Introduction
Congenital coloboma is a very rare birth defect with
a prevalence of 0.5–0.7/10,000 live births (1). Congenital macular coloboma is
characterized by well-circumscribed, punched out atrophic lesions
in the macula (2–4). Macular coloboma should be
differentiate from other diseases, such as Best vitelliform macular
dystrophy (BVMD), advanced cone-rod dystrophy (CORD), congenital
toxoplasmosis macular scar, Leber's congenital amaurosis, and
central areolar choroidal dystrophy (5,6). It
is usually sporadic, although autosomal dominant or other
inheritance patterns may be followed. It is thought to be caused by
the failure of normal closure of the optic fissure between 5 and 7
weeks of development (1). Macular
coloboma can be classified into three types, namely pigmented
macular coloboma, non-pigmented macular coloboma, and macular
coloboma associated with abnormal vessels (7).
The genetic changes responsible for the pathogenesis
of congenital macular coloboma are not well studied. Identification
of genetic mutations in congenital macular coloboma is the first
step to unravel the pathogenesis of this disease and will be
helpful for genetic counseling. In this study, we aimed to
characterize the clinical presentation of a 28-year-old female
presented with bilateral large macular coloboma, and to identify
the underlying genetic changes in this patient.
Patients and methods
Study participants
One patient presented with bilateral large atrophy
at the macula in both eyes underwent complete ophthalmic
examinations in Zhongshan Ophthalmic Center. Visual acuity was
examined using the ETDRS chart (Precision Vision, La Salle, IL,
USA). Anterior segment photograph was obtained using a BX 900 Slit
Lamp (Haag-Streit, Bern, Switzerland). Anterior segment
measurements were taken by Pentacam HR version 70700 (Oculus,
Wetzlar, Germany). Fundus photograph were carried out using a
Heidelberg Retina Angiograph (Heidelberg Engineering, Inc.,
Heidelberg, Germany). OCT was carried out by Cirrus HD-OCT (Carl
Zeiss Meditec, Inc., Dublin, CA, USA). Physical examinations were
performed to exclude systemic diseases. Venous blood samples from
this patient, her unaffected family members, and 200 unrelated
control subjects from the same population were collected.
Target capture and next-generation
sequencing
A capture panel of inherited retinal-disease genes
was previously designed and assessed by our group. The capture
panel comprised 708,919 bp that covered all exons together with the
flanking exon and intron boundaries (±15 bp) of 175 genes,
including 138 genes causing common inherited nonsyndromic eye
diseases and 54 genes causing syndromic eye diseases that have been
previously reported and have been accepted by researcheres in this
field.
Genomic DNA from peripheral blood leucocytes was
extracted using the QIAamp DNABlood Midi Kit (Qiagen, Hilden,
Germany). Then the genomic DNA was fragmented by Covaris LE220
(Covaris, Inc., Woburn, MA, USA) to generate paired-end library
(200–250 bp). The library was enriched by array hybridization as
previously described (8), followed
by elution and post-capture amplification. The products were then
subjected to Agilent 2100 Bioanalyzer and ABI StepOne for
estimating the magnitude of enrichment. After quality control,
captured library sequencing was carried out on Illumina HiSeq2500
Analyzers (Illumina, San Diego, CA, USA) for 90 cycles per read to
generate paired-end reads. Image analysis, error estimation, and
base calling were performed using Illumina Pipeline software
(version 1.3.4) to generate raw data.
Data analysis and interpretation of
genetic variants
To detect the potential variants in the family, we
performed bioinformatics processing and data analysis after
receiving the primary sequencing data. We used previously published
filtering criteria to generate ‘clean reads’ for further analysis
(8). The ‘clean reads’ (with a
length of 90 bp) derived from targeted sequencing and filtering
were then aligned to the human genome reference (hg19) using the
BWA (Burrows Wheeler Aligner) Multi-Vision software package
(9). After alignment, the output
files were used to perform sequencing coverage and depth analysis
of the target region, single-nucleotidevariants (SNVs) and INDEL
calling. We used SOAPsnp software (9) and Samtools (10) to detect SNVs and indels. All SNVs
and indels were filtered and estimated via multiple databases,
including NCBI dbSNP, HapMap, 1,000 human genome dataset and a
database of 200 Chinese healthy adults.
To predict the effect of missense variants, SIFT and
PolyPhen were used to predict the possible impact of an amino acid
substitution on the protein structure and function using
straightforward physical and comparative considerations. Variants
were predicted to be pathogenic only when at least one of the two
programs predicted deleterious effect of the amino acid
substitution on the protein structure and function. The Human Gene
Mutation Database (HGMD) was used to screen mutations reported in
published studies.
We also used PolyPhen to check whether the mutations
affected highly conserved amino acid residues.
Mutation validation
The two novel pathogenic mutations were validated
using conventional polymerase chain reaction (PCR) -based
sequencing methods (11–13). Exon 9 of the BEST1 gene and the
Exon 23 of RIMS1 were amplified by PCR with respective primers
(Table I). Briefly, PCR was
conducted in 50 µl reactions. The cycling profile included one
cycle at 94°C for 5 min, followed by 40 cycles at 94°C for 45 sec,
59–60°C for 45 sec, 72°C for 45 sec, and one cycle at 72°C for 10
min. The PCR products were sequenced from both directions with an
ABI3730 Automated Sequencer (PE Biosystems, Foster City, CA, USA).
The sequencing results were analyzed using Seqman (version 2.3;
Technelysium Pty, Ltd., Brisbane, QLD, Australia), and compared
with the reference sequences in the database at the National Center
for Biotechnology Information.
 | Table I.Primers used for the amplification of
the BEST1 and RIMS1 in this study. |
Table I.
Primers used for the amplification of
the BEST1 and RIMS1 in this study.
| Gene | Exon | Forward (5′-3′) | Reverse (5′-3′) | Product size
(bp) | Annealingt emperature
(°C) |
|---|
| BEST1 | 9 |
CAGGGAAACTGAGGTCCAGA |
AGGCTGTCCTTCGAGTAGCA | 539 | 60 |
| RIMS1 | 23 |
GGCGGATTCCAAACATCTTCC |
AGGTGCTTTACCAGAGTTGGC | 487 | 60 |
All experimental protocols were carried out
according to the guidelines approved by the Ethics Committee of
Zhongshan Ophthalmic Center, and in accordance with the Declaration
of Helsinki. Informed consent was obtained from all subjects. The
data generated or analyzed in the current study are included
herein.
Results
Clinical data
The patient studied in this report was from the
southern area of China. Patient is a 28-year-old female without
known familial history of ocular disease. Her best-corrected visual
acuity was 1.3 LogMAR in the right eye, and figure count/40 cm
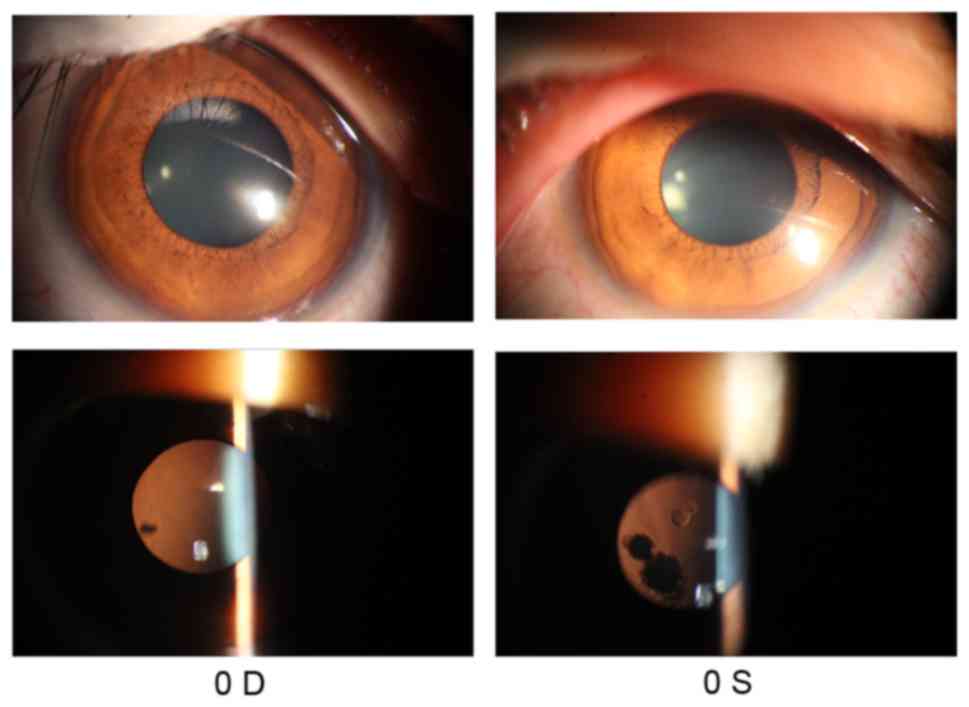
(FC/40 cm) in the left eye. Anterior segment photograph showed some
opacities in the lens of both eyes (Fig. 1). Fundus examination revealed
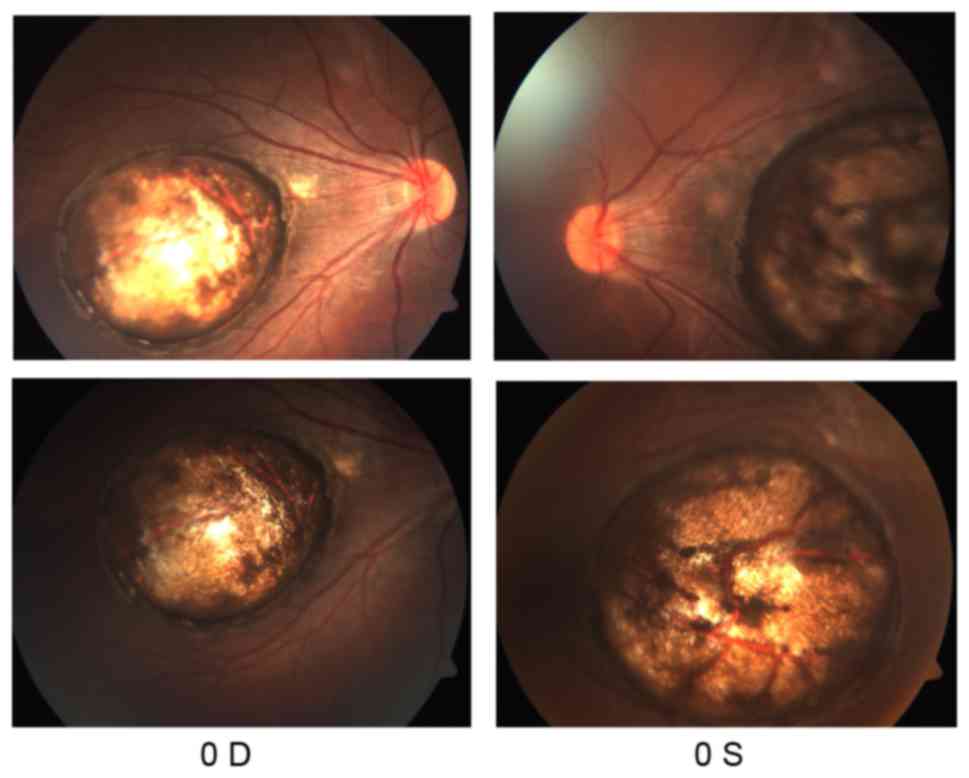
bilateral large atrophy in the macula of each eye with
well-circumscribed borders (Fig.
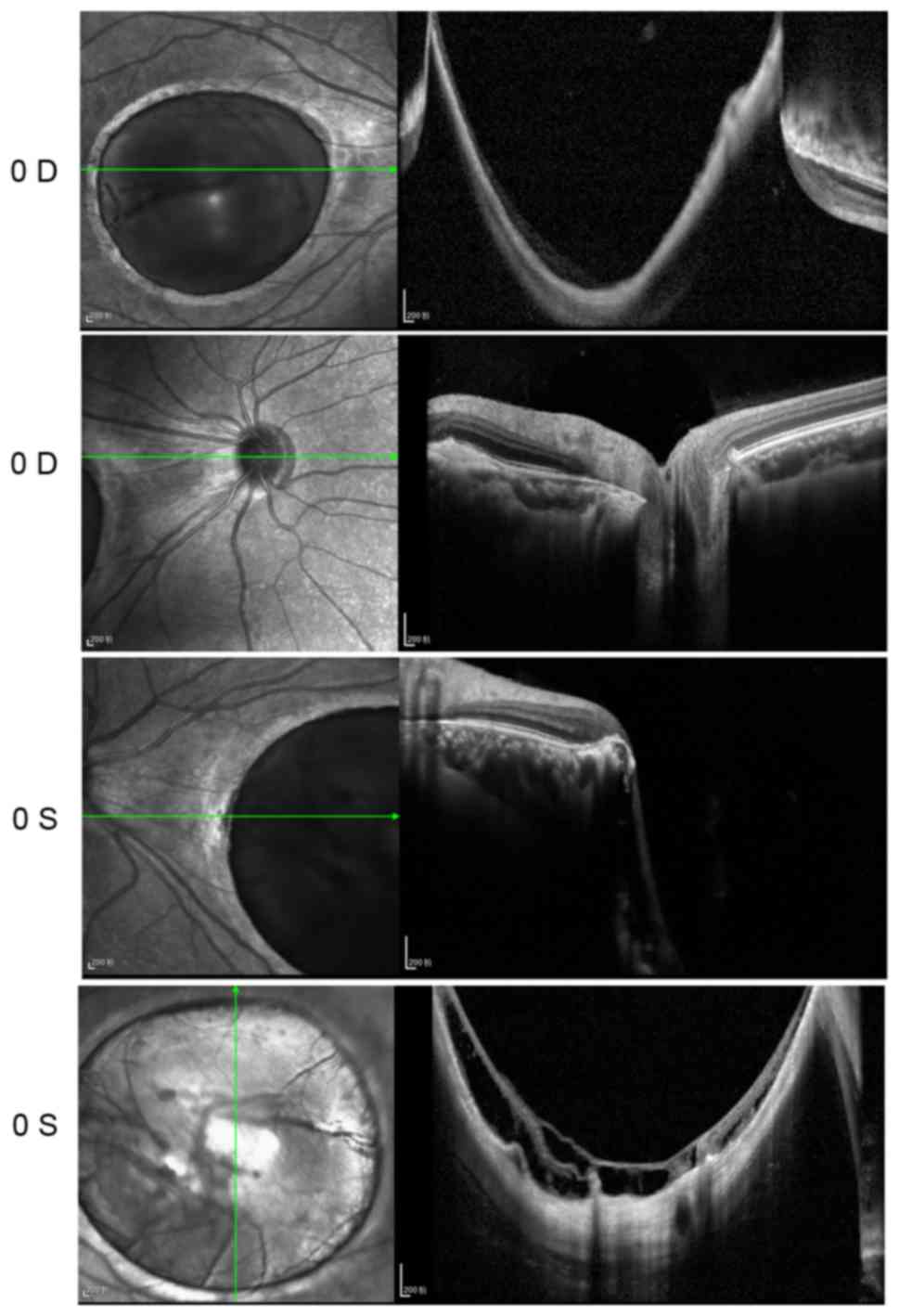
2). OCT showed that the foveal region of both eyes were
abnormally thin. A large cave in the macular area and retinal
schisis were observed in the left eye (Fig. 3).
Mutation screening
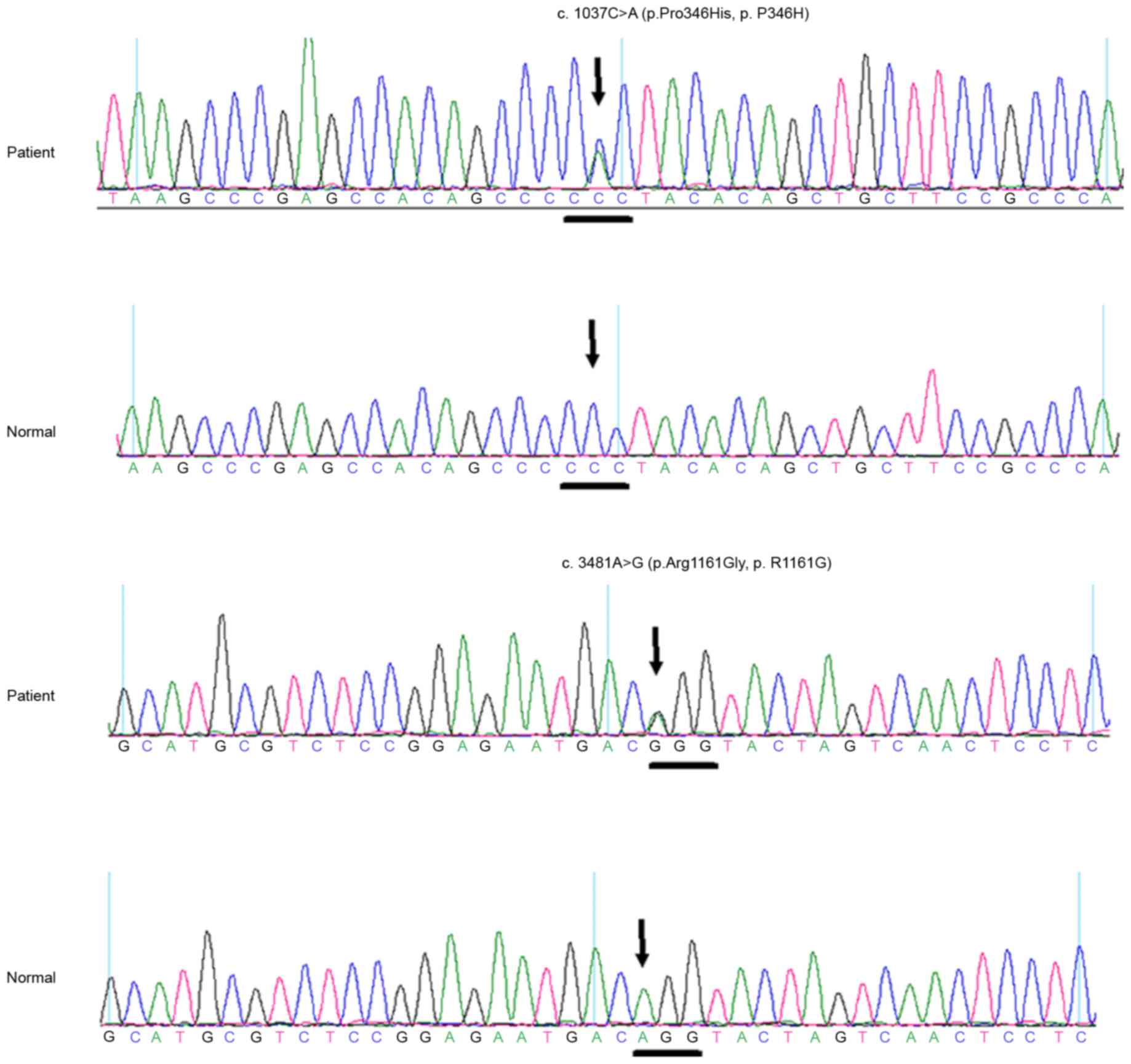
A heterozygous BEST1 mutation c.1037C>A
(p.Pro346His, p.P346H) in exon 9 and a heterozygous RIMS1 mutation
c.3481A>G (p.Arg1161Gly, p.R1161G) in exon 23 were identified in
the affected case, but not in any of the normal controls (Fig. 4). The first mutation we identified
has previously been reported in Japanese patients (14). Since we were unable to obtain
information of the patient's parents, we could not determine the
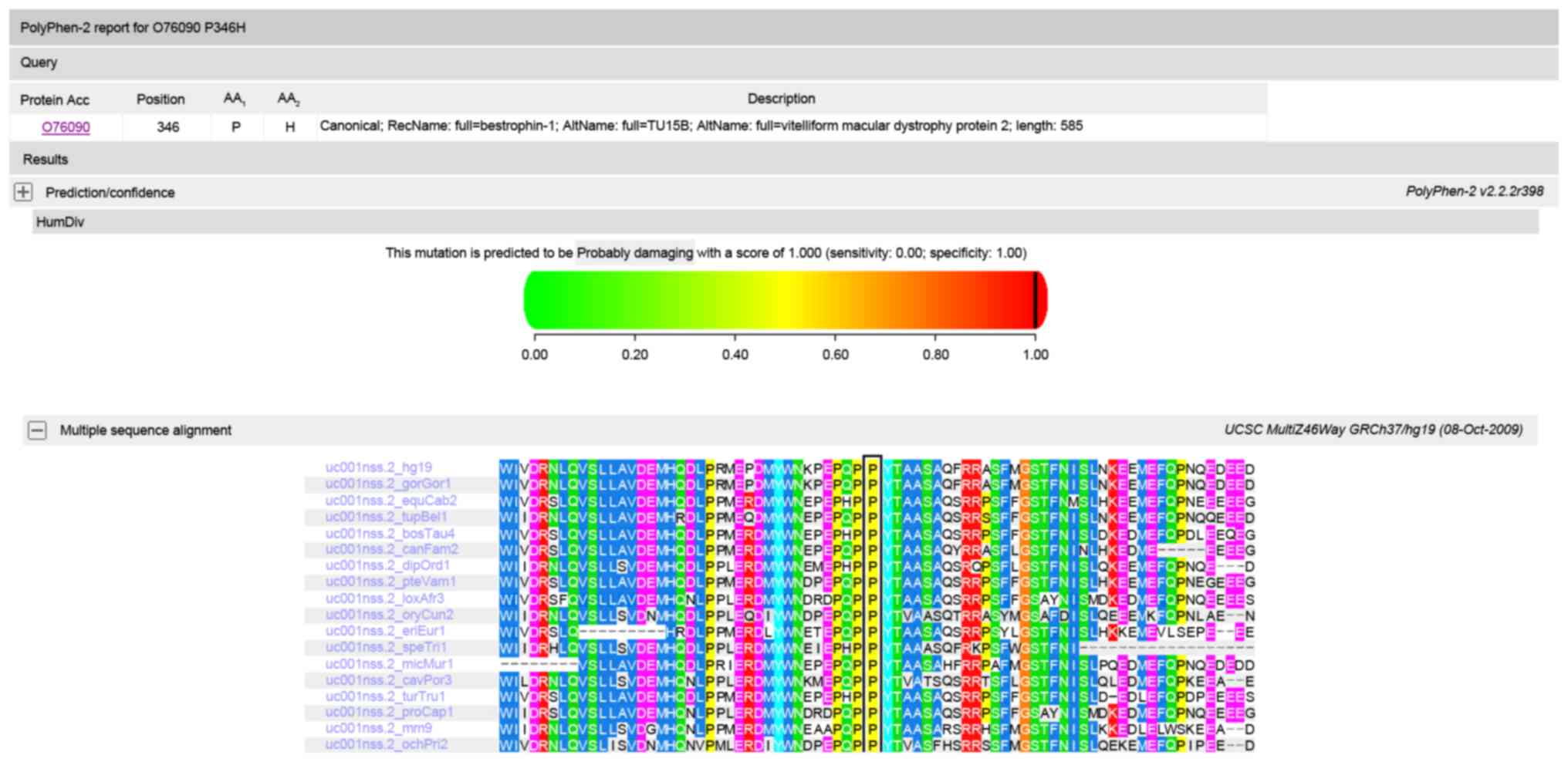
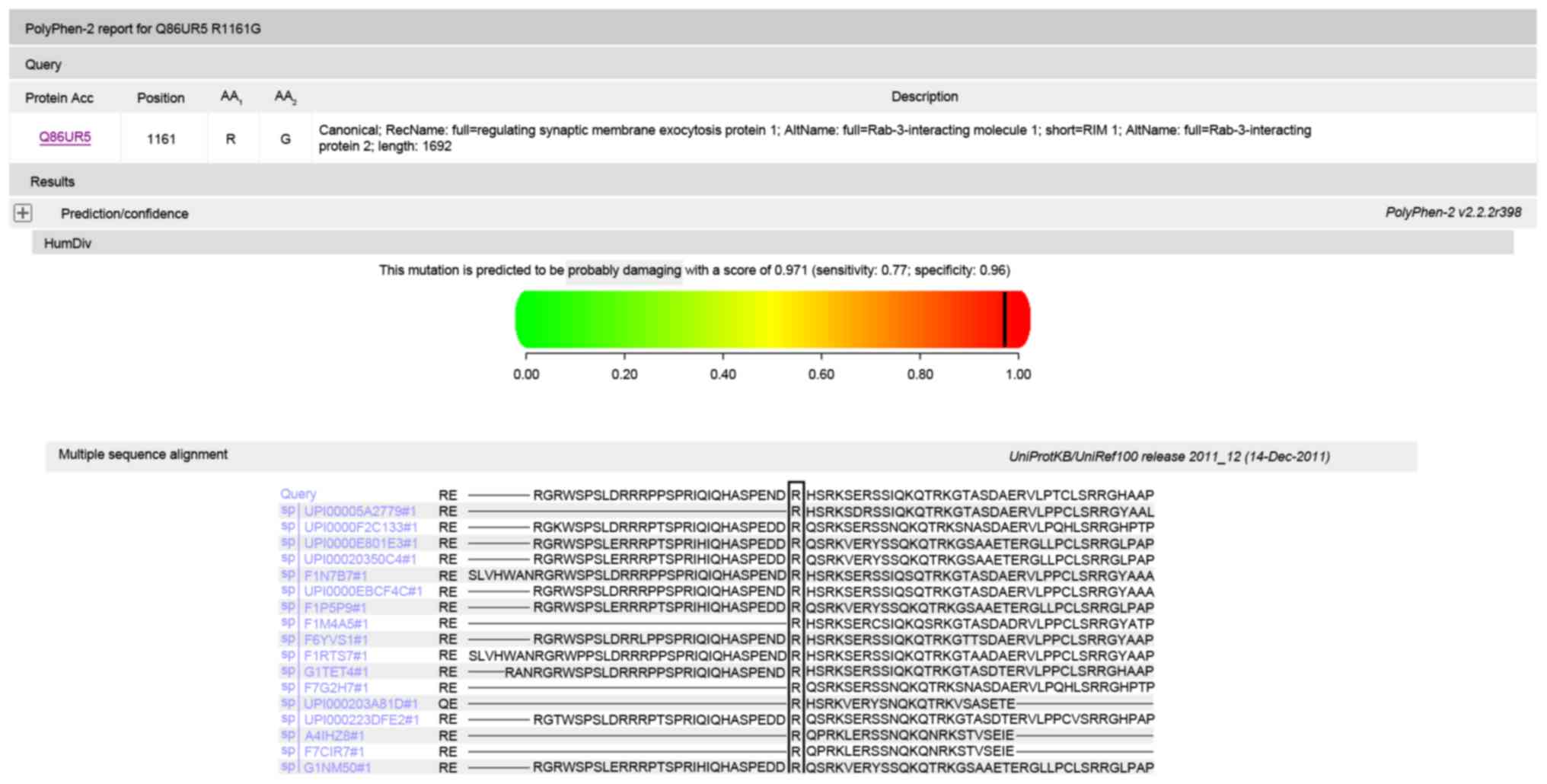
inheritance pattern of the mutations. Polyphen and SIFT predicted
that the amino acid substitution p.P346H in protein bestrophin 1 is
damaging (Fig. 5), and Polyphen
predicted that the amino acid substitution p.R1161G in protein RIM1
is damaging (Fig. 6). Multiple
sequenced alignment indicated that the residue at position 346 of
bestrophin-1 and the residue at position 1161 of RIM1 are highly
conserved across species.
Discussion
Macular coloboma may result from intrauterine
inflammation (15), and can be
associated with systemic developmental abnormalities. Notably, it
may be difficult to distinguish macular coloboma with macular
atrophy if medical history is not provided. The underlying
biological mechanism for development of macular coloboma is
unclear. Interestingly, we found that this patient had two
different mutations simultaneously, reminding us that some serious
congenital defects can be caused by multiple mutations on multiple
genes, making gene therapy more challenging.
BVMD is one of the most frequent form of autosomal
dominant macular dystrophy (16).
It is associated with mutations in the BEST1 gene (17) and results from dysfunction of the
retinal pigment epithelium (RPE) (18). Bestrophin-1 is the product of the
gene BEST1. This protein is mainly expressed in the basolateral
plasma membrane of the RPE (19).
This protein contains several domains with a high degree of
evolutionary conservation. The function of Bestrophin-1 remains
unclear, and some studies proposed that it acts as a Cl- channel
activated by intracellular Ca2+ and/or as a channel
regulator (20,21). A previous study conducted by
Katagiri et al (14).
identified a novel mutation p.P346H on BEST1 in a 38-year-old
patient who was in the vitelliruptive stage, which was less serious
than the patient in our study. It is likely that in BVMD, the
clinical manifestations of transheterozygous mutations may be more
serious than a single mutation.
CORD is one of the common forms of inherited retinal
degeneration with a prevalence of 1/40 000 (22,23).
CORD is characterized by the impairment of cone photoreceptors with
or without dysfunction of rod photoreceptors (24,25).
Clinical manifestations in CORD include photophobia, reduced visual
acuity, color vision defects, and central scotomata (26). At present, a total of 30 genes have
been found associated with CORD, including 10 genes related to
autosomal dominant CORD (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3,
PROM1, PRPH2, RIMS1, SEMA4A, UNC119) (27,28).
RIM1, the protein product of RIMS1, localizes to the presynaptic
active zones in brain and retinal tissue, and plays an important
role in regulating synaptic vesicle release and presynaptic
plasticity (29). RIM1 is a large
multi-domain protein that interacts with multiple molecules at
different regions (30).
In this case, we consider the patient had BVMD and
CORD simultaneously. Mutations on BEST1 and RIM1 affected the RPE
and photoreceptors, respectively. Therefore, the atrophy of the
retina was very serious with only a little retina tissue remained.
For these patients, transplantation of the retina stem cells or
retina cell membrane may be more feasible for treatment.
In summary, our study identified two mutations of
BEST1 and RIMS1 in one Chinese patient with bilateral macular
coloboma. These findings expand the mutation spectrums of BEST1 and
RIMS1, and will be valuable for genetic counseling and development
of therapeutic interventions for patients with macular
coloboma.
Acknowledgements
The authors are grateful to all of the patients,
their families, and the control volunteers for participating in
this study. This study was supported by the National Natural
Science Foundation of China (grant nos. 81500709, 81570862,
81371019 and 81670872), the Medical Scientific Research Foundation
of Guangdong Province (grant no. A2016460), and the Fundamental
Research Funds for the Universities (grant no. 13ykpy43).
References
|
1
|
Hornby SJ, Adolph S, Gilbert CE, Dandona L
and Foster A: Visual acuity in children with coloboma: Clinical
features and a new phenotypic classification system. Ophthalmology.
107:511–520. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Varghese M, Kavalakatt JA, Pandey S and
Kolath JJ: Macular coloboma. Oman J Ophthalmol. 9:67–68. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Primo SA: Macular coloboma. J Am Optom
Assoc. 61:373–377. 1990.PubMed/NCBI
|
|
4
|
Sharma S, Naqvi A and Cruess AF: Bilateral
macular colobomas. Can J Ophthalmol. 31:27–28. 1996.PubMed/NCBI
|
|
5
|
Ishaq M, Mukhtar A and Khan S: Macular
coloboma in a child with usher syndrome. J Ayub Med Coll
Abbottabad. 27:470–472. 2015.PubMed/NCBI
|
|
6
|
Izumikawa Y: Macular
coloboma-brachydactyly. Ryoikibetsu Shokogun Shirizu (34 Pt 2).
126–127. 2001.(In Japanese).
|
|
7
|
Jimenez-Sierra JM, Ogden TE and Van Boemel
GB: Inherited retinal diseasesA diagnostic guide. The C. V. Mosby
Company; St. Louis: 1989
|
|
8
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li R, Li Y, Fang X, Yang H and Wang J,
Kristiansen K and Wang J: SNP detection for massively parallel
whole-genome resequencing. Genome Res. 19:1124–1132. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G, Durbin R, et al: 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: PAX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.PubMed/NCBI
|
|
12
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
13
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.PubMed/NCBI
|
|
14
|
Katagiri S, Hayashi T, Ohkuma Y, Sekiryu
T, Takeuchi T, Gekka T, Kondo M, Iwata T and Tsuneoka H: Mutation
analysis of BEST1 in Japanese patients with Best's vitelliform
macular dystrophy. Br J Ophthalmol. 99:1577–1582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamaguchi K and Tamai M: Congenital
macular coloboma in Down syndrome. Ann Ophthalmol. 22:222–223.
1990.PubMed/NCBI
|
|
16
|
Low S, Davidson AE, Holder GE, Hogg CR,
Bhattacharya SS, Black GC, Foster PJ and Webster AR: Autosomal
dominant best disease with an unusual electrooculographic light
rise and risk of angle-closure glaucoma: A clinical and molecular
genetic study. Mol Vis. 17:2272–2282. 2011.PubMed/NCBI
|
|
17
|
Tian R, Yang G, Wang J and Chen Y:
Screening for BEST1 gene mutations in Chinese patients with
bestrophinopathy. Mol Vis. 20:1594–1604. 2014.PubMed/NCBI
|
|
18
|
Wong RL, Hou P, Choy KW, Chiang SW, Tam
PO, Li H, Chan WM, Lam DS, Pang CP and Lai TY: Novel and homozygous
BEST1 mutations in Chinese patients with Best vitelliform macular
dystrophy. Retina. 30:820–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin Y, Gao H and Liu Y, Liang X, Liu X,
Wang Z, Zhang W, Chen J, Lin Z, Huang X and Liu Y: Two novel
mutations in the bestrophin-1 gene and associated clinical
observations in patients with best vitelliform macular dystrophy.
Mol Med Rep. 12:2584–2588. 2015.PubMed/NCBI
|
|
20
|
Lin CF and Sarraf D: Best disease
presenting as a giant serous pigment epithelial detachment. Retin
Cases Brief Rep. 8:247–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Apushkin MA, Fishman GA, Taylor CM and
Stone EM: Novel de novo mutation in a patient with best macular
dystrophy. Arch Ophthalmol. 124:887–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hamel CP: Cone rod dystrophies. Orphanet J
Rare Dis. 2:72007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brodie S: Cone-rod dystrophy in Danon
disease. Graefes Arch Clin Exp Ophthalmol. 250:6332012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Burstedt MS, Ristoff E, Larsson A and
Wachtmeister L: Rod-cone dystrophy with maculopathy in genetic
glutathione synthetase deficiency: A morphologic and
electrophysiologic study. Ophthalmology. 116:324–331. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khan AO and Abu-Safieh L: Rod-Cone
dystrophy with initially preserved visual acuity despite early
macular involvement suggests recessive CERKL mutations. Ophthalmic
Genet. 36:369–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuehlewein L and Sadda SR: Rod-Cone
dystrophy associated with williams syndrome. Retin Cases Brief Rep.
9:298–301. 2015.PubMed/NCBI
|
|
27
|
Pras E, Abu A, Rotenstreich Y, Avni I,
Reish O, Morad Y, Reznik-Wolf H and Pras E: Cone-rod dystrophy and
a frameshift mutation in the PROM1 gene. Mol Vis. 15:1709–1716.
2009.PubMed/NCBI
|
|
28
|
Huang L, Xiao X, Li S, Jia X, Wang P, Sun
W, Xu Y, Xin W, Guo X and Zhang Q: Molecular genetics of cone-rod
dystrophy in Chinese patients: New data from 61 probands and
mutation overview of 163 probands. Exp Eye Res. 146:252–258. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weiss N, Sandoval A, Kyonaka S, Felix R,
Mori Y and De Waard M: Rim1 modulates direct G-protein regulation
of Ca(v)2.2 channels. Pflugers Arch. 461:447–459. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schoch S, Mittelstaedt T, Kaeser PS,
Padgett D, Feldmann N, Chevaleyre V, Castillo PE, Hammer RE, Han W,
Schmitz F, et al: Redundant functions of RIM1alpha and RIM2alpha in
Ca(2+)-triggered neurotransmitter release. EMBO J. 25:5852–5863.
2006. View Article : Google Scholar : PubMed/NCBI
|