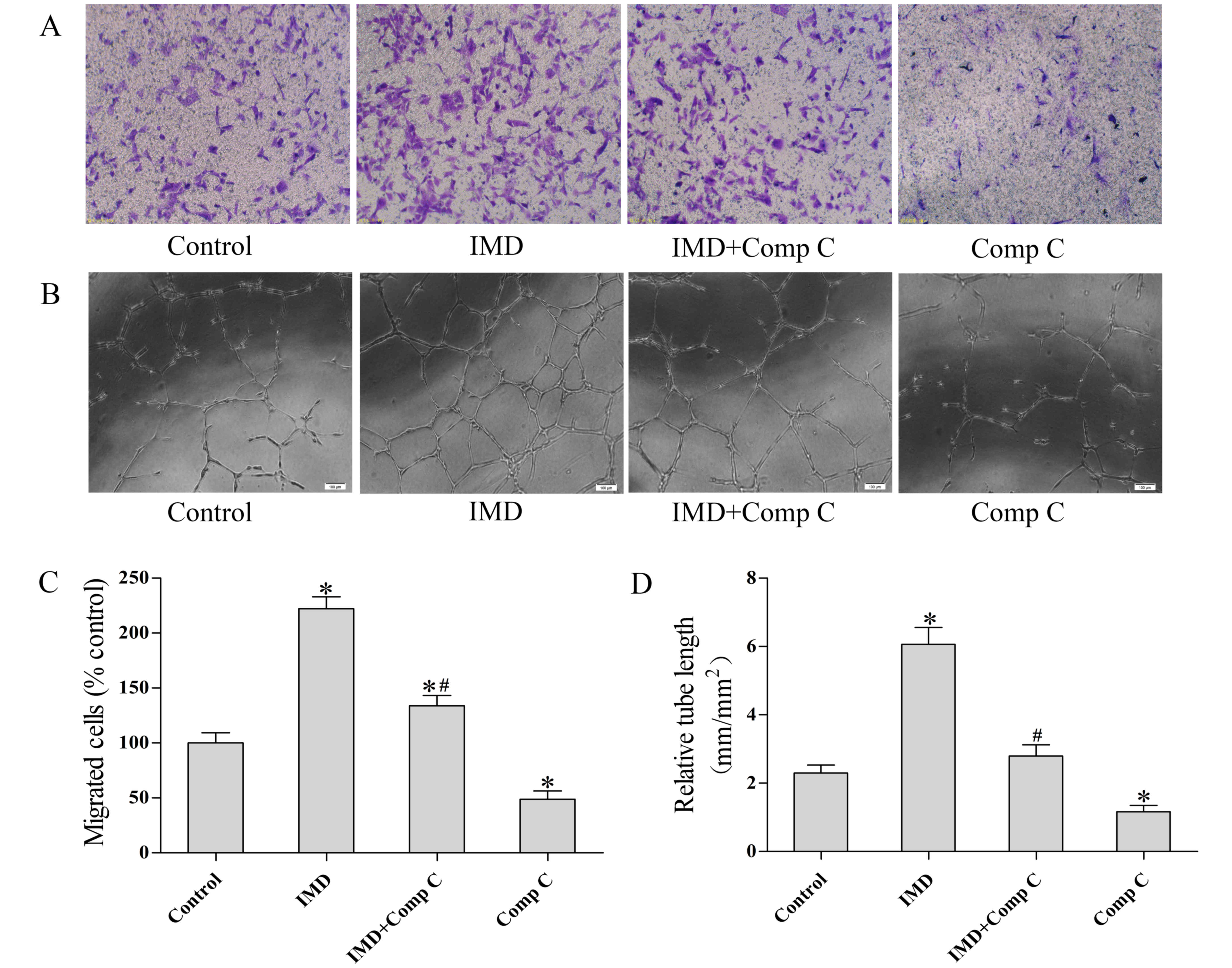
IMD1–53 promotes migration
and tube formation in MMVECs
The effects of IMD1–53 on endothelial
cell migration were further evaluated by a modified Boyden chamber
assay (Fig. 2). The results
indicated that the optimal dose (80 nmol, as shown in the MTT
assay) of IMD1–53 treatment stimulated the migration of
MMVECs (P<0.05 vs. control, Fig. 2A
and C). Co-treatment with Comp C (20 µmol) partially suppressed
the pro-migratory effect of IMD1–53 (P<0.05 vs. IMD,
Fig. 2A and C).
To examine the effects of IMD1–53 on the
differentiation of cardiovascular endothelial cells into vascular
structures, MMVECs were plated at 1×104 cells/well in
the presence or absence of IMD1–53 (80 nmol) and/or Comp
C (20 µmol) and tube formation was detected microscopically. A
total of 24 h after plating, the MMVECs treated with
IMD1–53 formed a significantly more extensive tube
network compared with the control group (P<0.05). The effects of
IMD1–53 on tube formation were reduced in MMVECs that
were also pretreated with Comp C (P<0.05 vs. IMD group).
Treatment with Comp C alone significantly decreased tube formation
as compared with the control group (P<0.05, Fig. 2B and D).
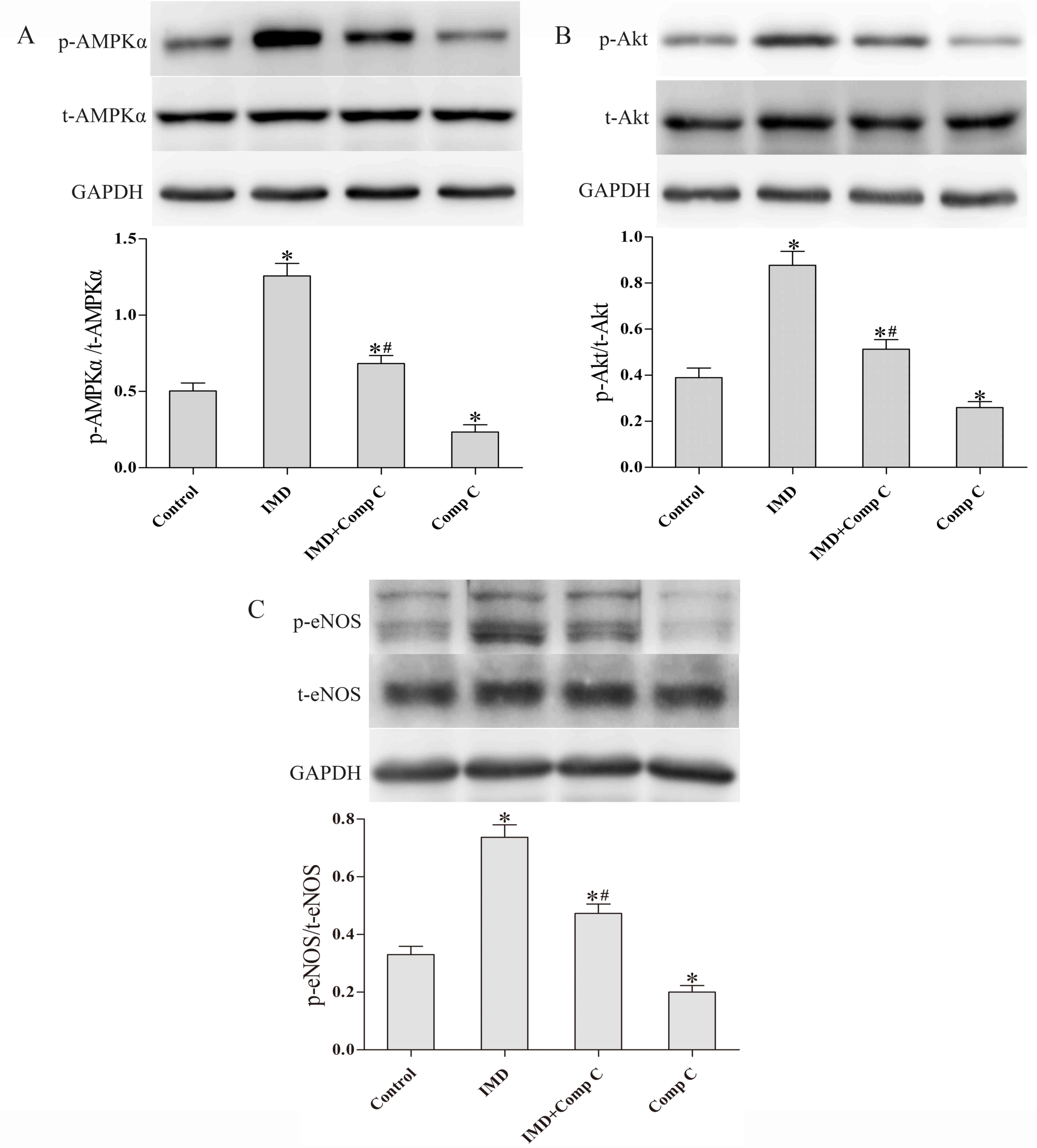
IMD1–53 stimulates
activation of the AMPK-Akt-eNOS signaling pathway in MMVECs
Since AMPK is involved in the regulation of
angiogenesis (14) and
IMD1–53 was recently reported to increase AMPK
activation in rat cardiomyocytes (9), the present study aimed to investigate
whether AMPK is involved in IMD1–53-induced angiogenesis
in MMVECs. MMVECs were exposed to IMD1–53 (80 nmol) for
2 h; subsequently, the total and phosphorylated protein levels of
AMPKα, Akt and eNOS were analyzed by western blot analysis. As
shown in Fig. 3, the expression
levels of p-AMPKα, p-Akt and p-eNOS were all significantly
increased by IMD1–53 treatment (P<0.05 vs. control,
Fig. 3). Co-treatment with the
Comp C suppressed the ability of IMD1–53 to activate
either Akt or eNOS, as measured by the relative expression levels
of p-Akt and p-eNOS (P<0.05 vs. IMD, Fig. 3), whereas treatment with Comp C
alone slightly but significantly decreased the phosphorylated
levels of Akt and eNOS (P<0.05, respectively; Fig. 3) and significantly decreased the
phosphorylated levels of AMPKα (P<0.05; Fig. 3).
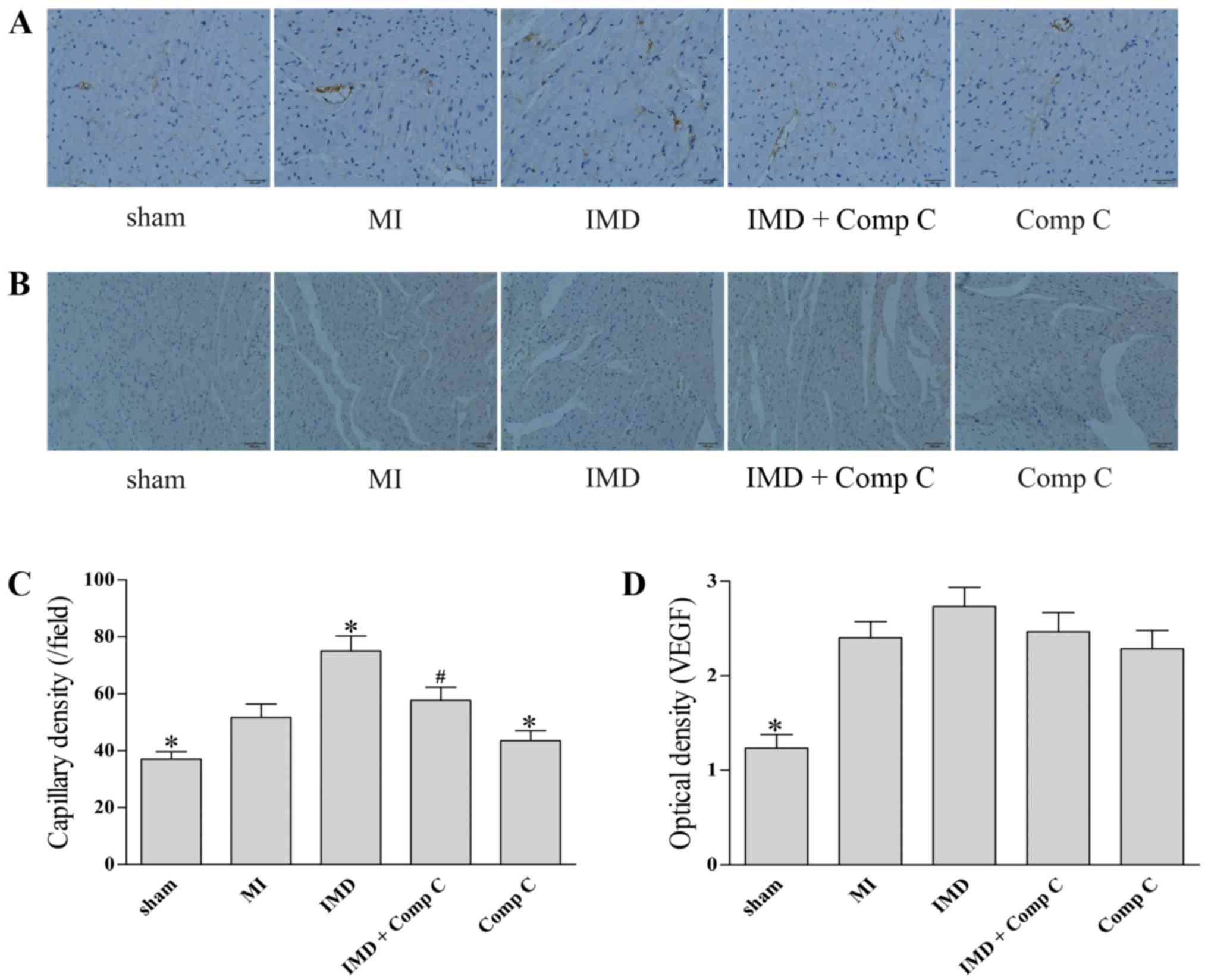
IMD1–53 increases capillary
density in post-MI rat heart
To assess the effects of IMB of angiogenesis in
vivo, myocardium capillary density tests were performed.
Compared with the sham group, induction of MI significantly
increased the capillary density in the peri-infarct zone
(P<0.05, Fig. 4A and C).
Treatment with IMD1–53 significantly increased capillary
density in the peri-infarct zone of post-MI rat myocardium
(P<0.05 vs. MI, Fig. 4A and C).
Co-administration of Comp C mitigated the effects of
IMD1–53 on capillary density in the peri-infarct zone
(P<0.05 vs. IMD, Fig. 4A and
C). Compared with the MI group, administration of Comp C alone
slightly decreased capillary density in the peri-infarct zone
(P<0.05, Fig. 4A and C).
Compared to the sham group, the expression levels of VEGF were
increased following induction of MI (P<0.05, Fig. 4B and D). However, the
administration of IMD1–53, Comp C or a combination of
the two had no significant effect on VEGF expression in the
peri-infarct zone post-MI (P>0.05 vs. MI group, Fig. 4B and D).
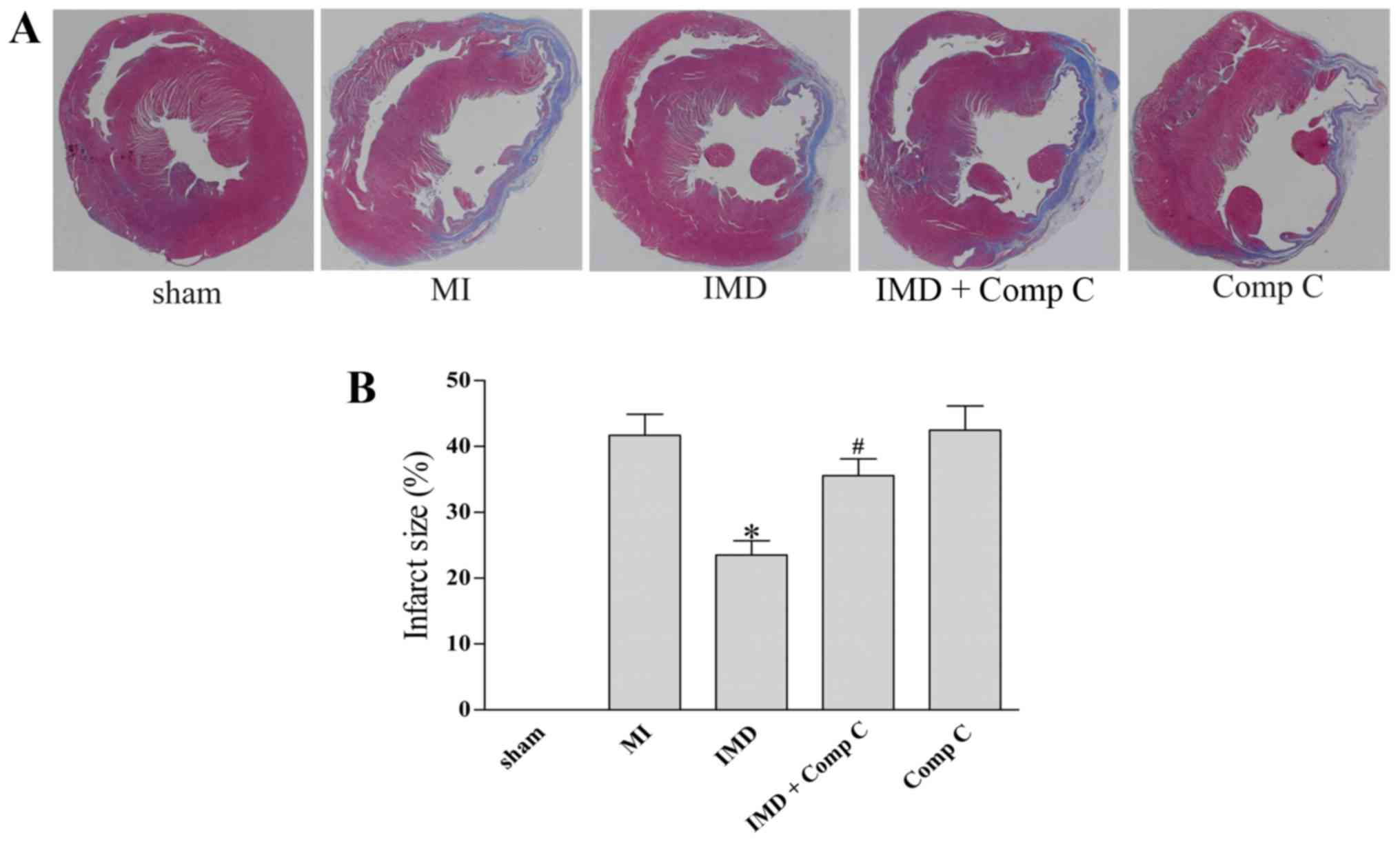
IMD1–53 attenuates
ventricular remodeling post-MI
To evaluate the effects of IMD1–53 on
ventricular remodeling post-MI, Masson's trichrome staining was
performed on all groups 4 weeks post-MI induction (Fig. 5). The results demonstrated that
treatment with IMD1–53 markedly suppressed elongation of
the infarct wall segment and dilation of the LV, as demonstrated by
cross-section morphology (Fig. 5A)
and calculated by infarct size post-MI (P<0.05 vs. MI group;
Fig. 5B). The effects of
IMD1–53 on ventricular remodeling were abrogated by
co-administration of Comp C (P<0.05 vs. IMD, Fig. 5), whereas Comp C alone had no
significant effect on myocardial infarct size post-MI (P>0.05
vs. MI, Fig. 5).
IMD1–53 improves cardiac
function post-MI
A total of 4 weeks after MI induction, cardiac
geometry and performance were evaluated using echocardiography
(Table I). Compared with the sham
group, significantly decreased LVEF and FS, along with increased
LVESD and LVEDD, were observed in the MI group (P<0.05, Table I). In IMD-treated MI rats, LVEF and
FS were significantly improved, whereas LVESD and LVEDD were
significantly reduced, compared with the MI group (P<0.05,
Table I). Co-administration of
Comp C abrogated the effects of IMD1–53 on LVEF, FS,
LVESD and LVEDD (P<0.05 vs. IMD, Table I), whereas Comp C alone had no
significant effects on cardiac geometry and performance (P>0.05,
Table I). Neither
IMD1–53 nor Comp C administration had significant
effects on survival rate of rats post-MI (P>0.05 vs. MI group;
Table I).
 | Table I.Representative motion-mode
echocardiograms obtained 4 weeks post-MI induction. |
Table I.
Representative motion-mode
echocardiograms obtained 4 weeks post-MI induction.
| Parameter | Sham | MI | IMD | IMD+Comp C | Comp C |
|---|
| Number | 10 | 15 | 15 | 15 | 15 |
| Survival | 10 | 11 | 12 | 11 | 10 |
| LVESD (mm) |
3.47±0.28a | 6.11±0.31 |
4.52±0.27a |
5.84±0.33b | 6.24±0.28 |
| LVEDD (mm) |
5.83±0.36a | 8.28±0.44 |
6.42±0.42a |
7.61±0.49b | 8.48±0.47 |
| LVEF (%) |
81.3±5.38a | 46.8±5.14 |
65.2±6.03a |
53.7±5.93b | 44.9±5.26 |
| FS (%) |
75.6±4.97a | 44.9±3.71 |
66.5±4.89a |
52.2±3.87b | 43.7±3.54 |
IMD1–53 stimulates
activation of the AMPK-Akt-eNOS signaling pathway in post-MI rat
myocardium
Western blot analysis was used to evaluate the
activation of AMPK, Akt and eNOS, and the expression levels of VEGF
in rat myocardium (Fig. 6). The
results indicated that chronic IMD1–53 treatment
resulted in significantly increased activation (as measured by
phosphorylation) of AMPK, Akt and eNOS in post-MI rat myocardium
(P<0.05 vs. MI, Fig. 6B-D),
whereas it had no significant effect on the expression of VEGF
(P>0.05 vs. MI, Fig. 6A).
Co-administration of Comp C abrogated the effects of
IMD1–53 on AMPK, Akt and eNOS phosphorylation (P<0.05
vs. IMD, Fig. 6B-D), whereas Comp
C alone slightly decreased levels of p-AMPK, p-Akt and p-eNOS
(P<0.05 vs. MI, Fig. 6B-D).
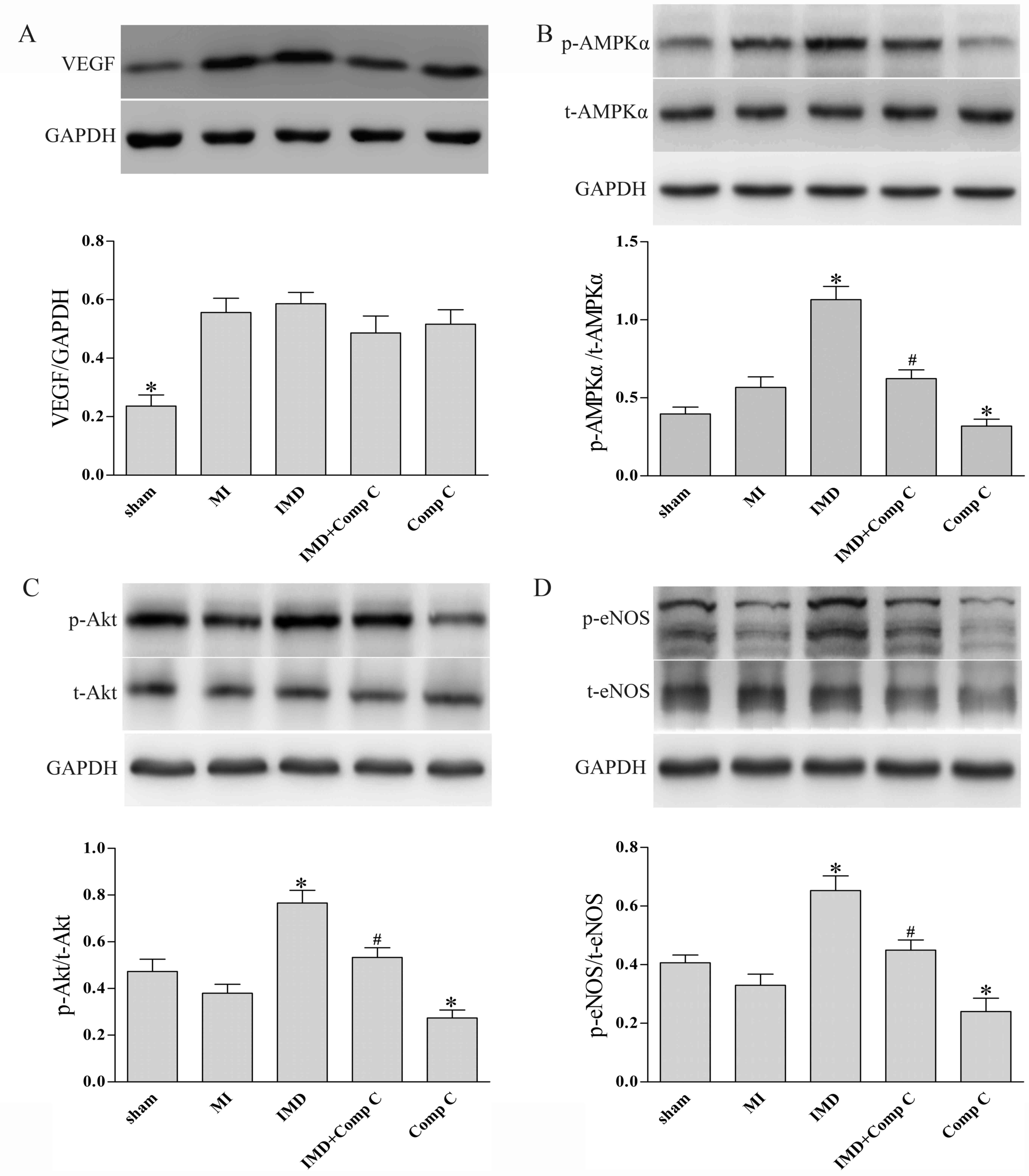 | Figure 6.Effects of IMD1–53 on
expression and phosphorylation of VEGF, AMPK, Akt and eNOS in rat
myocardium post-MI. The infarcted heart tissue from the LV free
wall or the non-infarcted heart tissue at the same site was
harvested 4 weeks after left anterior descending coronary artery
ligation. Representative western blots demonstrating (A) VEGF
protein expression, GAPDH is used as a loading control; (B) p-AMPK
over total AMPK protein expression; (C) p-Akt over total Akt
protein expression; and (D) p-eNOS over total eNOS protein
expression. Data are expressed as the mean ± standard deviation;
n=4 for sham group, n=6 for other groups. *P<0.05 vs. MI group;
#P<0.05 vs. IMD group. AMPK, AMP-activated protein
kinase; Comp C, compound C; eNOS, endothelial nitric oxide
synthase; IMD, intermedin; MI, myocardial infarction; LV, left
ventricle; p, phosphorylated; VEGF, vascular endothelial growth
factor. |
Discussion
Using in vitro and in vivo
experiments, the present study demonstrated that IMD1–53
promoted angiogenesis in primary cultured MMVECs, which was
demonstrated by an increase in proliferation, migration and tube
formation. In addition, experiments using an in vivo rat
model of MI demonstrated that treatment with IMD1–53
significantly increased the capillary density in ischemic
myocardium, attenuated LV remodeling and improved post-MI cardiac
function, as determined by CD31 staining, Masson trichrome staining
and echocardiography, respectively. Furthermore, the addition of
Comp C, an AMPK inhibitor, demonstrated that the in vitro
and the in vivo effects of IMD1–53 were at least
partially dependent on the activation of AMPK. These results
indicated that IMD1–53 attenuates post-infarct
myocardial remodeling via the promotion of AMPK-dependent
angiogenesis. To the best of our knowledge, this is the first
demonstration of the involvement of AMPK activation in IMD-induced
angiogenesis and is one of only a few studies (8,10,15)
to link the cardioprotective role of IMD to therapeutic
angiogenesis.
The human IMD gene encodes a prepropeptide of 148
amino acids (preproIMD), which can generate a series of mature
peptide fragments by proteolytic cleavage at the C-terminal in
vivo (16), such as
IMD1–47, IMD8–47 and IMD1–53. Yang
et al were the first to report that IMD1–53
stimulated L-Arg transport and increased NOS activity in the rat
aorta (15). Furthermore, using
direct gene delivery, Smith et al established IMD as a novel
angiogenic factor in a rodent hindlimb ischemia model and cultured
human umbilical vein endothelial cells (10). Consistent with these findings, the
present study demonstrated that IMD1–53 increased
proliferation of MMVECs in a dose-dependent manner with a maximal
effect at around 80 nmol, and the optimal dose of
IMD1–53 treatment promoted migration and tube formation
of MMVECs.
Cardiac hypertrophy is initiated as an adaptive
response during post-infarction remodeling to compensate for
increased load (17). However, the
capillary network within the infarcted heart is unable to support
the increased demands of the hypertrophied myocardium, resulting in
a gradual loss of healthy tissue, extension of the infarct and
fibrous replacement (18).
Therefore, promotion of angiogenesis in post-infarct myocardium may
be an effective approach to attenuate adverse ventricular
remodeling following MI, as demonstrated by several previous
studies (19–21). The present study demonstrated that
the in vivo administration of IMD1–53 promoted
angiogenesis in post-infarct rat myocardium, which was indicated by
increased capillary density in the peri-infarct zone. In addition,
chronic treatment with IMD1–53 preserved cardiac
geometry and improved cardiac performance post-MI, as evidenced by
Masson trichrome staining and echocardiography, suggesting that
IMD1–53 may attenuate adverse remodeling post-MI and
prevent subsequent heart failure by promoting angiogenesis.
IMD1–53 has previously been reported to protect against
myocardial ischemia/reperfusion injury (5) and to attenuate
pressure-overload-induced cardiac hypertrophy (9) through other mechanisms (6,8);
however, to the best of our knowledge, this is the first study to
link the cardioprotective role of IMD to therapeutic
angiogenesis.
AMPK is a key sensor and regulator of cellular
energy homeostasis, which modulates various physiological processes
that may favor the restoration of energy balance in several systems
(22). In endothelial cells, AMPK
was demonstrated to serve an essential role in angiogenesis in
response to stress (14,23). In addition, a series of agents,
including pravastatin, adiponectin and erythropoietin, and
cytokines have been reported to increase angiogenesis through
AMPK-dependent mechanisms (24–26).
Notably, CGRP was also reported to promote angiogenesis by
activating the AMPK-eNOS pathway in endothelial cells (27). The present study demonstrated that
treatment with IMD1–53 increased the expression levels
of p-AMPK in cultured MMVECs and in post-MI rat myocardium, and
notably, administration of Comp C, a known inhibitor of AMPK,
abrogated the effects of IMD1–53 on angiogenesis, as
evidenced by reduced migration and tube formation of MMVECs in
vitro, and lower capillary density at the peri-infarct zone
in vivo. These results demonstrated that the angiogenic
activity of IMD1–53 is at least partially dependent on
the activation of AMPK.
It has previously been reported that
IMD1–53 increased total NOS activity, but had little
effect on mRNA levels of either inducible NOS or eNOS in the rat
aorta (16). The results of the
current study demonstrated that IMD1–53 promoted the
activation of eNOS in vitro and in vivo. In addition,
the administration of Comp C significantly attenuated the effects
of IMD1–53 on the level of p-eNOS expression, suggesting
that AMPK is involved in the IMD1–53-induced activation
of eNOS. It is also worth noting that, in the present study,
treatment with IMD1–53 had no significant effect on the
expression of VEGF in post-infarct rat myocardium, indicating that
IMD1–53 may promote the activation of eNOS and
subsequent angiogenesis through VEGF-independent mechanisms.
Notably, the present study did not determine how IMD1–53
regulated AMPK signaling. In addition, although the α subunit of
AMPK was identified as the major target in the present study,
further evaluation is required regarding the specific subunit (α1
or α2) involved.
In conclusion, the present study demonstrated that
IMD1–53 could attenuate adverse ventricular remodeling
post-MI by promoting therapeutic angiogenesis, possibly through the
activation of AMPK signaling.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81100099).
Glossary
Abbreviations
Abbreviations:
|
Comp C
|
compound C
|
|
FS
|
fractional shortening
|
|
IMD
|
intermedin
|
|
LVEDD
|
left ventricular end-diastolic
diameter
|
|
LVEF
|
left ventricular ejection fraction
|
|
LVESD
|
left ventricular end-systolic
diameter; MMVECs;
|
|
MI
|
myocardial infarction
|
References
|
1
|
Pfeffer MA and Braunwald E: Ventricular
remodeling after myocardial infarction. Experimental observations
and clinical implications. Circulation. 81:1161–1172. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jessup M and Brozena S: Heart failure. N
Engl J Med. 348:2007–2018. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, De Ferranti S, Després JP, Fullerton HJ,
Howard VJ, et al: Heart disease and stroke statistics-2015 update:
A report from the American Heart Association. Circulation.
131:e29–e322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang JH, Jia YX, Pan CS, Zhao J, Ouyang M,
Yang J, Chang JK, Tang CS and Qi YF: Effects of intermedin(1–53) on
cardiac function and ischemia/reperfusion injury in isolated rat
hearts. Biochem Biophys Res Commun. 327:713–719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao L, Peng DQ, Zhang J, Song JQ, Teng X,
Yu YR, Tang CS and Qi YF: Extracellular signal-regulated kinase 1/2
activation is involved in intermedin1-53 attenuating myocardial
oxidative stress injury induced by ischemia/reperfusion. Peptides.
33:329–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang X, Zhang H, Jia Y, Ni L, Li G, Xue L
and Jiang Y: Effects of intermedin1-53 on myocardial fibrosis. Acta
Biochim Biophys Sin (Shanghai). 45:141–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen H, Wang X, Tong M, Wu D, Wu S, Chen
J, Wang X, Wang X, Kang Y, Tang H, et al: Intermedin suppresses
pressure overload cardiac hypertrophy through activation of
autophagy. PLoS One. 8:e647572013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu WW, Zhao L, Zhang JS, Hou YL, Yu YR,
Jia MZ, Tang CS and Qi YF: Intermedin1-53 protects against cardiac
hypertrophy by inhibiting endoplasmic reticulum stress via
activating AMP-activated protein kinase. J Hypertens. 33:1676–1687.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith RS Jr, Gao L, Bledsoe G, Chao L and
Chao J: Intermedin is a new angiogenic growth factor. Am J Physiol
Heart Circ Physiol. 297:H1040–H1047. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang W, Wang LJ, Xiao F, Wei Y, Ke W and
Xin HB: Intermedin: A novel regulator for vascular remodeling and
tumor vessel normalization by regulating vascular
endothelial-cadherin and extracellular signal-regulated kinase.
Arterioscler Thromb Vasc Biol. 32:2721–2732. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Derumeaux G, Mulder P, Richard V,
Chagraoui A, Nafeh C, Bauer F, Henry JP and Thuillez C: Tissue
Doppler imaging differentiates physiological from pathological
pressure-overload left ventricular hypertrophy in rats.
Circulation. 105:1602–1608. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang F, Huang D, Zhu W, Li S, Yan M, Wei M
and Li J: Selective inhibition of PKCβ2 preserves cardiac function
after myocardial infarction and is associated with improved
angiogenesis of ischemic myocardium in diabetic rats. Int J Mol
Med. 32:1037–1046. 2013.PubMed/NCBI
|
|
14
|
Nagata D, Mogi M and Walsh K:
AMP-activated protein kinase (AMPK) signaling in endothelial cells
is essential for angiogenesis in response to hypoxic stress. J Biol
Chem. 278:31000–31006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang JH, Pan CS, Jia YX, Zhang J, Zhao J,
Pang YZ, Yang J, Tang CS and Qi YF: Intermedin1-53 activates
L-arginine/nitric oxide synthase/nitric oxide pathway in rat
aortas. Biochem Biophys Res Commun. 341:567–572. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roh J, Chang CL, Bhalla A, Klein C and Hsu
SY: Intermedin is a calcitonin/calcitonin gene-related peptide
family peptide acting through the calcitonin receptor-like
receptor/receptor activity-modifying protein receptor complexes. J
Biol Chem. 279:7264–7274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sutton MG and Sharpe N: Left ventricular
remodeling after myocardial infarction: Pathophysiology and
therapy. Circulation. 101:2981–2988. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kocher AA, Schuster MD, Szabolcs MJ,
Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM and Itescu S:
Neovascularization of ischemic myocardium by human
bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis,
reduces remodeling and improves cardiac function. Nat Med.
7:430–436. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Zhang Y, Li C, Xie J, Liu Y, Zhu W,
Zhang X, Jiang S, Liu L and Ding Z: HSPA12B attenuates cardiac
dysfunction and remodelling after myocardial infarction through an
eNOS-dependent mechanism. Cardiovasc Res. 99:674–684. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vandoorne K, Vandsburger MH, Raz T, Shalev
M, Weisinger K, Biton I, Brumfeld V, Raanan C, Nevo N, Eilam R, et
al: Chronic Akt1 deficiency attenuates adverse remodeling and
enhances angiogenesis after myocardial infarction. Circ Cardiovasc
Imaging. 6:992–1000. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao X, Balaji P, Pachon R, Beniamen DM,
Vatner DE, Graham RM and Vatner SF: Overexpression of cardiomyocyte
α1A-Adrenergic receptors attenuates postinfarct remodeling by
inducing angiogenesis through heterocellular signaling.
Arterioscler Thromb Vasc Biol. 35:2451–2459. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grahame Hardie D: AMP-activated protein
kinase: A key regulator of energy balance with many roles in human
disease. J Intern Med. 276:543–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahluwalia A and Tarnawski AS: Activation
of the metabolic sensor-AMP activated protein kinase reverses
impairment of angiogenesis in aging myocardial microvascular
endothelial cells. Implications for the aging heart. J Physiol
Pharmacol. 62:583–587. 2011.PubMed/NCBI
|
|
24
|
Izumi Y, Shiota M, Kusakabe H, Hikita Y,
Nakao T, Nakamura Y, Muro T, Miura K, Yoshiyama M and Iwao H:
Pravastatin accelerates ischemia-induced angiogenesis through
AMP-activated protein kinase. Hypertens Res. 32:675–679. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimano M, Ouchi N, Shibata R, Ohashi K,
Pimentel DR, Murohara T and Walsh K: Adiponectin deficiency
exacerbates cardiac dysfunction following pressure overload through
disruption of an AMPK-dependent angiogenic response. J Mol Cell
Cardiol. 49:210–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su KH, Yu YB, Hou HH, Zhao JF, Kou YR,
Cheng LC, Shyue SK and Lee TS: AMP-activated protein kinase
mediates erythropoietin-induced activation of endothelial nitric
oxide synthase. J Cell Physiol. 227:3053–3062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng S, Li W, Xu M, Bai X, Zhou Z, Han J,
Shyy JY and Wang X: Calcitonin gene-related peptide promotes
angiogenesis via AMP-activated protein kinase. Am J Physiol Cell
Physiol. 299:C1485–C1492. 2010. View Article : Google Scholar : PubMed/NCBI
|