Introduction
Mitochondria are cytoplasmic organelles that are
responsible for most of the energy supply of cells. Furthermore,
they are important in regulating various cellular processes,
including calcium homeostasis, signaling pathways, apoptosis and
the production of reactive oxygen species (ROS) (1). Within cells, mitochondria are highly
dynamic organelles, transitioning from a giant tubular network to
small round organelles through reversible fission and fusion
(2). These opposing processes work
in concert to maintain the shape, size and number of mitochondria,
as well as their physiological function. Alterations in
mitochondrial dynamics have an effect on the majority of aspects of
mitochondrial function, including the stability of mitochondrial
DNA, respiratory capacity, apoptosis, responses to cellular stress
and mitophagy (3–7). Mitochondrial dynamics are regulated
by mitochondrial dynamics-related GTPase proteins. Dynamin 1 like
(Dnm1l, also known as Drp1) is one of the GTPase proteins that
promotes mitochondrial fragmentation, whereas the expression of a
dominant-negative form of Drp1 inhibits mitochondrial fission and
thereby apoptosis (8). Other
studies have demonstrated that the inhibition of mitochondrial
fission protects against neurotoxicity (9,10)
and neurodegeneration in the cerebellum (11). In addition, numerous reports have
indicated that excessive mitochondrial fission contributes to
Alzheimer's disease (AD) pathogenesis via synaptic damage and
neuronal death (12,13). Therefore, the protection of
mitochondria from fragmentation may provide a novel therapeutic
target for neurodegenerative disorders, such as AD (14,15).
Hydrogen sulfide (H2S) has been
identified as a biological gaseous transmitter alongside nitric
oxide and carbon monoxide (16).
In mammals, H2S is endogenously produced by
cystathionine-β-synthase, cystathionine-γ-lyase and
3-mercaptopyruvate sulfurtransferase, with L-cysteine as the
dominant substrate (17,18). H2S is recognized as a
biological mediator that serves various functions, including
neuromodulation, the regulation of vascular tone, cytoprotection,
anti-inflammation, oxygen sensing and angiogenesis, in addition to
mitochondrial functions (19–22).
A number of recent studies focused on the effects of H2S
on mitochondrial function. H2S has been demonstrated to
suppress mitochondrial ROS production, increase ATP production and
improve the loss of mitochondrial membrane potential under stress
conditions (23). However, since
mitochondrial dynamics are important for essential mitochondrial
functions, and since mitochondrial fission is abnormally increased
in affected neurons in neurodegenerative diseases, inhibitors of
mitochondrial fission may be promising therapeutic targets for the
treatment of neurodegenerative diseases (24). Therefore, the present study focused
on the effects of H2S on mitochondrial fission and
fusion and the potential underlying mechanisms.
Materials and methods
Materials
Sodium hydrosulfide (NaHS), an H2S donor,
was obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany)
and dissolved in sterile water. PD98059, an extracellular
signal-regulated kinase (ERK) 1/2 signal pathway inhibitor, was
purchased from Abcam (Cambridge, UK) and dissolved in sterile
water.
Cell culture and lentiviral
transduction
Neuro-2a (N2a) mouse neuroblastoma cells were
purchased from the Cell Bank of Type Culture Collection of Chinese
Academy of Sciences (Shanghai, China). The cells were cultured in
Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% heat-inactivated
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin and streptomycin, and were maintained at 37°C with 95%
humidified air and 5% CO2. The cells (2×105)
were plated in 35 mm dishes and incubated for 48 h. On the third
day, the medium was removed and the cells were washed with PBS.
Subsequently, the cells were exposed to serum-free culture medium
for 24 h. The experimental treatments were also performed in
serum-free culture medium. Ready-to-use lentiviral particles for a
Drp1-overexpressing lentiviral vector (LV-Drp1) and a control
vector (LV-empty) were purchased from GeneChem Co., Ltd. (Shanghai,
China; cat. nos. V20140402006 and V20140402007 respectively). The
LV-Drp1 and the LV-empty virus-containing media were added to the
N2a cells with a multiplicity of infection (MOI) of 5.
Non-transduced cells were used as controls. To calculate the amount
of virus required for a certain MOI, the following formula was
used: Total ml of virus required=number of cells xdesired
MOI/(plaque forming units/ml). All of the media was changed 12 h
post-transduction. Three days after transduction, western blot
analysis was performed to evaluate transduction efficiency.
Transmission electron microscopy
To determine whether mitochondrial morphology was
altered in the N2a cells, transmission electron microscopy was
performed. The N2a cells were prefixed in a 2.5% glutaraldehyde
solution overnight at 4°C and post-fixed in cold 1% aqueous osmium
tetroxide for 1 h at 4°C. The samples were rinsed three times with
PBS, dehydrated in a graded series of 25–100% ethanol, embedded in
fresh resin and polymerized at 60°C for 24 h. The samples were
sectioned on a Leica EM UC6 ultramicrotome at 60–80 nm and
collected on pioloform-coated Cu2*1 oval slot grids (Electron
Microscopy Sciences, Hatfield, PA, USA). The sections were
subsequently examined under a Hitachi-7500 transmission electron
microscope (Hitachi, Ltd., Tokyo, Japan). The number of
mitochondria per cell was counted in ≥10 cells in each group. The
proportion of ‘elongated’ mitochondria was estimated as the number
of mitochondria with a length that was at least twice the width,
divided by the total number of mitochondria counted (25).
Determination of cell viability
Cell viability was assessed by the reduction of MTT.
Briefly, 20 µl MTT (5 mg/ml; Sigma-Aldrich; Merck KGaA) was added
to each well and the cells were incubated at 37°C for 1 h. The
cells were washed with PBS and the formazan dye was dissolved in
dimethyl sulfoxide. The amount of converted formazan dye was
measured at 570 nm on a PowerWave reader (Biotek Instruments, Inc.,
Winooski, VT, USA). This method detects complex II-dependent
mitochondrial activity and is often used to estimate mitochondrial
function and/or cell viability.
Measurement of ATP levels
ATP levels were determined using an ATP
Bioluminescence Assay kit CLS II (Sigma-Aldrich; Merck KGaA)
following the manufacturer's instructions (26). Briefly, cells were harvested using
the provided lysis buffer, incubated on ice for 15 min and
centrifuged at 13,000 × g for 10 min at 4°C. ATP levels were
measured using a luminescence plate reader (Molecular Devices, LLC,
Sunnyvale, CA, USA) with an integration time of 10 sec.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cells using TRIzol
reagent (Takara Biotechnology Co., Ltd., Dalian, China) according
to the manufacturer's protocol. After evaluating the concentration
of total RNA by a UV spectrophotometer, total RNA was
reverse-transcribed using the PrimeScript RT reagent kit (Takara
Biotechnology Co., Ltd.). qPCR was performed using SYBR Premix Ex
Taq II (Takara Biotechnology Co., Ltd.) and a CFX96 Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
mRNA expression was normalized to β-actin and relative expression
was calculated using the 2(−Delta Delta C(q)) method (27). The primer sequences (Sangon Biotech
Co., Ltd., Shanghai, China) were as follows: Drp1, forward
5′-GGCATTACAAGGAGCCAGTC and reverse 5′-CAGCAGGTTCAAGTCAGCAA;
β-actin, forward 5′-CACCCGCGAGTACAACCTTC and reverse
5′-CCCATACCCACCATCACACC. Each experiment was performed in
triplicate.
Western blot assay
Cells were harvested by scraping and centrifugation
at 220 × g at room temperature for 5 min, washed twice with
ice-cold PBS and re-suspended in lysis buffer (50 mM Tris, 150 mM
NaCl, 1% NP-40, 0.1% SDS, 10 mM EDTA, 1 mM PMSF and 0.5% sodium
deoxycholate), according to the manufacturer's protocol (KeyGEN
Biotech Co., Ltd., Nanjing, China). Soluble proteins were collected
by centrifugation at 12,000 × g for 15 min at 4°C. Protein
concentrations were determined using the bicinchoninic acid method.
Protein (30 µg) was separated by 10% SDS-PAGE and subsequently
transferred onto polyvinylidene difluoride membranes. After the
membranes were blocked with 5% skim milk for 1 h at room
temperature, they were incubated with antibodies against Drp1
(1:1,000; cat. no. ab56788; Abcam, Cambridge),
phosphorylated-(p)-ERK1/2 (1:1,000, cat. no. sc-81492; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), total (t)-ERK1/2 (1:1,000,
cat. no. sc-514302; Santa Cruz Biotechnology, Inc.) and β-actin
(1:1,000, cat. no. sc-130301; Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Subsequently, the membranes were incubated with a
secondary horseradish peroxidase-conjugated antibody (1:2,000; cat.
no. A0216; Beyotime Instistute of Biotechnology, Shanghai, China)
for 2 h at room temperature and the SuperSignal West Pico
Chemiluminescent Substrate (Thermo Fisher Scientific, Inc.) was
used for detection. Densitometric quantification of blots was
performed using Alphapart11 Ease version 5.0 (ProteinSimple;
Bio-Techne, Minneapolis, MS, USA) and expression levels were
calculated as a ratio relative to the control level.
Statistical analysis
All data are expressed as the mean + standard error
of the mean. Differences between groups were analyzed by ANOVA
followed by the Student-Newman-Keuls test. All statistical analyses
were performed using SPSS version 17.0 (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
The effect of NaHS treatment on
mitochondrial fission
The present study used transmission electron
microscopy to determine changes in the number and morphology of
mitochondria in the cell bodies of N2a cells. Control untreated and
NaHS-treated N2a cells were collected for transmission electron
microscopy imaging (Fig. 1). In
the present study, NaHS was used as a donor for H2S,
since it readily releases H2S upon its suspension in
water. Initially, the cells were treated with different
concentrations of NaHS for 16 h (Fig.
1A-C). A dose-dependent increase in the % of elongated
mitochondria was observed following NaHS treatment compared with
control (Fig. 1B; control,
38.75±2.39%; NaHS 100 µM, 48.50±3.01%; NaHS 200 µM, 61.75±1.97%;
NaHS 400 µM, 68.75±2.39%). By contrast, the number of mitochondria
per cell was decreased in a dose-dependent manner following NaHS
treatment compared with control (Fig.
1C; control, 39.25±3.25; NaHS 100 µM, 32.75±1.10, NaHS 200 µM,
25.5±1.32; NaHS 400 µM, 21.25±1.31). Next, cells were treated with
400 µM NaHS for different durations (Fig. 1D-F). Treatment with 400 µM NaHS for
8 and 16 h resulted in an increased % of elongated mitochondria
(Fig. 1E; control, 42.50±2.15%; 8
h, 52.75±2.35%; 16 h, 63.75±1.75%) and a reduced number of
mitochondria per cell (Fig. 1F;
control, 32.75±1.25; 8 h, 22.50±1.89; 16 h, 18.75±1.25), compared
with control. The differences between the control group and the 400
µM NaHS groups at 8 and 16 h were statistically significant
(P<0.01). These results indicate that NaHS treatment may inhibit
mitochondrial fission in a dose- and time-dependent manner.
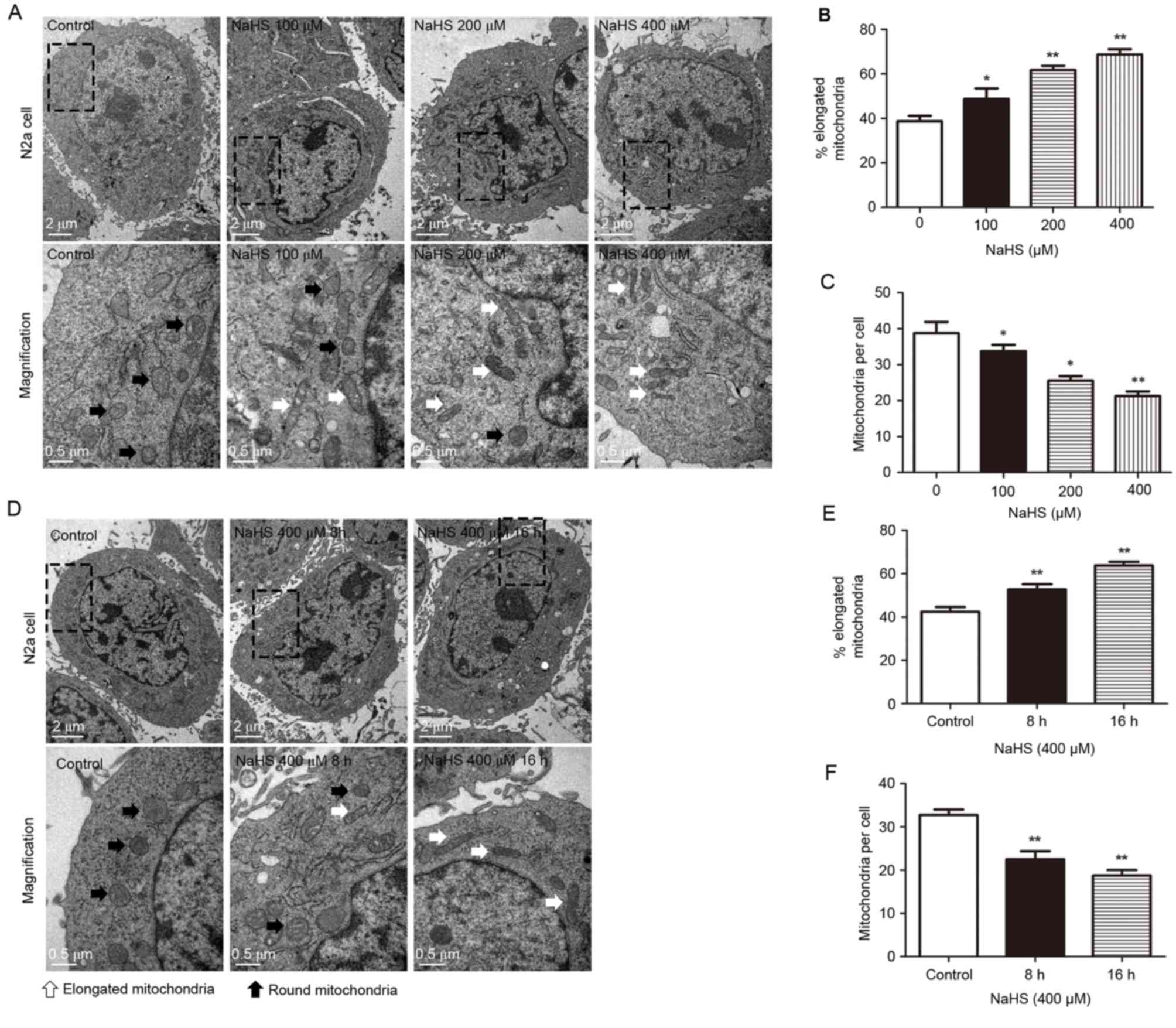 | Figure 1.Transmission electron microscopy
analysis of mitochondria within the cell body demonstrates an
increased number of elongated mitochondria in the NaHS-treated
cells. (A) Representative images of the cell bodies are presented
for N2a cells treated with 0, 100, 200 and 400 µM NaHS for 16 h.
Images on the bottom row are magnified images of the boxed areas in
the top row. (B) Percentage of elongated mitochondria and (C)
number of mitochondria per cell were calculated for N2a cells
treated with 0, 100, 200 and 400 µM NaHS. (D) Representative images
of the cell bodies are presented for N2a cells treated with 400 µM
NaHS for 8 and 16 h, and control cells. Images on the bottom row
are magnified images of the boxed areas in the top row. (E)
Percentage of elongated mitochondria and (F) number of mitochondria
per cell were calculated for N2a cells treated with 400 µM NaHS for
8 and 16 h, and control cells. White arrows, elongated
mitochondria; black arrows, round mitochondria. Data are presented
as the mean + standard error of the mean of three independent
experiments (n≥10 images per group). *P<0.05 and **P<0.01 vs.
control. NaHS, sodium hydrosulfide; N2a, neuro-2a. |
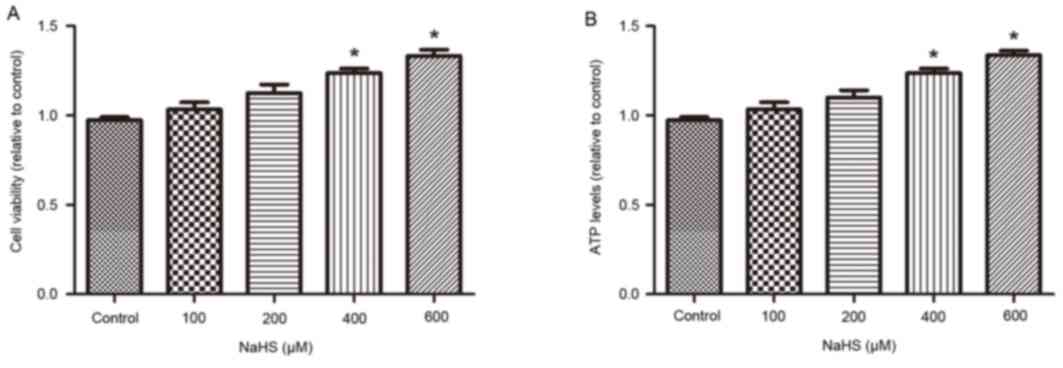
The effect of NaHS treatment on cell
viability and mitochondrial ATP synthesis
The MTT assay was used to evaluate the effect of
H2S on cell viability. As demonstrated in Fig. 2A, NaHS treatment for 16 h increased
cell viability in a dose-dependent manner (control, 0.97±0.01; NaHS
100 µM, 1.03±0.04; NaHS 200 µM, 1.13±0.04; NaHS 400 µM, 1.24±0.02;
NaHS 600 µM, 1.33±0.03) compared with control. In addition, the
rate of ATP production was gradually increased following NaHS
treatment for 16 h, compared with control (Fig. 2B; control, 0.97±0.02; NaHS 100 µM,
1.03±0.04; NaHS 200 µM, 1.10±0.04; NaHS 400 µM, 1.24±0.02; NaHS 600
µM, 1.34±0.02). These results indicated that H2S
increased cell viability and mitochondrial function in a
dose-dependent manner.
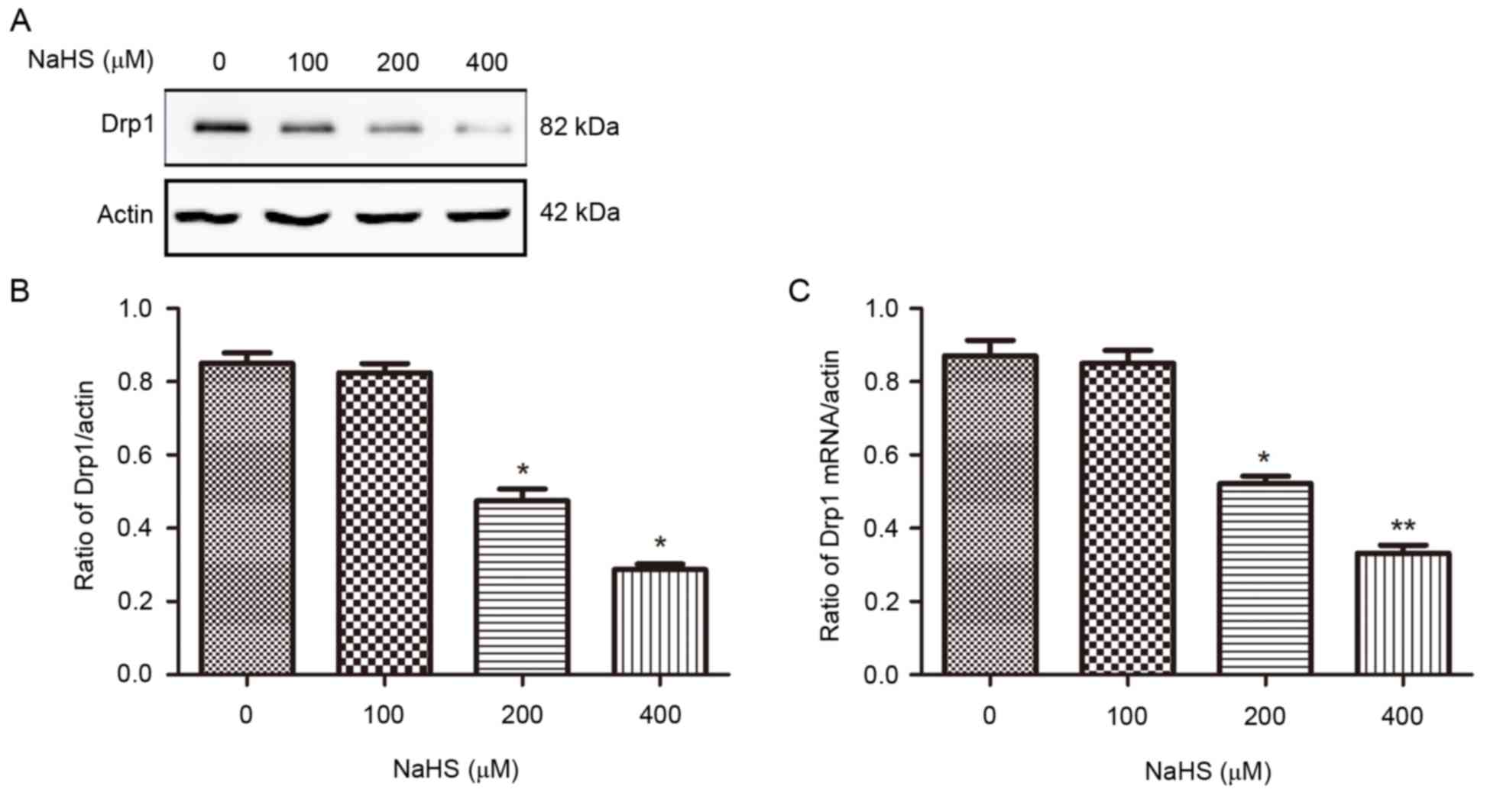
NaHS reduces the mRNA and protein
expression of Drp1
To further understand the effects of H2S
on mitochondrial morphology changes, the protein and mRNA
expression levels of Drp1, an important regulator of mitochondrial
fission, were examined. Using RT-qPCR and western blotting, the
results demonstrated that treatment with NaHS (200 and 400 µM) for
16 h significantly decreased Drp1 mRNA and protein expression
levels compared with control (Fig.
3), consistent with the reduction of mitochondrial fission
observed in NaHS-treated N2a cells (Fig. 1). These results indicate that
H2S may inhibit mitochondrial fission by inhibiting Drp1
mRNA and protein expression.
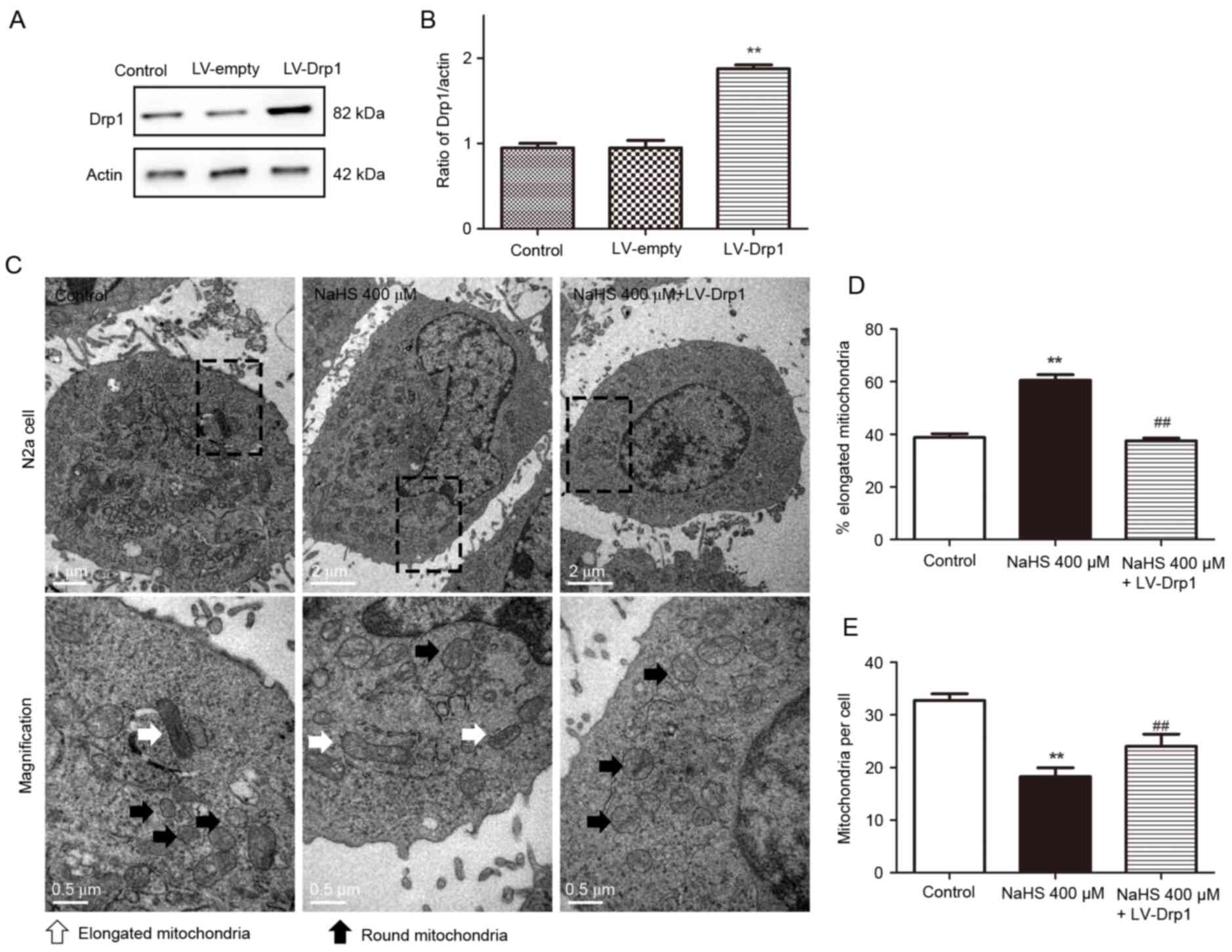
Overexpression of Drp1 reverses the
mitochondrial morphology changes induced by H2S
To determine whether H2S affects
mitochondrial morphology by affecting Drp1 expression, Drp1 was
overexpressed in N2a cells using a lentivirus encoding Drp1 cDNA
(LV-Drp1). Western blotting confirmed that the cells in the LV-Drp1
group stably overexpressed Drp1 compared with the control group and
the empty vector group (LV-empty; Fig.
4A and B). Transmission electron microscopy demonstrated that
NaHS treatment for 16 h inhibited mitochondrial fission and that
this inhibition was reversed by NaHS and LV-Drp1 co-treatment
(Fig. 4C). In the NaHS-treated
group, mitochondrial fission was significantly inhibited compared
with the control group; the % of elongated mitochondria was
increased (control, 38.75±1.49%; NaHS 400 µM, 60.50±2.10%), and the
number of mitochondria per cells was decreased (control,
32.75±1.25; NaHS 400 µM, 18.25±1.70). However, this effect was
reversed by LV-Drp1 trasduction. Compared with the NaHS-treated
group, the NaHS and LV-Drp1 co-treated group displayed
significantly fewer elongated mitochondria (NaHS 400 µM,
60.50±2.10%; NaHS 400 µM + LV-Drp1, 37.50±1.04%) and higher number
of mitochondria per cell (NaHS 400 µM, 18.25±1.70; NaHS 400 µM +
LV-Drp1, 24.00±2.34). These results provide further evidence that
H2S may inhibit mitochondrial fission by inhibiting Drp1
expression.
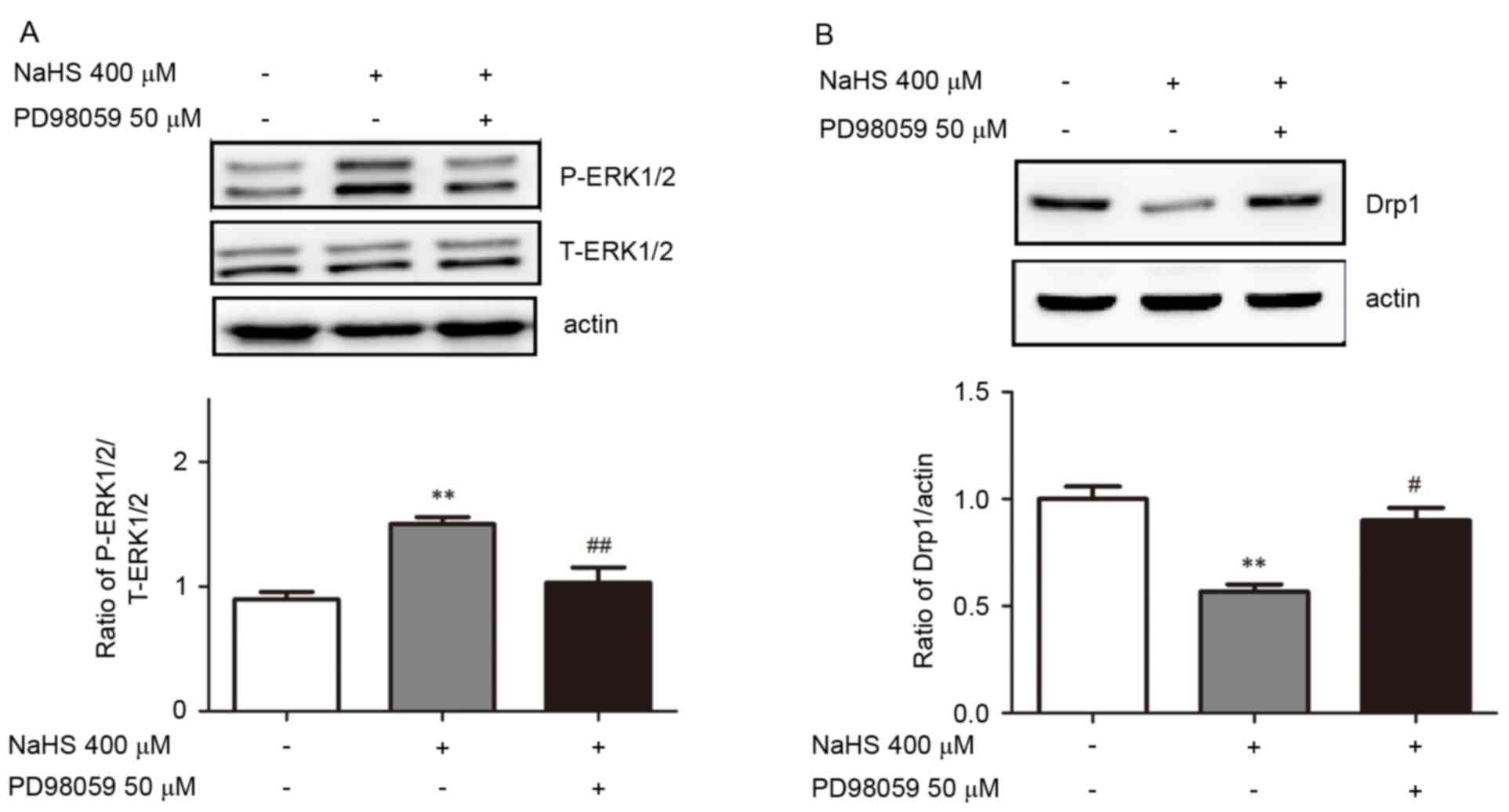
The ERK1/2 signaling pathway is
involved in the NaHS-induced decrease in Drp1 expression
To further understand the molecular mechanisms of
the H2S-induced reduction of mitochondrial fission and
Drp1 expression, lysates were obtained from cells that were treated
with 400 µM NaHS for 16 h and subjected to western blot analysis
for p-ERK1/2 and t-ERK1/2. Treatment with 400 µM NaHS significantly
increased the phosphorylation of ERK1/2 compared with untreated
cells (Fig. 5A). The cells were
then preincubated with PD98059 (50 µM) for 30 min before they were
treated with 400 µM NaHS for 16 h. As presented in Fig. 5A, the NaHS-induced increase in
ERK1/2 phosphorylation was reversed by PD98059 pretreatment. The
effect of PD98059 on the NaHS-induced changes in Drp1 protein
expression were similar to its effect on ERK1/2 phosphorylation.
Pretreatment with PD98059 (50 µM) reversed the NaHS-mediated Drp1
downregulation by ~45% compared with NaHS treatment alone (Fig. 5B). These findings indicate that the
ERK1/2 signaling pathway may be involved in the NaHS-induced
decrease in mitochondrial fission and Drp1 expression.
Discussion
H2S has been identified as the third
gasotransmitter, and various functions of H2S have been
demonstrated. H2S facilitates the induction of
hippocampal long-term potentiation (28), modulates inflammation by
suppressing leukocyte adherence and infiltration and edema
formation (29), and suppresses
the release of insulin by stimulating ATP-sensitive K+
channels (30). Progress has also
been made towards understanding the effects of H2S on
mitochondrial function. H2S stimulates mitochondrial
respiration, suppresses mitochondrial ROS production and increases
ATP production (20). However,
there have been no reports regarding the effects of H2S
on mitochondrial dynamics, which provide the foundation for the
maintenance of normal mitochondrial functioning.
To the best of our knowledge, the present study is
the first to demonstrate that H2S inhibits mitochondrial
fission. The current study used transmission electron microscopy,
which is the most accurate method of detecting changes in
mitochondrial morphology. To ensure the validity of the results,
previously described methods were used to analyze the number of
mitochondria as well as changes in their shape (25,31).
The results confirmed that H2S inhibited mitochondrial
fission in a dose- and time-dependent manner. In addition, the
present study evaluated the effect of NaHS on cell viability and
ATP generation and demonstrated that 400–600 µM NaHS increased cell
viability and ATP generation, indicating that NaHS may enhance
mitochondrial function. This finding was consistent with previous
studies (32,33). Subsequently, it was investigated
whether H2S may also affect the expression of Drp1,
thereby affecting mitochondrial dynamics. To test this hypothesis,
RT-qPCR and western blotting were performed to detect the mRNA and
protein levels of Drp1 in N2a cells following treatment with 400 µM
NaHS. The results demonstrated that H2S significantly
affected the mRNA and protein expression levels of Drp1. Drp1 mRNA
and protein expression in the cells in the 400 µM NaHS-treated
group were 60% lower compared with control levels. Based on this
result, we hypothesized that the impact of H2S on
mitochondrial morphology changes may be achieved through reduced
Drp1 expression. In a previous study, Barsoum et al
(34) observed that Drp1
overexpression leads to mitochondrial fragmentation and that
dominant-negative Drp1 inhibits mitochondrial fission in primary
neurons. The current study also demonstrated that Drp1
overexpression reversed the NaHS-induced inhibition of
mitochondrial fission. This finding further supports our hypothesis
that H2S affects mitochondrial fission by reducing Drp1
expression.
To further elucidate the molecular mechanisms of the
effects of H2S on Drp1 expression and mitochondrial
fission, the ERK1/2 pathway was analyzed. In a previous study,
Zhang et al (35)
demonstrated that H2S increases ERK1/2 phosphorylation,
and downregulates β-site amyloid precursor protein cleaving enzyme
1 expression and amyloid β1–42 release in PC12 cells. Gan et
al (36) also reported that
inhibiting ERK1/2 activation attenuates aberrant mitochondrial
morphology and function in AD hybrid cells. The results of these
studies indicate that there may be a specific association between
H2S, ERK1/2 activation and changes in mitochondrial
morphology. To investigate this, the present study measured ERK1/2
phosphorylation in N2a cells following treatment with NaHS and the
ERK1/2 signaling pathway inhibitor PD98059. The results
demonstrated that H2S increased ERK1/2 phosphorylation
and that PD98059 blocked this effect, indicating that
H2S does affect ERK1/2 activation. In addition, the
present study investigated whether PD98059 affects
H2S-induced mitochondrial fission and observed that
PD98059 partially suppressed the effect of H2S on
mitochondrial fission. These results indicate that the ERK1/2
signaling pathway may be crucial for the H2S-induced
decrease in mitochondrial fission. The effects of inhibitor
pretreatment on Drp1 protein levels were similar, supporting our
hypothesis that H2S hindered mitochondrial fission by
reducing Drp1 expression. However, the effects of H2S on
other signaling pathways, including the phosphoinositide
3-kinase/AKT serine-threonine kinase and c-Jun N-terminal kinase
pathways, require further investigation.
In conclusion, the results of the present study
support the hypothesis that H2S may downregulate Drp1
expression and inhibit mitochondrial fission, and that these
effects may involve the ERK1/2 signaling pathway. These results lay
a foundation for an improved understanding of the effects of the
novel gaseous signaling molecule H2S on mitochondrial
function. However, further studies in vivo are required to
confirm this hypothesis.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81271222). The authors thank
the Laboratory Research Center of the First Affiliated Hospital of
Chongqing Medical University (Chongqing, China) for providing
equipment support.
References
|
1
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Westermann B: Mitochondrial fusion and
fission in cell life and death. Nat Rev Mol Cell Biol. 11:872–884.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen H, Chomyn A and Chan DC: Disruption
of fusion results in mitochondrial heterogeneity and dysfunction. J
Biol Chem. 280:26185–26192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Vermulst M, Wang YE, Chomyn A,
Prolla TA, McCaffery JM and Chan DC: Mitochondrial fusion is
required for mtDNA stability in skeletal muscle and tolerance of
mtDNA mutations. Cell. 141:280–289. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gomes LC, Di Benedetto G and Scorrano L:
During autophagy mitochondria elongate, are spared from degradation
and sustain cell viability. Nat Cell Biol. 13:589–598. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karbowski M: Mitochondria on guard: Role
of mitochondrial fusion and fission in the regulation of apoptosis.
Adv Exp Med Biol. 687:131–142. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi X, Qvit N, Su YC and Mochly-Rosen D: A
novel Drp1 inhibitor diminishes aberrant mitochondrial fission and
neurotoxicity. J Cell Sci. 126:789–802. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie N, Wang C, Lian Y, Wu C, Zhang H and
Zhang Q: Inhibition of mitochondrial fission attenuates Aβ-induced
microglia apoptosis. Neuroscience. 256:36–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen H, McCaffery JM and Chan DC:
Mitochondrial fusion protects against neurodegeneration in the
Cerebellum. Cell. 130:548–562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baloyannis SJ: Mitochondrial alterations
in Alzheimer's disease. J Alzheimers Dis. 9:119–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bossy-Wetzel E, Barsoum MJ, Godzik A,
Schwarzenbacher R and Lipton SA: Mitochondrial fission in
apoptosis, neurodegeneration and aging. Curr Opin Cell Biol.
15:706–716. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonda DJ, Wang X, Perry G, Smith MA and
Zhu X: Mitochondrial dynamics in Alzheimer's disease: Opportunities
for future treatment strategies. Drugs Aging. 27:181–192. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Su B, Wang X, Bonda D, Perry G, Smith M
and Zhu X: Abnormal mitochondrial dynamics-a novel therapeutic
target for Alzheimer's disease? Mol Neurobiol. 41:87–96. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Whiteman M, Le Trionnaire S, Chopra M, Fox
B and Whatmore J: Emerging role of hydrogen sulfide in health and
disease: Critical appraisal of biomarkers and pharmacological
tools. Clin Sci (Lond). 121:459–488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fiorucci S, Distrutti E, Cirino G and
Wallace JL: The emerging roles of hydrogen sulfide in the
gastrointestinal tract and liver. Gastroenterology. 131:259–271.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shibuya N, Tanaka M, Yoshida M, Ogasawara
Y, Togawa T, Ishii K and Kimura H: 3-Mercaptopyr-uvate
sulfurtransferase produces hydrogen sulfide and bound sulfane
sulfur in the brain. Antioxid Redox Signal. 11:703–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kimura Y, Goto Y and Kimura H: Hydrogen
sulfide increases glutathione production and suppresses oxidative
stress in mitochondria. Antioxid Redox Signal. 12:1–13. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whiteman M, Armstrong JS, Chu SH, Jia-Ling
S, Wong BS, Cheung NS, Halliwell B and Moore PK: The novel
neuromodulator hydrogen sulfide: An endogenous peroxynitrite
‘scavenger’? J Neurochem. 90:765–768. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mikami Y, Shibuya N, Kimura Y, Nagahara N,
Yamada M and Kimura H: Hydrogen sulfide protects the retina from
light-induced degeneration by the modulation of Ca2+ influx. J Biol
Chem. 286:39379–39386. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Elrod JW, Calvert JW, Morrison J, Doeller
JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Szabo C, Ransy C, Módis K, Andriamihaja M,
Murghes B, Coletta C, Olah G, Yanagi K and Bouillaud F: Regulation
of mitochondrial bioenergetic function by hydrogen sulfide. Part I.
Biochemical and physiological mechanisms. Br J Pharmacol.
171:2099–2122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reddy PH: Inhibitors of mitochondrial
fission as a therapeutic strategy for diseases with oxidative
stress and mitochondrial dysfunction. J Alzheimers Dis. 40:245–256.
2014.PubMed/NCBI
|
|
25
|
Calkins MJ, Manczak M, Mao P, Shirendeb U
and Reddy PH: Impaired mitochondrial biogenesis, defective axonal
transport of mitochondria, abnormal mitochondrial dynamics and
synaptic degeneration in a mouse model of Alzheimer's disease. Hum
Mol Genet. 20:4515–4529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Du H, Guo L, Yan S, Sosunov AA, McKhann GM
and Yan SS: Early deficits in synaptic mitochondria in an
Alzheimer's disease mouse model. Proc Natl Acad Sci USA.
107:18670–18675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abe K and Kimura H: The possible role of
hydrogen sulfide as an endogenous neuromodulator. J Neurosci.
16:1066–1071. 1996.PubMed/NCBI
|
|
29
|
Zanardo RC, Brancaleone V, Distrutti E,
Fiorucci S, Cirino G and Wallace JL: Hydrogen sulfide is an
endogenous modulator of leukocyte-mediated inflammation. FASEB J.
20:2118–2120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kaneko Y, Kimura Y, Kimura H and Niki I:
L-cysteine inhibits insulin release from the pancreatic beta-cell:
Possible involvement of metabolic production of hydrogen sulfide, a
novel gasotransmitter. Diabetes. 55:1391–1397. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nisoli E, Falcone S, Tonello C, Cozzi V,
Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini
M, et al: Mitochondrial biogenesis by NO yields functionally active
mitochondria in mammals. Proc Natl Acad Sci USA. 101:16507–16512.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen Y, Guo W, Wang Z, Zhang Y, Zhong L
and Zhu Y: Protective effects of hydrogen sulfide in hypoxic human
umbilical vein endothelial cells: A possible mitochondria-dependent
pathway. Int J Mol Sci. 14:13093–13108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wen YD, Wang H, Kho SH, Rinkiko S, Sheng
X, Shen HM and Zhu YZ: Hydrogen sulfide protects HUVECs against
hydrogen peroxide induced mitochondrial dysfunction and oxidative
stress. PLoS One. 8:e531472013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Barsoum MJ, Yuan H, Gerencser AA, Liot G,
Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, et al:
Nitric oxide-induced mitochondrial fission is regulated by
dynamin-related GTPases in neurons. EMBO J. 25:3900–3911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang H, Gao Y, Zhao F, Dai Z, Meng T, Tu
S and Yan Y: Hydrogen sulfide reduces mRNA and protein levels of
β-site amyloid precursor protein cleaving enzyme 1 in PC12 cells.
Neurochem Int. 58:169–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gan X, Huang S, Wu L, Wang Y, Hu G, Li G,
Zhang H, Yu H, Swerdlow RH, Chen JX and Yan SS: Inhibition of
ERK-DLP1 signaling and mitochondrial division alleviates
mitochondrial dysfunction in Alzheimer's disease cybrid cell.
Biochim Biophys Acta. 1842:220–231. 2014. View Article : Google Scholar : PubMed/NCBI
|