Introduction
Breast cancer is one of the most common female
malignant tumors, and its incidence is increasing every year
(1,2). However, the global mortality rate of
breast cancer is falling due to the established system of breast
cancer screening and early diagnosis, as well as improvements in
molecular biological techniques and comprehensive diagnosis and
treatments (3). The first known
use of endocrine therapy to treat breast cancer was in 1896 by the
British scholar Beatson, who treated premenopausal advanced breast
cancer using oophorectomy, and it has been recognized to be a
postoperative adjuvant treatment of early breast cancer and as a
treatment of advanced metastatic breast cancer (4).
Tamoxifen (TAM), the earliest selective estrogen
receptor (ER) modulator, has been widely used for the treatment or
prevention of estrogen receptor-positive (ER+) breast cancer, as
well as for chemoprevention in women at high risk of developing
breast cancer (5). Tamoxifen is
composed of a triphenylethylene backbone structure and functions by
combining with ERα (6) and
inhibiting the estradiol (E2)-ER genetic pathway, which inhibit
proliferation and induce apoptosis in breast cancer cells. In all
breast tumor types, ~75% are ERα positive and thus tamoxifen is the
most widely used therapy for breast cancer, leading to tumor
stabilization in ~50% of all previously untreated patients with
metastatic breast cancer (7,8).
There are two possible antitumor mechanisms
underlying TAM, which may produce these effects. TAM is able to
inhibit activation function 2 (AF2) of the ER, produce antiestrogen
function E and alter its structure by combining with the ER
(9). In addition, TAM may induce
apoptosis in breast cancer cells through the membrane receptor
pathway, the release of cytochrome c (Cyt C) and the
biochemical pathways activated by caspase family proteins (10). TAM also inhibits protein kinase C
activity and the extracellular signal-regulated kinase 1/2
signaling pathway to modulate the growth of breast cancer cells
(11). However, the exact
mechanism underlying the effect of TAM on breast cancer cell growth
and metastasis remains unclear.
The present study was designed to investigate the
role of TAM on the growth and invasion of MCF-7 (ER+) cells in
vitro. TAM significantly induced MCF-7 cell apoptosis, and
inhibited cell growth, migration and invasion. In addition, the
effect of TAM on the mitochondrial membrane potential (MMP), energy
metabolism (ATP) and reactive oxygen species (ROS) in MCF-7 cells
was investigated.
Materials and methods
Cell culture
The human breast cancer cell line MCF-7 was obtained
from the Shanghai Cell Bank (Shanghai, China). Cells were cultured
in Dulbecco's modified Eagle's medium (DMEM; Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine
serum (FBS; Hyclone; GE Healthcare Life Sciences) at 37°C in a
humid environment with 5% CO2.
Cell survival assay
Human breast cancer cells MCF-7 were inoculated in a
96-well plate with 100 µl culture medium/well at density of
5×103 cells. The cells were treated with 0, 0.25, 0.50,
0.75, 1.0, 1.25, 1.5, 2.0, 2.5, 3.0 and 4.0 µM TAM (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) for 24 h at 37°C. Alternatively,
cells were pretreated with 5 mM N-acetylcysteine (NAC;
Sigma-Aldrich; Merck KGaA) for 1 h at 37°C, and then treated with
0.5, 1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0 and 10.0 µM TAM for 24 h at
37°C. Cell proliferation was measured using a Cell Counting kit-8
(CCK-8) assay (Beyotime Institute of Biotechnology, Haimen, China).
A total of 10 µl CCK-8 solution was added to each well and the
cells were incubated for 2 h at 37°C. The optical density (OD) was
measured at 450 nm with a microplate reader (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cell survival fraction was
calculated as the Survival
fraction=ODExperimental/ODControl. Three
independent experiments were performed in quadruplicate.
Flow cytometric analysis of the cell
cycle and apoptosis
MCF-7 cells were plated in 6-well plates at a cell
density of 3×105 cells and incubated until the anchoring
rate was between 70 and 80%. The cells were treated with 0, 1, 2
and 4 µM TAM for 48 h at 37°C. Cells from each experimental group
were collected and fixed with 70% ice-cold ethanol at 4°C
overnight. Cells were centrifuged at 150 × g for 5 min at room
temperature and resuspended with 500 µl propidium iodide (PI; 100
µg/ml) for 30 min in the dark at room temperature, prior to
analysis. The cell cycle profiles were assayed using the FACScan
ESP flow cytometer (BD Biosciences, San Jose, CA, USA) at 488 nm,
and data were analyzed using MultiCycle AV software for Windows
32-bit (Phoenix Flow Systems, San Diego, CA, USA). For analysis of
apoptosis, cells from each experimental group were collected and
processed as described in the Annexin V-Fluorescein Isothiocyanate
(FITC) Apoptosis Detection kit I manual (BD Biosciences) and
analyzed by FACScan flow cytometry using FlowJo.7.6.2 software (BD
Biosciences).
Wound-healing assay in vitro
Cells were plated in 6-well plates and allowed to
form a confluent monolayer for 24 h. The monolayer was scratched
with the tip of a 200 µl pipette and then washed twice with PBS to
remove the floating and detached cells. Fresh serum-free medium,
which contained 0, 1 and 2 µM TAM, was added and images of the
wound area were captured at 0, 24 and 48 h to assess cell migration
using an Olympus IX71 microscope (Olympus Corporation, Tokyo,
Japan).
Transwell invasion assay
Cell invasion was assessed using 24-well Matrigel
invasion chambers (Corning Incorporated, NY, USA). The inserts were
pre-coated with 40 µl Matrigel (dilution, 1:4; BD Biosciences).
Cells were suspended in serum-free DMEM, which contained 0 or 1 µM
TAM. Subsequently, 1×104 cells were added to the upper
chambers. The lower chambers were filled with medium containing 10%
FBS. Following incubation for 24 h at 37°C, the chambers were fixed
with 3.7% paraformaldehyde for 10 min, and stained with 2% crystal
violet for 30 min at room temperature. The penetration of cells
through the membrane was quantified by counting the number of cells
that penetrated the membrane in 10 microscopic fields/filter
(×100).
MMP test
MCF-7 cells were plated in 6-well plate and
incubated until the anchoring rate reached 70 to 80%. Cells
(1×106) were treated with 0, 1, 2 and 4 µM TAM for 24 h
and trypsinized to obtain single-cell suspensions. Subsequently,
cells were incubated in 0.5 ml 1X JC-1 (Thermo Fisher Scientific,
Inc.) for 15 min at 37°C and analyzed using a FACSCalibur flow
cytometer with FlowJo.7.6.2 software.
ROS analysis
MCF-7 cells were plated in 6-well plates at a cell
density of 3×105 cells and allowed to attach overnight.
Cells were treated with 0, 1, 2 and 4 µM TAM for 24 h. Cells were
harvested, resuspended in serum-free DMEM containing 10 µmol/l
chloromethyl-2′,7′-dichlorofluorescein diacetate (Beyotime
Institute of Biotechnology) and incubated at 37°C in the dark for
30 min. The cells were subsequently washed three times with PBS and
the intensity of fluorescence was assayed using the FC500 flow
cytometer at 488 nm.
ATP determination
MCF-7 cells were plated in 6-well plates at a cell
density of 1×105 and allowed to attach overnight. Cells
were treated with 0, 1, 2 and 4 µM TAM for 24 h and lysed using 200
µl lysis buffer (Beyotime Institute of Biotechnology). A total of
100 µl ATP Assay kit (Beyotime Institute of Biotechnology) was
added into each well of a 96-well plate and incubated at room
temperature for 3 to 5 min to remove the background ATP.
Subsequently, 100 µl of sample was added to each well and ATP was
analyzed on a flow cytometer following mixing.
Western blot analysis
Cells were washed with PBS and lysed in cell lysis
buffer containing 1X phenylmethylsulfonyl fluoride (Beyotime
Institute of Biotechnology, China) for 30 min. Protein
concentration was determined by BCA Protein Assay kit (Beyotime
Institute of Biotechnology). Proteins (50 µg) from each group were
separated by 10% SDS-PAGE, transferred onto nitrocellulose
membranes and blocked with 5% skimmed milk for 1 h at room
temperature. Following blocking, the membranes were incubated with
antibodies against β-actin (1:1,000, #3700), apoptosis regulator
Bcl-2 (1:1,000, #15071), apoptosis regulator BAX (1:1,000, #5023)
or caspase-3 (1:1,000, #9662) (all from Cell Signaling Technology,
Inc., Danvers, MA, USA) over night at 4°C. Blots were subsequently
incubated with horseradish peroxidase-conjugated anti-mouse
(1:2,000, #7076) or anti-rabbit IgG antibodies (1:2,000, #7074)
(both from Cell Signaling Technology, Inc.) for 1 h at room
temperature. Protein bands were visualized with enhanced
chemiluminescence solution (Beyotime Institute of
Biotechnology).
Statistical analysis
The data are presented as the mean ± standard
deviation. SPSS software (version 19; IBM SPSS, Armonk, NY, USA)
was used to perform the statistical analysis. The data were
analyzed using a paired Student's t-test. P<0.05 was considered
to indicate a statistically significant difference. Each experiment
was repeated three times.
Results
TAM suppresses breast cancer cell
proliferation
Breast cancer cells were treated with varying
concentrations of TAM to investigate the effect of TAM on cell
proliferation. As presented in Fig.
1, the proliferation of these cells was markedly suppressed by
TAM in a dose-dependent manner.
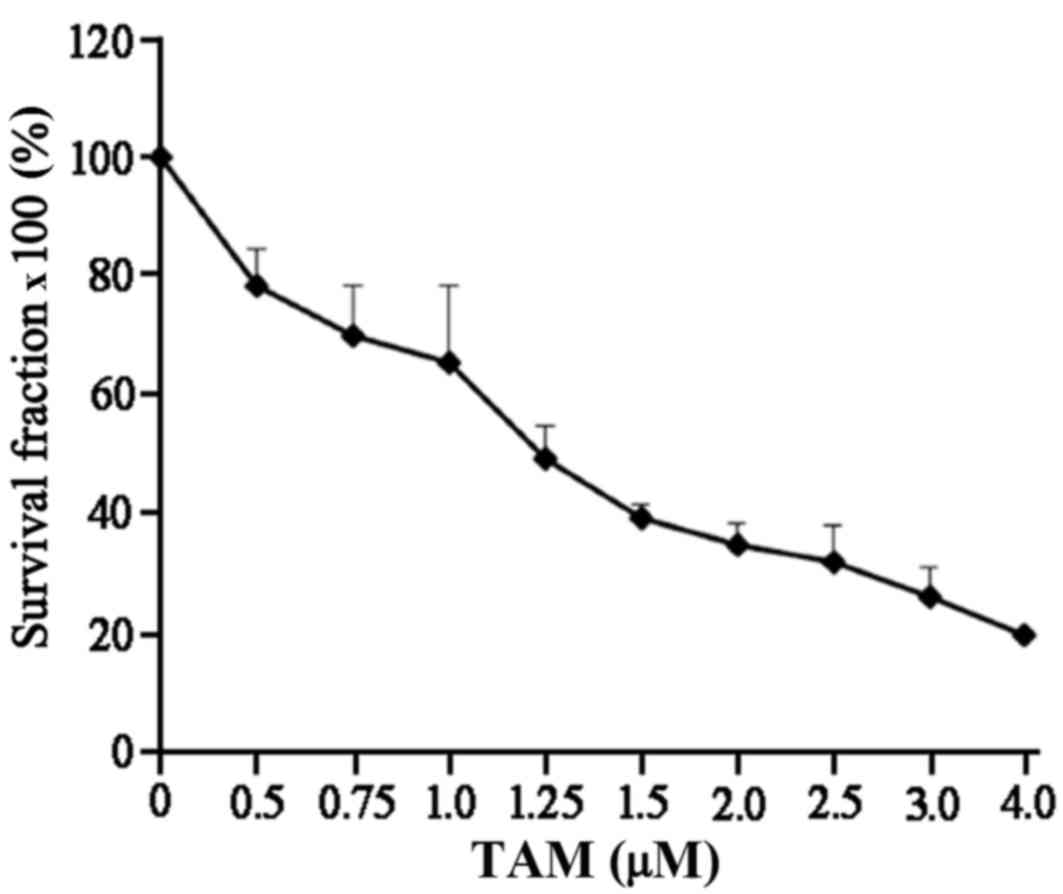 | Figure 1.TAM suppresses breast cancer cell
proliferation. Estrogen receptor-positive breast cancer MCF-7 cells
were cultured in 96-well plates at 5×103 cells/well, and
treated with 0, 0.25, 0.50, 0.75, 1.0, 1.25, 1.5, 2.0, 2.5, 3.0 and
4.0 µM TAM for 24 h. Cell proliferation was subsequently assessed
using a Cell Counting kit-8 assay. TAM, tamoxifen. |
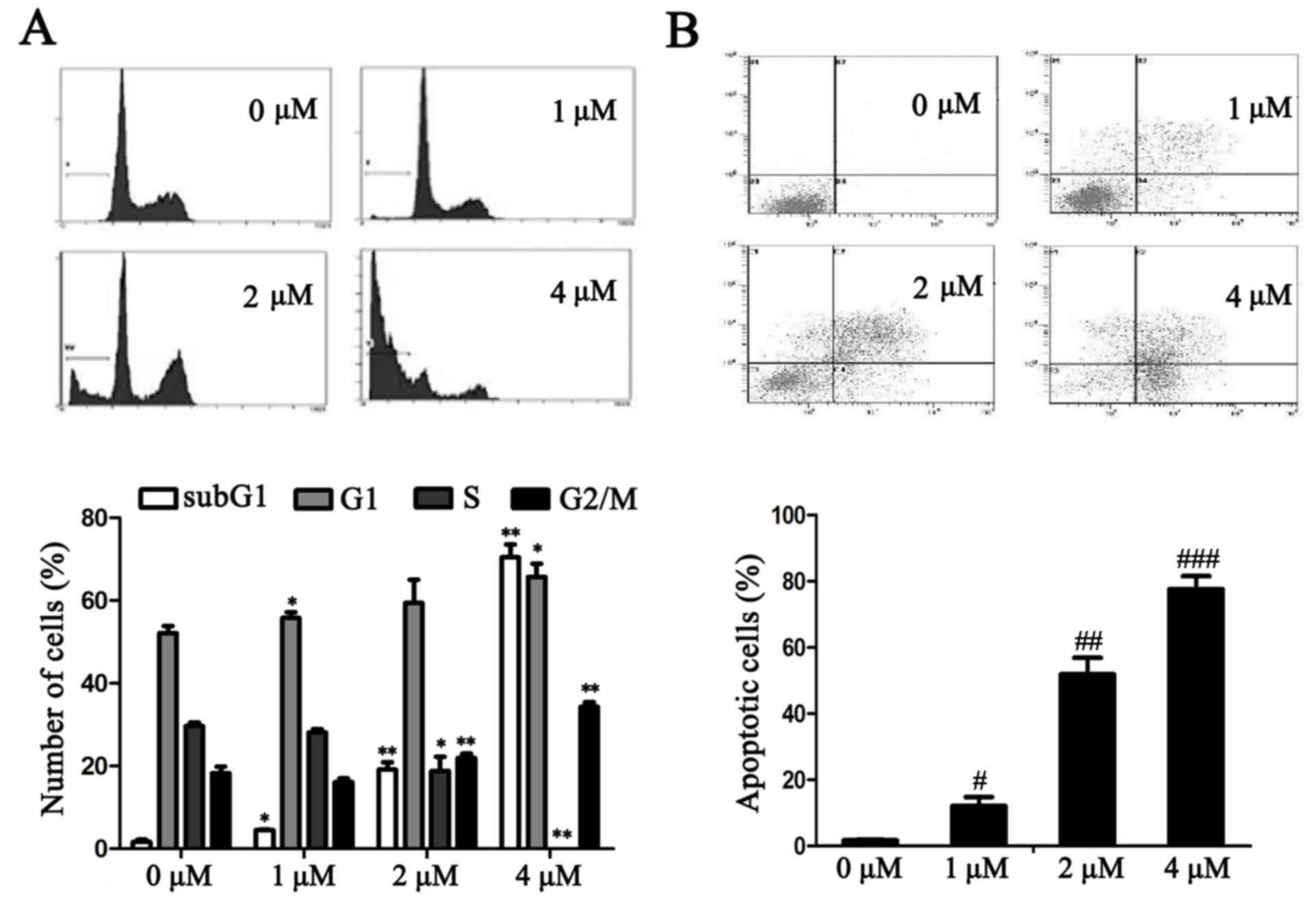
TAM induces cell cycle arrest and
promotes cell apoptosis in breast cancer cells
The effects of TAM on the MCF-7 cell cycle were
further investigated. Following treatment with TAM for 24 h, the
number of MCF-7 cells in the G0/G1 and G2/M phases increased when
compared with the untreated cells. By contrast, in the S phase the
number of cells decreased. In addition, as the concentration of TAM
increased, the SubG1 Peak also significantly increased (P<0.05;
Fig. 2A).
The effects of TAM on cell apoptosis were determined
using an Annexin V-FITC and PI staining assay. As presented in
Fig. 2B, TAM induced MCF-7 cell
apoptosis and the percentage of apoptotic cells significantly
increased in a dose-dependent manner (P<0.05).
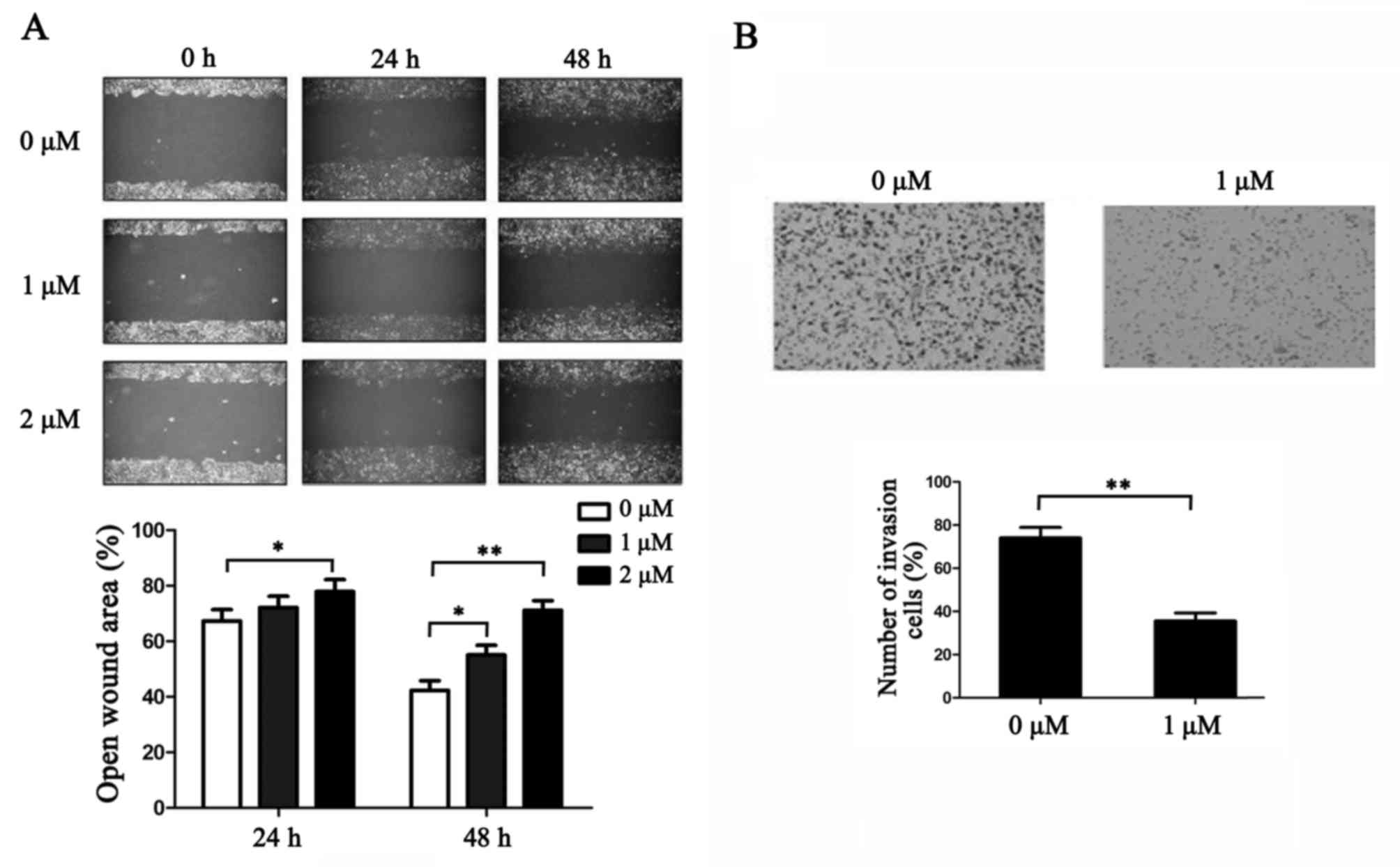
TAM inhibits breast cancer cell
migration and invasion
Cancer metastasis is a complex process that involves
cell migration and invasiveness. To examine the effect of TAM on
the migration of breast cancer cells, MCF-7 cells were subjected to
in vitro wound healing assays. Confluent cell cultures were
scraped to create a wound and were subsequently treated with three
concentrations of TAM. Cell motility was determined at different
time points. As presented in Fig.
3A, TAM reduced the rate of wound healing in MCF-7 cell. The
effect of inhibition was enhanced with increased concentrations of
TAM.
Matrigel invasion assays were performed to explore
the effect of TAM on the invasiveness of breast cancer cells.
Following treatment for 24 h, the migratory cells at the bottom
surface of the membrane were stained with crystal violet and cell
penetration through the membrane was calculated manually. As
presented in Fig. 3B, TAM
treatment significantly decreased the ability of MCF-7 cells to
invade through the Matrigel when compared with the control cells
(P<0.01).
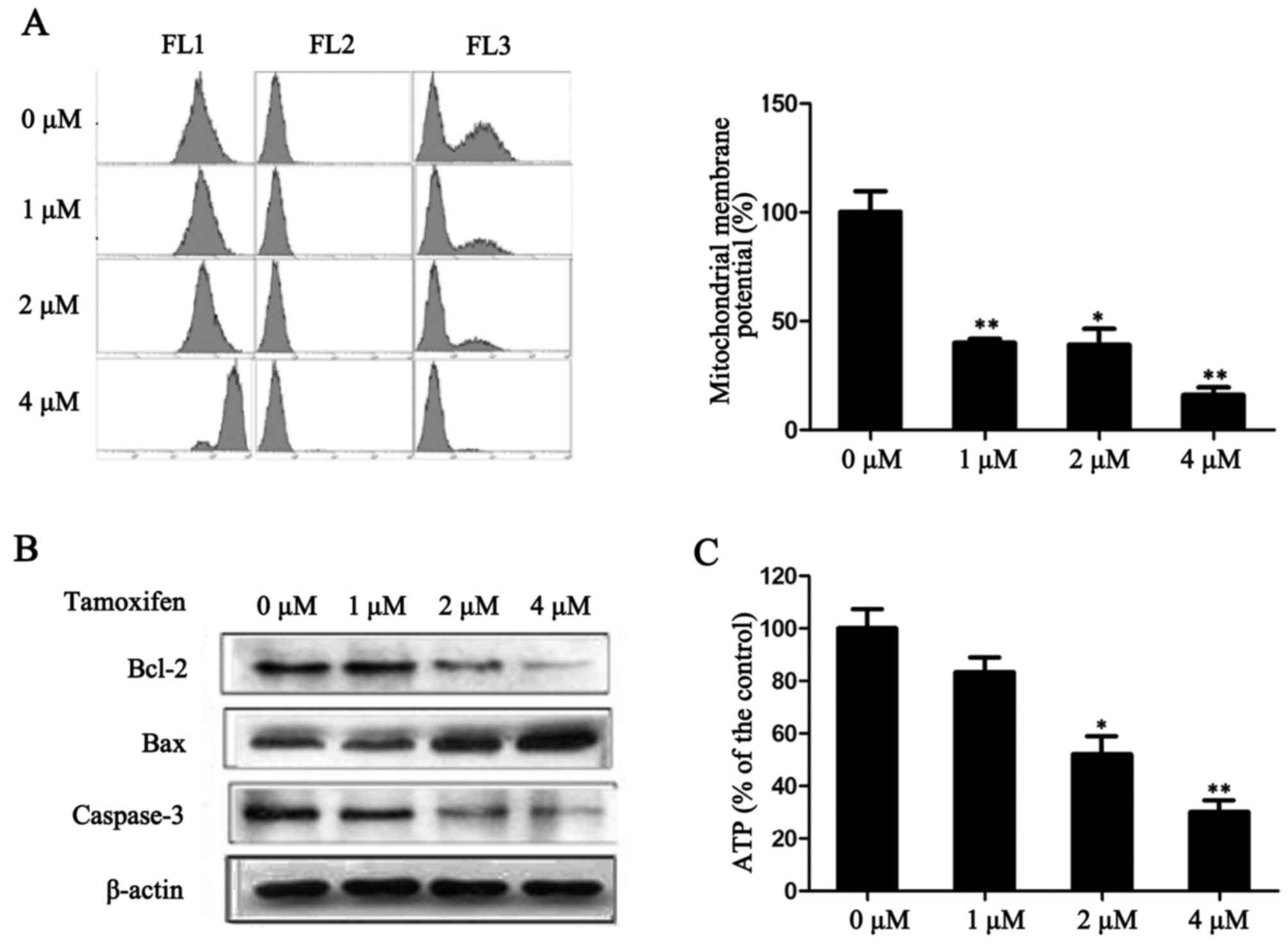
TAM reduces MMP in ER-positive breast
cancer cells
A decrease in MMP is a sign of early cell apoptosis.
Once the MMP has decreased, apoptosis is irreversible (12). The fluorescent dye JC-1 is
widely-used for detecting the functionality of mitochondria.
Following treatment with TAM for 24 h, the fluorescence intensity
significantly decreased in MCF-7 (P<0.05; Fig. 4A), which indicates that TAM may
mediate MMP dysfunction. In addition, the expression levels of
anti-apoptotic factor Bcl-2 and pro-apoptotic factors Bax and
caspase-3, which are involved in the mitochondrial apoptosis
pathway, were detected. As presented in Fig. 4B, TAM reduced the expression of
Bcl-2 and caspase-3, and increased the expression of Bax. These
results indicate that the promotion of cell apoptosis by TAM may be
mediated by the mitochondrial apoptosis pathway.
To investigate the variation of energy metabolism in
cells following TAM-induced suppression of proliferation, the level
of ATP in MCF-7 cells was measured. As presented in Fig. 4C, following treatment with 1, 2 and
4 µM TAM, the production of ATP significantly decreased in a
dose-dependent manner (P<0.05). These results indicate that TAM
may have reduced the production of ATP and thus, energy production
was not able to be compensated due to mitochondrial dysfunction.
This may be one of the important mechanisms underlying TAM-induced
ER-positive breast cancer cell death.
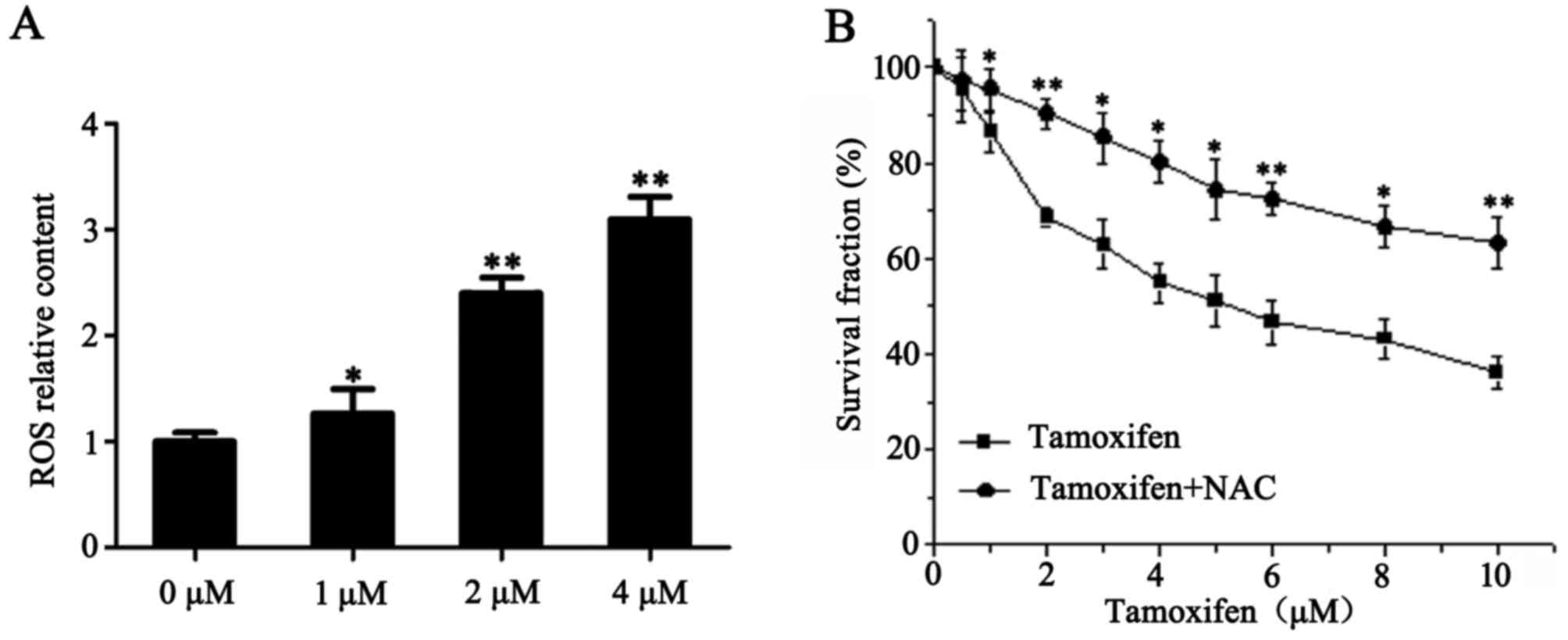
TAM promotes the formation of ROS in
ER-positive breast cancer cells
Accumulation of intracellular ROS may activate a
number of pathways that are important for the induction of
apoptosis and inhibition of invasion (13,14).
Therefore, the present study examined the involvement of ROS in
TAM-induced ER-positive breast cancer cell death. Following
treatment with 1, 2 and 4 µM TAM for 24 h, the levels of ROS in
MCF-7 cells were 1.3, 2.4 and 3.1 times higher when compared with
the control group, respectively (P<0.05; Fig. 5A). To further elucidate the role of
ROS in TAM-induced cell death, NAC (a type of ROS inhibitor) was
used in conjunction with TAM to treat MCF-7 cells, then cell
proliferation was determined using a CCK-8 assay. As presented in
Fig. 5B, NAC prevented TAM-induced
MCF-7 cell death. These results indicate that TAM promotes the
formation of ROS in ER-positive breast cancer cells, thereby
inducing cell death.
Discussion
TAM, the earliest synthetic selective estrogen
receptor modulator non-steroidal antiestrogen drug, has been widely
used in endocrine therapies for breast cancer (5). Estrogen specifically binds with ER
forming E2-ER complexes to activate a number of transcription
factors including, c-Myc, transforming growth factor (TGF)-α and
cathepsin D, which promote cell growth and metastasis (15–17).
TAM competes with estrogen to bind the ER, thereby preventing
proliferative stimulation by estrogens (18). A low concentration of TAM activates
ER-dependent pathways, while a high concentration of TAM induces
the oxidative stress reaction and activates non-ER pathways to
induce cell apoptosis (19,20).
TAM may participate in the regulation of cell apoptosis-associated
signaling pathways and directly control the mitochondrial apoptosis
pathway by regulating the expression of Bcl-2/Bcl-2-like protein 1
and Bax/Bcl-2 homologous antagonist killer family of proteins
(21). TAM may also regulate cell
cyclin proteins (22–24), calmodulin (25) and TGF expression (26). However, the exact mechanism of the
effect of TAM on breast cancer cell growth remains unclear, and the
role of TAM on breast cancer metastasis is unknown.
In the present study, the effect of TAM on the
growth, cell cycle progression and apoptosis of ER+ MCF-7 cells was
investigated. The results demonstrated that there is a
dose-dependent association between TAM and its effect on MCF-7 cell
growth. In addition, TAM induced G0/G1 and G2/M phase arrest in
MCF-7 cells and significantly promoted MCF-7 cell apoptosis.
A high degree of invasiveness is an important
feature of malignant tumors and is the basic cause of tumor
recurrence. Due to the development of radiotherapy and
chemotherapy, the local control of breast cancer has improved;
however, distant metastasis has been the primary cause of treatment
failure. In the present study, following treatment with TAM, the
migration and invasiveness of MCF-7 cells was markedly reduced.
Since Warburg's initial hypothesis (27) the role of mitochondrial dysfunction
in tumorigenesis has been investigated extensively using multiple
approaches (12,28). The results of the present study
indicate that the MMP became unstable and decreased in MCF-7 cells,
observed as a decrease in JC-1 accumulation within mitochondria. In
addition, the level of ATP decreased with increasing concentrations
of TAM, which indicated that the cell energy metabolism was
inhibited and the decline in ATP could not be compensated by the
tricarboxylic acid cycle. Stabilization of the mitochondrial
membrane is also controlled by Bcl-2 family members. This family is
composed of a number of pro-apoptotic and anti-apoptotic proteins
that heterodimerize and modulate each other's function (29). In the present study, the
anti-apoptotic protein Bcl-2 was downregulated and the
pro-apoptotic protein Bax was upregulated following MCF-7 cell
treatment with TAM. In addition, the expression of caspase-3, a
protein associated with the mitochondrial apoptosis pathway,
decreased with increasing TAM concentrations.
Mitochondria are an important source of ROS and are
also the target of ROS (30).
Previously, studies have demonstrated that elevated ROS may induce
damage to the mitochondrial membrane, causing cell cycle
progression arrest (31) and cell
apoptosis (13). The involvement
of ROS signaling in tumor metastasis has also been revealed
(14). In the present study,
following treatment with TAM for 24 h, the level of ROS
significantly increased in MCF-7 cells. Notably, NAC (a type of ROS
inhibitor) was able to prevent TAM-induced MCF-7 cell death.
In conclusion, the present study demonstrated that
TAM was able to specifically inhibit the growth and invasion of ER+
MCF-7 breast cancer cells, and induce G0/G1 and G2/M phase arrest,
and cell apoptosis in MCF-7 cells. The level of ATP reduced and the
MMP decreased, in conjunction with an increase in ROS, contributing
to apoptosis and the inhibition of invasion in MCF-7 cells. These
results support the theoretical foundations for the targeted
therapy of breast cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81502758), the
Science and Technology Foundation of Suzhou (grant no. SYS201417),
and a Project Funded by the Priority Academic Program Development
of Jiangsu Higher Education Institutions.
References
|
1
|
Printz C: American Cancer Society reports
progress in reducing cancer deaths: However, some groups still lag
behind this trend. Cancer. 117:4573–4574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lash TL, Fox MP and Silliman RA: Reduced
mortality rate associated with annual mammograms after breast
cancer therapy. Breast J. 12:2–6. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Montemurro F, Del Mastro L, De Laurentiis
M and Puglisi F: Endocrine therapy in premenopausal women with
breast cancer: A critical appraisal of current evidence. Expert Rev
Anticancer Ther. 16:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fisher B, Costantino JP, Wickerham DL,
Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N,
Atkins J, et al: Tamoxifen for prevention of breast cancer: Report
of the National Surgical Adjuvant Breast and Bowel Project P-1
Study. J Natl Cancer Inst. 90:1371–1388. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jordan VC: Tamoxifen: A most unlikely
pioneering medicine. Nat Rev Drug Discov. 2:205–213. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ring A and Dowsett M: Mechanisms of
tamoxifen resistance. Endocr Relat Cancer. 11:643–658. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaiyesimi IA, Buzdar AU, Decker DA and
Hortobagyi GN: Use of tamoxifen for breast cancer: Twenty-eight
years later. J Clin Oncol. 13:513–529. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Glaros S, Atanaskova N, Zhao C, Skafar DF
and Reddy KB: Activation function-1 domain of estrogen receptor
regulates the agonistic and antagonistic actions of tamoxifen. Mol
Endocrinol. 20:996–1008. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Carrier L and Rowan BG:
Methylseleninic acid synergizes with tamoxifen to induce
caspase-mediated apoptosis in breast cancer cells. Mol Cancer Ther.
7:3056–3063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Wang N, Fang J, Huang J, Tian F, Li
C and Xie F: Role of PKC-ERK signaling in tamoxifen-induced
apoptosis and tamoxifen resistance in human breast cancer cells.
Oncol Rep. 27:1879–1886. 2012.PubMed/NCBI
|
|
12
|
Cavalli LR and Liang BC: Mutagenesis,
tumorigenicity, and apoptosis: Are the mitochondria involved? Mutat
Res. 398:19–26. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Storz P: Reactive oxygen species in tumor
progression. Front Biosci. 10:1881–1896. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Katzenellenbogen BS, Fang H, Ince BA,
Pakdel F, Reese JC, Wooge CH and Wrenn CK: William L. McGuire
Memorial Symposium. Estrogen receptors: Ligand discrimination and
antiestrogen action. Breast Cancer Res Treat. 27:17–26. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katzenellenbogen BS, Montano MM, Le Goff
P, Schodin DJ, Kraus WL, Bhardwaj B and Fujimoto N: Antiestrogens:
Mechanisms and actions in target cells. J Steroid Biochem Mol Biol.
53:387–393. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amin S, Kumar A, Nilchi L, Wright K and
Kozlowski M: Breast cancer cells proliferation is regulated by
tyrosine phosphatase SHP1 through c-jun N-terminal kinase and
cooperative induction of RFX-1 and AP-4 transcription factors. Mol
Cancer Res. 9:1112–1125. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mandlekar S and Kong AN: Mechanisms of
tamoxifen-induced apoptosis. Apoptosis. 6:469–477. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mamay CL, Mingo-Sion AM, Wolf DM, Molina
MD and Van Den Berg CL: An inhibitory function for JNK in the
regulation of IGF-I signaling in breast cancer. Oncogene.
22:602–614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montano MM and Katzenellenbogen BS: The
quinone reductase gene: A unique estrogen receptor-regulated gene
that is activated by antiestrogens. Proc Natl Acad Sci USA.
94:2581–2586. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nazarewicz RR, Zenebe WJ, Parihar A,
Larson SK, Alidema E, Choi J and Ghafourifar P: Tamoxifen induces
oxidative stress and mitochondrial apoptosis via stimulating
mitochondrial nitric oxide synthase. Cancer Res. 67:1282–1290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Umekita Y, Ohi Y, Sagara Y and Yoshida H:
Overexpression of cyclinD1 predicts for poor prognosis in estrogen
receptor-negative breast cancer patients. Int J Cancer. 98:415–418.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Altucci L, Addeo R, Cicatiello L, Dauvois
S, Parker MG, Truss M, Beato M, Sica V, Bresciani F and Weisz A:
17beta-Estradiol induces cyclin D1 gene transcription,
p36D1-p34cdk4 complex activation and p105Rb phosphorylation during
mitogenic stimulation of G(1)-arrested human breast cancer cells.
Oncogene. 12:2315–2324. 1996.PubMed/NCBI
|
|
24
|
Fu XD, Cui YH, Lin GP and Wang TH:
Non-genomic effects of 17beta-estradiol in activation of the
ERK1/ERK2 pathway induces cell proliferation through upregulation
of cyclin D1 expression in bovine artery endothelial cells. Gynecol
Endocrinol. 23:131–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pawar P, Ma L, Byon CH, Liu H, Ahn EY,
Jhala N, Arnoletti JP, McDonald JM and Chen Y: Molecular mechanisms
of tamoxifen therapy for cholangiocarcinoma: Role of calmodulin.
Clin Cancer Res. 15:1288–1296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carthy JM, Sundqvist A, Heldin A, van Dam
H, Kletsas D, Heldin CH and Moustakas A: Tamoxifen inhibits
TGF-β-mediated activation of myofibroblasts by blocking non-smad
signaling through ERK1/2. J Cell Physiol. 230:3084–3092. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shay JW and Werbin H: Are mitochondrial
DNA mutations involved in the carcinogenic process? Mutat Res.
186:149–160. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Modica-Napolitano JS and Singh KK:
Mitochondrial dysfunction in cancer. Mitochondrion. 4:755–762.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boonstra J and Post JA: Molecular events
associated with reactive oxygen species and cell cycle progression
in mammalian cells. Gene. 337:1–13. 2004. View Article : Google Scholar : PubMed/NCBI
|