Introduction
The human fibroblast growth factors (FGFs) protein
family consists of 22 members, which share a high affinity for
heparin, as well as high-sequence homology within a central core
domain of 120 amino acids (1).
FGFs are essential in biological functions, such as angiogenesis,
mitogenesis, cell differentiation and wound repair. FGF17 is a
member of the heparin binding growth factor family (2), which is structurally the most
homologous to FGF8 and FGF18. FGF8, FGF17 and FGF18 are highly
conserved between human and mice, sharing 93% identity (2,3).
Mouse FGF17 has three isoforms, while human FGF17 has just two:
FGF17a and FGF17b, the latter of which has been selected as the
canonical sequence (4). FGF17 is
preferentially expressed in the embryonic brain and is highly
associated with the nervous system (5).
Numerous studies have indicated that FGF17 may serve
as a therapeutic agent to potentially treat certain types of
disease. There is an increasing demand in the market to produce the
FGF17 protein, and the large-scale production of bioactive human
FGF17 is a challenging rate-limiting step. Given these factors, the
development of a process that may enable significant preparation of
sufficient, highly bioactive recombinant human (rh)FGF17 is
considered to be a high priority for further investigations of the
underlying mechanisms and clinical pathology. With the development
of biotechnology, various expression systems are currently being
used for expressing recombinant proteins for industrial production,
as well as in research for structural and biochemical studies
(6). Escherichia coli, with
a short growth cycle, low cost, high stability and high
transformation efficiency, is suitable for large-scale manufacture
(7). In addition, E. coli
are the most frequently used expression system for high-scale
production of recombinant proteins (8–10).
As human (h)FGF17 is an important growth factor and,
to the best of our knowledge, its non-tag expression in E.
coli has not been reported, an rhFGF17 expression vector
pET3a-rhFGF17, with a high expression level of rhFGF17 protein with
soluble protein and inclusion bodies, was constructed in the
present study. Furthermore, the high purity of rhFGF17 protein was
obtained via heparin affinity and SP Sepharose Fast Flow
chromatography. In addition, the biological activity of rhFGF17,
which may significantly increase the proliferative activity of
NIH3T3 cells was examined. This novel expression strategy markedly
enhanced the yield of rhFGF17 with high biological activity, which
may meet the demand for fundamental research and therapeutic
applications.
Materials and methods
Reagents and bacterial strain
The PCR purification, gel extraction and plasmid
miniprep kits, and DNA Marker were purchased from Takara
Biotechnology Co., Ltd. (Dalian, China). Goat anti-FGF17 polyclonal
antibody (cat. no. sc-16826) and mouse anti-goat IgG-HRP (cat. no.
sc-2354) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Heparin Sepharose column, SP Sepharose Fast Flow
and AKTA purifier were purchased from GE Healthcare Life Sciences
(Shanghai, China). The E. coli DH5α and BL21(DE3)pLysS
component cells were purchased from Beijing Solarbio Science &
Technology Co., Ltd. (Beijing, China).
Construction of rhFGF17 expression
vector
The coding sequence of rhFGF17 (GenBank reference,
NM_001304478.1) was obtained from the pUC57-FGF17 vector,
previously constructed by our lab (unpublished data), using a
Veriti™ Thermal Cycler (cat. no. 4375786; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with Phusion® High-Fidelity PCR
Master Mix (cat. no. M0531S; New England BioLabs, Inc., Ipswich,
MA, USA). Amplification conditions were as follows: Initial
denaturation at 98°C for 30 sec, followed by 30 cycles at 98°C for
10 sec, at 65°C for 20 sec and at 72°C for 20 sec, with a final
extension step at 72°C for 10 min. The DNA fragment rhFGF17 was
subsequently cloned into pET3a vector, using the NdeI and
BamHI restriction enzymes, to create the recombinant
expression vector, pET3a-hFGF17, according to the manufacturer's
protocol. NdeI (cat. no. R0111S) and BamHI (cat. no.
R0136S) were purchased from New England BioLabs, Inc.
Production of rhFGF17
The recombinant vector pET3a-hFGF17 was transformed
into BL21(DE3)PLysS component cells. Briefly, 50 ng pET3a-hFGF17
vector were added to 100 µl thawed BL21(DE3)PLysS component cells.
Cells were incubated for 30 min on ice, heat shocked at 42°C for 90
sec, and then plated on a pre-warmed LB agar plate for further
culture. The transformed colonies were cultured in 5 ml LB medium
(tryptone 10 g/l, yeast extract 5 g/l, NaCl 5 g/l, in
ddH2O) containing 100 µg/ml ampicillin and 35 µg/ml
chloramphenicol at 37°C. When the optical density
(OD)600 reached 0.6–0.8, isopropyl
β-D-1-thiogalactopyranoside (IPTG) was added to a final
concentration of 1 mM. The cultures were incubated at 37°C for 4 h
under agitation (speed, 200 rpm). The colony with the greatest
expression level was selected as the seed strain in subsequent
experiments.
Optimizing the expression conditions
for rhFGF17
The IPTG concentration for rhFGF17 expression yield
was evaluated at 37°C and 16°C. Detection of soluble rhFGF17 was
performed as follows: The seed strain was cultured overnight with
agitation at 200 rpm in 20 ml LB medium containing 10 µg/ml
ampicillin and 35 µg/ml chloramphenicol at 37°C. Subsequently, the
culture (6 ml) was transferred into two bottles, each containing
600 ml fresh LB medium with 100 µg/ml ampicillin and 35 µg/ml
chloramphenicol for further growth. When OD600 reached
0.6–0.8, the IPTG was added to final concentrations of 0.4 and 1
mM, and cultured for 4 h at 37°C (speed, 200 rpm) and 24 h at 16°C
(speed, 180 rpm). Cells were collected by centrifugation at 15,000
× g for 20 min at 4°C. The cell pellets were resuspended in
improved lysis buffer [20 mM Tris-HCl, 200 mM Nacl, 1% Triton
X-100, 0.2% deoxysodium cholate, 1 mM EDTA, 5% glycerol, 0.2 M
sucrose and 1 mM phenylmethylsulfonyl fluoride (PMSF; pH 7.5)].
Cells were lysed by sonication for 10 min in an ice bath. Following
centrifugation at 15,000 × g for 20 min at 4°C, the sediment and
the supernatant were separated by 12% SDS-PAGE and the protein
expression levels of rhFGF21 were determined, using western blot
analysis.
Purification of soluble rhFGF17
The bacteria cells were harvested and lysed in lysis
buffer (pH 7.5) containing 50 mM Tris-HCl, 2 mM EDTA, 300 mM NaCl,
1% Triton X-100, 0.2% deoxysodium cholate, 5–10% glycerol, 0.01 M
sucrose and 1 mM PMSF. Supernatants were collected for subsequent
purification. The following steps were all performed at 4°C: First,
the heparin-sepharose column was equilibrated with five bed volumes
of binding buffer (20 mM Tris-HCl buffer, 25 mM NaCl and 1 mM EDTA;
pH 7.5) at a rate of 1 ml/min. Subsequently, the supernatant was
applied to the column. Following binding, the column was washed
with binding buffer with gradients of 0.4, 0.6, 0.8 and 1.0 M NaCl.
Further purification was performed using an SP Sepharose Fast Flow,
where the methodology was the same as the heparin-sepharose
purification. Finally, fractions were collected from the column
according to the ultraviolet absorption peaks and conductivity
curve. Then the elution fractions were determined using 12%
SDS-PAGE.
Isolation and refolding of rhFGF17
inclusion bodies
Following fermentation, bacteria were harvested by
centrifugation at 10,000 × g at 4°C for 15 min, and wet bacteria (1
g) was resuspended in 20 ml lysis buffer. The inclusion bodies were
collected following centrifugation at 10,000 × g at 4°C for 15 min
and resuspended in wash buffer (20 mM Tris-HCl, 200 mM NaCl, 1%
Triton X-100 and 1 mM EDTA; pH10) by centrifugation at 10,000 × g
at 4°C for 15 min after ultrasonication in an ice bath.
Subsequently, inclusion bodies (1 g) were resuspended in 20 ml
denaturing buffer (8 M urea, 20 mM Tris, 150 mM Nacl, 3 mM EDTA, 5
mM DTT and 0.5 M arginine; pH 7.5). The protein was then refolded
by a combination of dialysis and slow dilution. First, the
denaturing buffer was dialyzed in dialysis buffer (20 mM Tris, 50
mM NaCl, 15% glycerol, 0.5 M arginine and 4 M urea; pH 7.5) until
the urea concentration reached 4 M; subsequently, the buffer in the
dialysis bag were collected by centrifugation at 10,000 × g at 4°C
for 15 min and then slowly diluted into appropriate volumes of
renaturing buffer (20 mM Tris-HCl, 50 mM NaCl, 30% glycerol and 0.5
M arginine; pH 7.5). Following centrifugation at 15,000 × g at 4°C
for 20 min, the supernatant was retained and prepared for inclusion
in the heparin-sepharose column. Refolding of the protein was
performed at 4°C.
Purification of rhFGF17 inclusion
bodies
According to the heparin affinity of rhFGF17, a
heparin-sepharose column was selected for the purification.
Purification procedures were the same as soluble rhFGF17. Refolding
rhFGF17 protein was loaded onto the column that was equilibrated
with wash buffer (the same as the soluble fraction) at a speed of 1
ml/min. The flow-through was collected. The protein was
subsequently eluted using 0.4–1.0 M NaCl gradient in wash buffer at
a speed of 1 ml/min. The elution fractions were collected and
determined by Coomassie blue staining of 12% SDS-PAGE.
Western blot analysis
Protein concentration was determined using the Lowry
protein assay. Purified rhFGF17 proteins (50 ng) were separated by
12% SDS-PAGE and transferred onto polyvinylidene difluoride
membranes. The membranes were blocked with 5% non-fat milk for 20
min at room temperature, and then incubated with primary antibodies
at 4°C overnight, followed by incubation with the secondary
antibody at room temperature for 30 min. Protein bands were
visualized by enhanced chemiluminescence using the ChemiDoc™ MP
Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Goat anti-FGF17 polyclonal antibody served as the primary antibody
(dilution, 1:1,000) and mouse anti-goat IgG-HRP was used as the
secondary antibody (dilution, 1:8,000). The molecular sizes of the
obtained protein were verified by comparison with the migration of
pre-stained protein markers (cat. no. 26616; Thermo Fisher
Scientific, Inc.).
Mitogenic activity of rhFGF17
assay
NIH3T3 cells (2×103 cells/well) were
cultured in Dulbecco's modified Eagle's medium (DMEM) containing
10% fetal bovine serum (Thermo Fisher Scientific, Inc.) in a
96-well plate at 37°C for 24 h. The medium was then replaced with
DMEM supplemented with 1% FBS and cells were starved overnight. The
cells were treated with different concentrations of rhFGF17 or
commercial rhFGF17 (R&D Systems China Co., Ltd., Shanghai,
China) for 48 h and the number of viable cells was determined by
adding 25 µl MTT (5 mg/ml) per well for 4 h. Finally, the medium
was discarded and 150 µl dimethyl sulfoxide was added to each well
to dissolve the crystals by agitation at room temperature for 10
min; the absorbance was immediately measured at a wavelength of 600
nm using the GENESYS™ 10S UV–Vis Spectrophotometer (Thermo Fisher
Scientific, Inc.).
Results
Construction of the rhFGF17 expression
vector
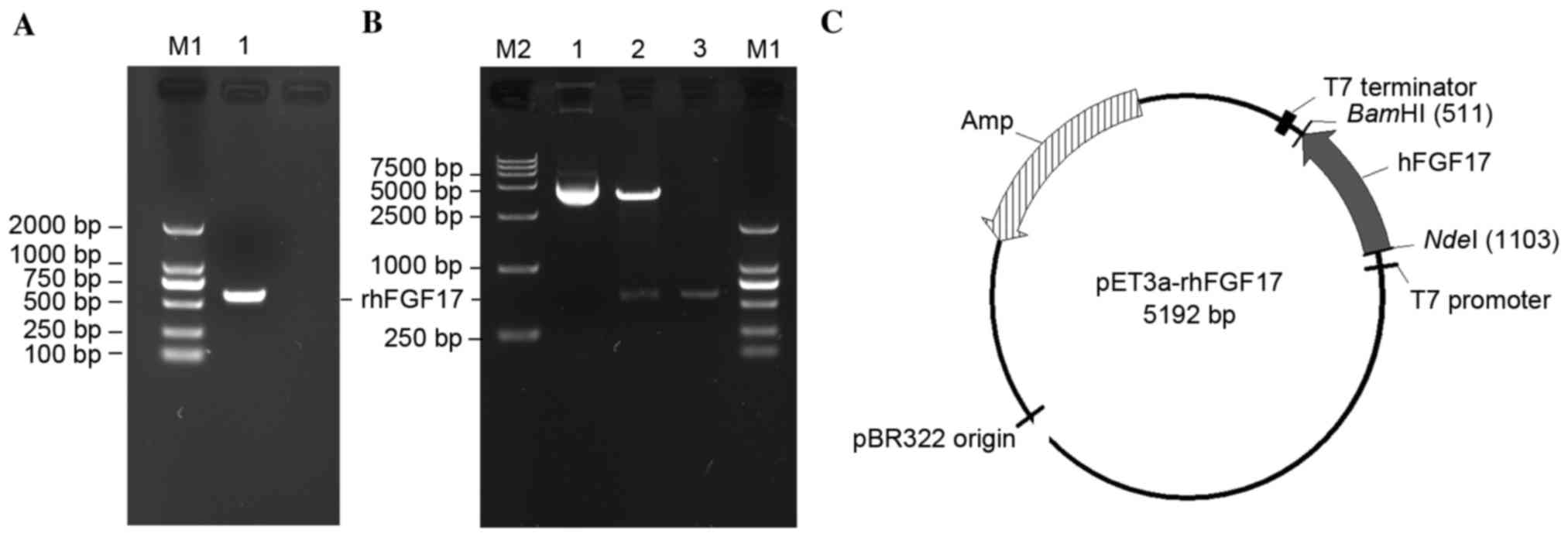
To produce the rhFGF17 protein, an expression vector
containing the optimized hFGF17 gene was constructed. The hFGF17
fragment was obtained (Fig. 1A),
then digested with NdeI and BamHI and cloned into the
pET3a vector to create the pET3a-rhFGF17 recombinant plasmids,
which were then confirmed by restriction enzymatic analysis
(Fig. 1B and C) and automated DNA
sequencing.
Expression of rhFGF17 in
BL21(DE3)pLysS
The recombinant plasmid was transformed into
BL21(DE3)pLysS. The SDS-PAGE demonstrated that rhFGF17 was induced
by 1 mM IPTG and the apparent molecular band was ~23 kDa,
corresponding to the predicted molecular weight (22.6 kDa; Fig. 2A). The greatest expression level of
rhFGF17 was ~30% of total protein.
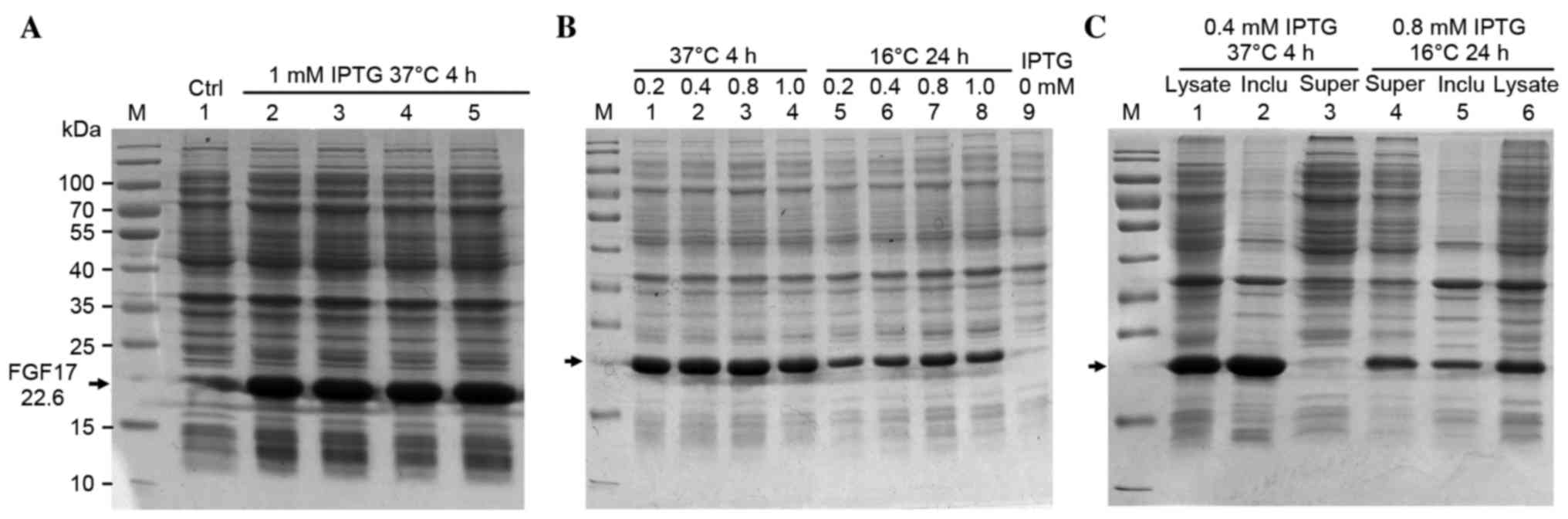 | Figure 2.Optimizing the expression conditions
of rhFGF17. (A) SDS-PAGE analysis of rhFGF17 expression in
BL21(DE3)PLysS induced by 1 mM IPTG for 4 h at 37°C. Lane 1, served
as a control and was not induced with IPTG; lanes 2–5, induced with
IPTG; (B) Optimizing the expression conditions of rhFGF17. Lanes
1–4 and 5–8: 0.2, 0.4, 0.8, 1.0 mM IPTG induced at 37°C for 4 h and
16°C for 24 h, respectively. (C) Distribution of rhFGF17. Lanes
1–3, 37°C culture (lane 1, induced BL21(DE3)PLysS/pET3a-rhFGF17;
lane 2, inclusion bodies of bacteria following ultrasonication; and
lane 3, supernatant). Lanes 4–6, 16°C culture (lane 4, supernatant
of bacteria following ultrasonication; lane 5, inclusion bodies;
and lane 6, induced BL21(DE3)PLysS/pET3a-rhFGF17. rhFGF17,
recombinant human fibroblast growth factor 17; IPTG, isopropyl
β-D-1-thiogalactopyranoside; Ctrl, control. |
Optimizing the expression of soluble
rhFGF17
To establish the optimal culture conditions, the
following concentrations of IPTG were evaluated: 0.2, 0.4, 0.8 and
1.0 mM at 37°C or 16°C, under agitation at 180 rpm. When rhFGF17
was induced by 0.4 mM IPTG at 37°C or 0.8 mM IPTG at 16°C, the
rhFGF17 yield reached the highest level with ratios of ~30 and ~20%
of the total protein, respectively according to the SDS-PAGE
results (Fig. 2B). Following
fermentation in a 2-liter flask under the above-mentioned optimized
conditions, the yield of bacteria was ~9 g/l at 37°C and 6 g/l at
16°C. Soluble detection was performed by lysis. SDS-PAGE analysis
of the lysate supernatant and sediment indicated that the
recombinant protein was marginally soluble at 16°C, but inclusion
bodies appeared to be formed at 37°C (Fig. 2C).
Purification of soluble rhFGF17
The soluble product was purified with improved lysis
buffer and more soluble proteins were obtained with almost no
sediment (Fig. 3A).
Heparin-affinity column chromatography combined with SP-Sepharose
column chromatography was used for purification of the soluble
fraction of proteins. rhFGF17 was eluted with 1.0 M NaCl in elution
buffer from the two columns (Fig. 3B
and C), and the yield was 1 mg/g (1 mg rhFGF17 from 1 g
bacteria cells).
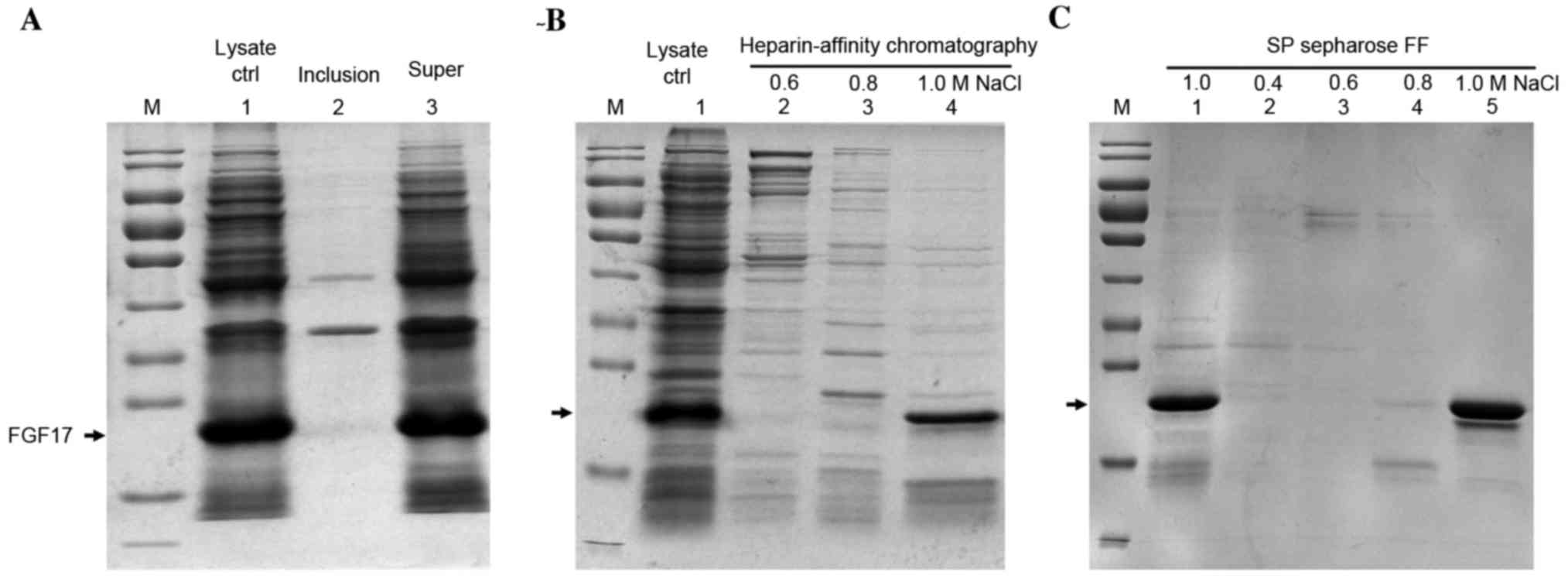 | Figure 3.SDS-PAGE analysis of the purification
of soluble rhFGF17 using improved lysis buffer. (A) Lane 1, lysate
control; lane 2, inclusion bodies of bacteria following
ultrasonication; lane 3, supernatant. (B) Purification of soluble
rhFGF17 with heparin-affinity chromatography (lanes 2–4). Lane 1,
supernatant of bacteria after ultrasonication; lane 2–4 eluted with
0.6, 0.8, 1.0 M NaCl from heparin-affinity chromatography, (C)
Lanes 1–5, SP Sepharose Fast Flow of rhFGF17 eluted with different
NaCl concentrations. Lane1 and 5, 1.0 M NaCl; lanes 2–4, 0.4, 0.6
and 0.8 M NaCl. The arrow indicates the rhFGF17 band site. rhFGF17,
recombinant human fibroblast growth factor 17; Ctrl, control; FF,
fast flow. |
Purification and identification of
rhFGF17 inclusion bodies
rhFGF17 inclusion bodies were predominantly produced
from the culture condition of 37°C for 4 h. rhFGF17 was denatured
by urea and refolded in the dialysis buffer by dialysis and then
renaturing buffer by dilution at pH 7.5 (Fig. 4A). As indicated in Fig. 4A denaturing buffer dissolved the
majority of the rhFGF17. The concentration of total protein in the
denaturing buffer was ~41 mg/ml and in the renaturing buffer prior
to applying it to the heparin-sepharose column, total protein was
decreased to ~2 mg/ml. As demonstrated in Fig. 4B the fractions containing rhFGF17
were finally eluted by heparin affinity chromatography using 20 mM
Tris-HCl containing 1.0 M NaCl. The purified rhFGF17 protein yield
reached 8 mg/g (8 mg rhFGF17 from 1 g bacteria cells).
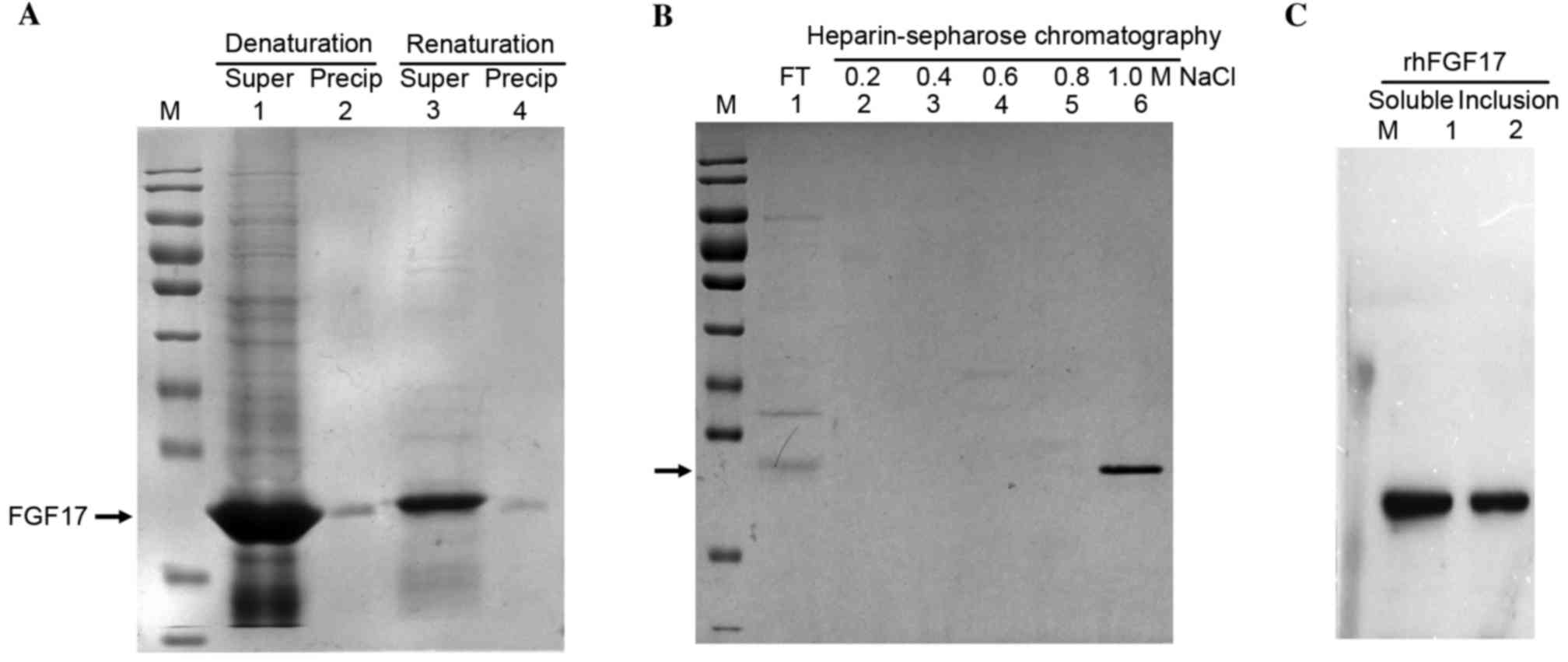 | Figure 4.SDS-PAGE analysis of the purification
of rhFGF17 inclusion bodies. (A) Lanes 1 and 3, Supernatant rhFGF17
and precipitation rhFGF17 in denaturation and renaturation buffer;
lanes 2 and 4, Precipitation rhFGF17 in denaturation and
renaturation buffer. (B) Heparin-sepharose chromatography. Lane 1,
FT; lanes 2–6, eluted rhFGF17 with 20 mM Tris-HCl containing 0.2,
0.4, 0.6, 0.8 and 1.0 M NaCl, respectively. (C) Western blot
analysis of rhFGF17. Lane 1, purified soluble rhFGF17; lane 2,
purified rhFGF17 inclusion bodies. rhFGF17, recombinant human
fibroblast growth factor 17; FT, flow through. |
The purified soluble rhFGF17 and rhFGF17 inclusion
bodies were homogenous and their purity was >95%. Western blot
analysis demonstrated that the purified rhFGF17 had good
immunoreactivity with the anti-human FGF17 antibody (Fig. 4C).
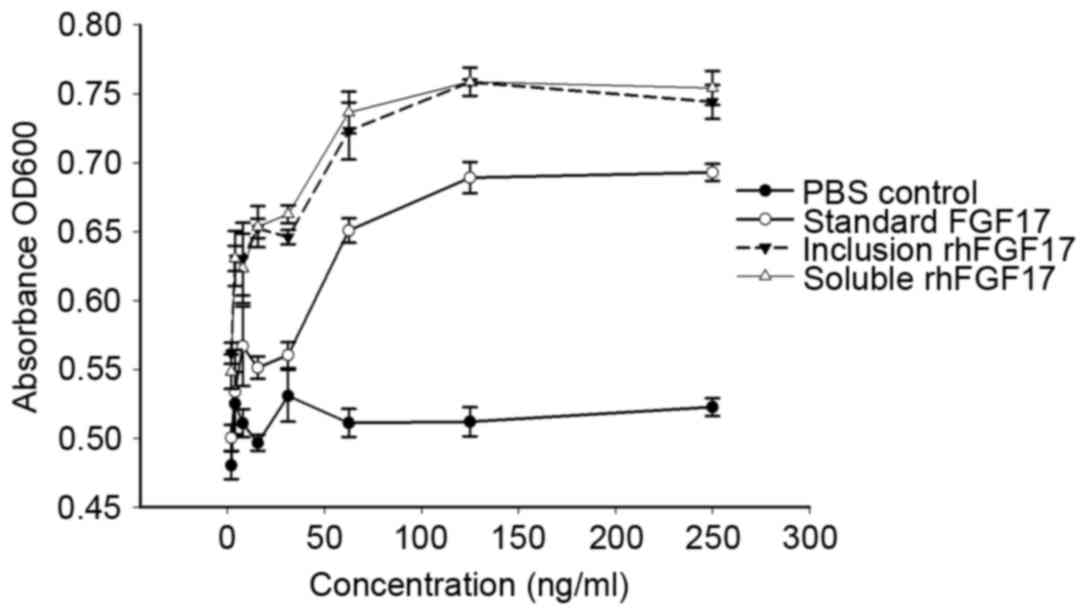
Mitogenic activity of rhFGF17
To assess the biological activity of purified
rhFGF17, the proliferative effect of rhFGF17 was determined using a
standard MTT assay on NIH3T3 cells and compared with commercial
rhFGF17 (the positive control). As shown in Fig. 5, the two purified soluble forms and
inclusion bodies of rhFGF17 demonstrate similar mitogenic activity
in NIH3T3 cells, which is consistent with the findings of a
previous study (11).
Additionally, compared with the commercial rhFGF17, the rhFGF17
protein formed during the present study exhibited improved
biological activity. Furthermore, rhFGF17 was found to have a
dose-dependent effect on the viability of NIH3T3 cells, whereas the
negative control did not. Finally, the results demonstrated that
the soluble and inclusion body forms of rhFGF17 had a marked
biological effect on NIH3T3 cells.
Discussion
As a novel member of the FGF family, numerous
pharmacological studies have demonstrated that FGF17 is a key
factor in neuropsychiatric diseases due to its important roles in
the patterning of the cerebellum and cortex (12). As a potential carcinogen, FGF17 is
predominantly associated with prostate cancer (13) and hematopoietic tumors (14). Therefore, it is necessary to
investigate FGF17 and develop strategies for abundant production of
FGF17 with high bioactivity. As reported previously, the
recombinant form of FGF17 protein was produced using insect cells;
however, compared with prokaryotic expression systems, eukaryotic
systems are considered unsuitable for large-scale purification
(15). To date, there are few
reports regarding the expression of hFGF17, particularly in E.
coli expression systems. The low level of soluble production
and difficulty purifying inclusion bodies, particularly denaturing
and refolding, has restricted further research and application.
There are certain methods used to overcome these limitations,
including fusion systems to enhance target protein expression, such
as Halo-tag fusion (16). However,
the method for obtaining target protein requires that the fused tag
must be removed, which involves an expensive cleavage restriction
enzyme and may impact the bioactivity of the target protein.
Low temperatures increase the expression levels of
soluble proteins and reduce the aggregation of recombinant
proteins, thus reducing the formation of inclusion bodies (17). In addition, low agitation speeds
will reduce the speed of bacteria proliferation, but increase the
amount of soluble proteins. Therefore, in the current study, the
culture conditions at 37°C and 200 rpm, and 16°C and 180 rpm would
yield high levels of inclusion bodies and soluble proteins,
respectively. To further improve the production levels of the
target protein, the expression conditions were optimized according
to the IPTG concentration, thus the expression level of inclusion
bodies and soluble protein reached >30 and >20% of total
protein, respectively with 0.4 mM IPTG for 4 h at 37°C and 1 mM
IPTG for 24 h at 16°C. Meanwhile, the lysis buffer was improved by
the addition of more Tris, glycerol and sucrose; thus, a soluble
protein was obtained with almost no sediment (Fig. 3A). Taken together, the conditions
were improved and high production levels of target protein were
achieved for purification.
Based on an isoelectric point of 10.43 and the
heparin binding ability for rhFGF17, the non-fusion rhFGF17 protein
was efficiently separated by heparin-sepharose chromatography and
SP Sepharose Fast Flow (18,19).
The purified rhFGF17 proteins were biologically active in
vitro and exerted a dose-dependent effect on the proliferation
of NIH3T3 cells; inclusion-bodies were demonstrated to have a
biological activity similar to the soluble proteins. Thus, the
soluble proteins and inclusion bodies obtained using the culture
conditions at 37°C and 200 rpm, and 16°C and 180 rpm, respectively,
are efficiently produced and are characterized by high levels of
bioactivity. Furthermore, FGF17 has previously been reported to be
involved in Kallmann syndrome (20) and causes tamoxifen resistance in
vitro (21). Therefore,
whether there is a direct association between FGF17 and breast
cancer requires further investigation.
In conclusion, soluble and inclusion bodies of
rhFGF17 were successfully expressed in E. coli. The current
study indicates that the non-tagged expression of either soluble
proteins or inclusion bodies of rhFGF17 is simple, viable and
highly effective, making it convenient for high-efficiency
expression and purification of proteins, whilst preserving the high
biological activity levels.
Acknowledgements
The present study was supported by Zhejiang
Extremely Key Subject of Pharmacology and Biochemical Pharmaceutics
and the National ‘863’ High Technology Research and Development
Program (grant No. 2011AA02A113) and Initiation Founding of
Xingxiang Medical University (grant nos. XYBSKYZZ201512 and
201513).
References
|
1
|
Ornitz DM and Itoh N: The Fibroblast
Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol.
4:215–266. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoshikawa M, Ohbayashi N, Yonamine A,
Konishi M, Ozaki K, Fukui S and Itoh N: Structure and expression of
a novel fibroblast growth factor, FGF17, preferentially expressed
in the embryonic brain. Biochem Biophys Res Commun. 244:187–191.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohbayashi N, Hoshikawa M, Kimura S,
Yamasaki M, Fukui S and Itoh N: Structure and expression of the
mRNA encoding a novel fibroblast growth factor, FGF-18. J Biol
Chem. 273:18161–18164. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu J, Lawshe A, MacArthur CA and Ornitz
DM: Genomic structure, mapping, activity and expression of
fibroblast growth factor 17. Mech Dev. 83:165–178. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Leary DD, Chou SJ and Sahara S: Area
patterning of the mammalian cortex. Neuron. 56:252–269. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dong X, Tang B, Li J, Xu Q, Fang S and Hua
Z: Expression and purification of intact and functional soybean
(Glycine max) seed ferritin complex in Escherichia coli. J
Micro Biotech. 18:299–307. 2008.
|
|
7
|
Jana S and Deb JK: Strategies for
efficient production of heterologous proteins in Escherichia
coli. Appl Microbiol Biotechnol. 67:289–298. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Derynck R, Roberts AB, Winkler ME, Chen EY
and Goeddel DV: Human transforming growth factor-alpha: Precursor
structure and expression in E. coli. Cell. 38:287–297. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Verdon J, Girardin N, Marchand A, Héchard
Y and Berjeaud JM: Purification and antibacterial activity of
recombinant warnericin RK expressed in Escherichia coli.
Appl Microbiol Biotechnol. 97:5401–5412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ajikumar PK, Xiao WH, Tyo KE, Wang Y,
Simeon F, Leonard E, Mucha O, Phon TH and Pfeifer B: Isoprenoid
pathway optimization for Taxol precursor overproduction in
Escherichia coli. Science. 330:70–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song L, Huang Z, Chen Y, Li H, Jiang C and
Li X: High-efficiency production of bioactive recombinant human
fibroblast growth factor 18 in Escherichia coli and its
effects on hair follicle growth. Appl Microbiol Biotechnol.
98:695–704. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tabarés-Seisdedos R and Rubenstein JL:
Chromosome 8p as a potential hub for developmental neuropsychiatric
disorders: Implications for schizophrenia, autism and cancer. Mol
Psychiatry. 14:563–589. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heer R, Douglas D, Mathers ME, Robson CN
and Leung HY: Fibroblast growth factor 17 is over-expressed in
human prostate cancer. J Pathol. 204:578–586. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nezu M, Tomonaga T, Sakai C, Ishii A,
Itoga S, Nishimura M, Matsuo Y, Tagawa M and Nomura F: Expression
of the fetal-oncogenic fibroblast growth factor-8/17/18 subfamily
in human hematopoietic tumors. Biochem Biophys Res Commun.
335:843–849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hoshikawa M, Ohbayashi N, Yonamine A,
Konishi M, Ozaki K, Fukui S and Itoh N: Structure and expression of
a novel fibroblast growth factor, FGF-17, preferentially expressed
in the embryonic brain. Biochem Biophys Res Commun. 244:187–191.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun C, Li Y, Taylor SE, Mao X, Wilkinson
MC and Fernig DG: Halo Tag is an effective expression and
solubilisation fusion partner for a range of fibroblast growth
factors. Peer J. 3:e10602015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
de Groot NS and Ventura S: Effect of
temperature on protein quality in bacterial inclusion bodies. FEBS
Lett. 580:6471–6476. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berman B, Ostrovsky O, Shlissel M, Lang T,
Regan D, Vlodavsky I, Ishai-Michaeli R and Ron D: Similarities and
differences between the effects of heparin and glypican-1 on the
bioactivity of acidic fibroblast growth factor and the keratinocyte
growth factor. J Biol Chem. 274:36132–36138. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee YF, Schmidt M, Graalfs H, Hafner M and
Frech C: Modeling of dual gradient elution in ion exchange and
mixed-mode chromatography. J Chromatogr A. 1417:64–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miraoui H, Dwyer AA, Sykiotis GP, Plummer
L, Chung W, Feng B, Beenken A, Clarke J, Pers TH, Dworzynski P, et
al: Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are
identified in individuals with congenital hypogonadotropic
hypogonadism. Am J Hum Genet. 92:725–743. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meijer D, Sieuwerts AM, Look MP, van
Agthoven T, Foekens JA and Dorssers LC: Fibroblast growth factor
receptor 4 predicts failure on tamoxifen therapy in patients with
recurrent breast cancer. Endocr Relat Cancer. 15:101–111. 2008.
View Article : Google Scholar : PubMed/NCBI
|