Introduction
Redox homeostasis is essential for normal
intracellular metabolism (1). It
is well established that oxidative stress is critical in the
pathophysiology of several diseases (2). Reductive stress is the counterpart of
oxidative stress, and is defined as an abnormal increase of
reducing equivalents (3). An
increasing number of studies have focused on the deleterious
effects of reductive stress in unicellular eukaryotic and mammalian
cells (4,5). Potent, exogenous reductants,
including dithiothreitol (DTT), are widely used to disrupt
disulfide bond formation and abrogate oxidative protein folding in
the endoplasmic reticulum (ER), which triggers reductive stress and
the ER stress response (6).
Previous findings describing experimental mice found associations
with the dysregulation of glutathione homeostasis and protein
aggregation cardiomyopathy (5).
However, despite decades of studies on redox biology, the molecular
and cellular mechanisms underlying reductive stress remain to be
fully elucidated.
Maintenance of the glutathione redox couple, reduced
glutathione (GSH)/oxidized glutathione (GSSG), is achieved by
recycling via the pentose phosphate pathway and GSH biosynthesis.
N-acetylcysteine (NAC), a precursor of GSH, is a widely used
thiol-containing antioxidant and modulator of the intracellular
redox state. NAC has attracted interest for its antioxidant
property, and increasing evidence has demonstrated that repletion
of the levels of GSH through NAC can protect against oxidative
stress-induced cell death though scavenging free radicals (7,8).
Previous studies have demonstrated that NAC can
induce apoptosis in vascular smooth muscle cells, enhance
fisetin-induced apoptosis in colorectal carcinoma cell lines, and
induce hypoxia-induced apoptosis in murine embryonic fibroblasts
(9–11). Our previous study demonstrated that
excess GSH from NAC treatment in H9c2 cells caused a further
reduction of glutathione redox potential (GSSG/2GSH), increased
mitochondrial oxidation and caused cytotoxicity in the presence of
lower reactive oxygen species (ROS) levels (12). However, the molecular mechanisms
have not been investigated. In the present study, the mechanisms of
NAC-induced cytotoxicity in H9c2 cells was investigated, and it was
found that NAC induced H9c2 cell apoptosis through the intrinsic
mitochondrial pathway but not via endoplasm reticulum stress. This
is, to the best of our knowledge, the first demonstration of GSH
repletion-induced apoptosis via the mitochondrial pathway.
Materials and methods
Reagents and antibodies
NAC, N-ethylmaleimide (NEM, cat. no. E3876)
and trichloroacetic acid (cat. no. T0699) were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Tunicamycin (cat.
no. T7765) was dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich; Merck KGaA) as a 1 mg/ml stock solution and stored
at −20°C. AlamarBlue® (cat. no. DAL1025) was from
Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Rabbit antibodies against cleaved caspase-9 (Asp353, cat. no.
9507), cleaved caspase-3 (Asp175, cat. no. 9661), cytochrome
c (cat. no. 4272), B-cell lymphoma 2 (Bcl-2)-associated X
protein (Bax, cat. no. 2772), binding immunoglobulin protein (BiP,
cat. no. 3183) and C/EBP homologous protein (CHOP; cat. no. 2895)
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Cell culture and cell viability
The H9c2 cell line was obtained from American Type
Culture Collection (cat. no. CRL-1446; ATCC, Manassas, VA, USA).
The H9c2 cells were grown in DMEM (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal calf serum (FCS;
Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
and 100 µg/ml streptomycin in a 5% CO2 humidified
atmosphere at 37°C. Cell viability was determined using non-toxic
alamarBlue®. The subconfluent, exponentially growing
H9c2 cells at a density of 1×105/ml and 100 µl medium
per well were incubated with NAC for 6, 12 and 24 h. A 1/10th
volume of alamarBlue® reagent was added directly to the
cells in the culture medium 2 h prior to reading fluorescence
(excitation at 540±35 nm and emission at 600±40 nm) using an Flx800
plate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Measurement of lactate dehydrogenase
(LDH) activity
The LDH activity was measured using a kit from
Cayman Chemical Co. (Ann Arbor, MI, USA), which used a coupled
two-step reaction. In the first step, LDH catalyzes the reduction
of NAD+ to NADH and H+ by the oxidation of
lactate to pyruvate. In the second step of the reaction, diaphorase
uses the newly-formed NADH and H+ to catalyze the
reduction of a tetrazolium salt to highly-colored formazan, which
absorbs at 490–520 nm. Following treatment, culture medium was
collected to measure LDH activity. All the determinations were
normalized to protein content, determined using the method of Lowry
et al (13). The absorbance
was recorded at 405 nm using a plate reader every 5 min for 30
min.
Immunofluorescence microscopy
The H9c2 cells at a density of 2×105/well
were grown on a coverslip in six-well plates for 24 h and treated
with NAC and H2O2 for the indicated
durations. The cells were then stained using Hoechst 33342 and
propidium iodide (PI), which is permeant stains only dead cells.
The staining pattern resulting from the simultaneous use of these
dyes makes it possible to distinguish normal and dead cell
populations using fluorescence microscopy.
Annexin V/PI double-staining analysis
of apoptosis
Cell apoptosis was determined using Annexin V-FITC
and PI double staining (Kaiji Biotechnology, Nanjing, China)
according to the manufacturer's instructions. The H9c2 cells were
seeded in six-well plates at a density of 1×105/well and
treated with different concentrations of NAC for 24 h. Following
treatment, the H9c2 cells were harvested with 0.25% trypsin and
washed twice in ice-cold PBS, following which they were resuspended
in 300 µl of binding buffer containing 1 µg/ml PI and 0.05 µg/ml
Annexin V-FITC. The samples were incubated for 15 min at room
temperature in the dark and were analyzed using flow cytometry
(Beckman Coulter, Inc., Miami, FL, USA) at an excitation wavelength
of 488 nm. The emissions of annexin-V and PI were monitored at
wavelengths of 525 and 630 nm, respectively. The percentage of
apoptotic cells was determined using Multicycle software version
2.5 (Phoenix Flow Systems, San Diego, CA, USA).
Analysis of the activities of
caspase-3, −8, −9 and −12
Caspase activity within the treated cells was
determined fluorometrically using a Caspase-3 Fluorescence Assay
kit (cat. no. 10009135; Cayman Chemical Co.), Caspase-8
Fluorescence Assay kit (cat. no. K112; BioVision, Inc., Milpitas,
CA, USA), Caspase-9 Fluorescence Assay kit (cat. no. K118;
BioVision, Inc.) and Caspase-12 Fluorescence Assay kit (cat. no.
K139; BioVision, Inc.). These assays are based on detecting the
cleavage of substrates N-Ac-DEVD-N'-MC-R110, IETD-AFC, LEHD-AFC and
ATAD-AFC. The treated cells (5×105) were pelleted and
resuspended in 50 µl of chilled cell lysis buffer, and transferred
to a 96-well plate. Caspase buffer (50 µl) containing 50 µM
substrate was added to the sample and cleavage of substrate was
performed at 37°C using an Flx800 plate reader (BioTek Instruments,
Inc.).
Subcellular fractionation, SDS-PAGE
and immunoblotting
The whole cell lysate was extracted using 1X SDS
buffer. The cytosolic and mitochondrial fractions were prepared
using a Mitochondria/Cytosol Isolation kit (Abcam, Cambridge, UK).
The protein contents of the subcellular fractions and whole cell
lysate were determined by BCA protein assay kit and 30 µg of
samples were separated on a 12% glycine SDS-PAGE gel and
transferred onto a PVDF membrane. The membranes were blocked in 5%
dry milk in TBS with 0.1% Tween-20 (TBST) for 1 h at room
temperature, followed by incubation with the indicated primary
antibodies to cytochrome c (1:1,000), Bax (1:1,000), GAPDH
(1:2,000), VDAC (1:1,000), BiP (1:1,000) and CHOP (1:1,000) and
subsequent incubation with horseradish peroxidase goat anti-rabbit
IgG secondary antibody (cat. no. 7074, 1:10,000; Cell Signaling
Technology, Inc.) in TBST with 0.2% BSA for 1 h at room
temperature. The immunoblot signals were visualized using Super
Signal West Pico Chemiluminescent substrate (Pierce; Thermo Fisher
Scientific, Inc.).
NEM-alkylated redox western blot
analysis
For protein disulfide isomerase (PDI) redox
analysis, the cells were treated with NAC or 10 mM DTT for the
indicated time and washed twice with ice-cold PBS immediately
following treatment. The cells were then precipitated with chilled
trichloroacetic acid (10%) for 30 min at 4°C. The samples were
centrifuged at 12,000 × g for 10 min at room temperature and washed
twice with 100% acetone. The protein pellets were dissolved in
non-reducing buffer containing 100 mM Tris-HCl (pH 6.8), 2% SDS and
40 mM NEM. PDI redox forms were separated via 10% non-reducing
SDS-PAGE.
Measurement of changes in
mitochondrial membrane potential (∆ψm)
Mitochondrial transmembrane depolarization was
detected using JC-1 (Molecular Probes; Thermo Fisher Scientific,
Inc.). Following washing twice with pre-warmed PBS, 2 µg/ml JC-1
was added into each well and incubated at 37°C for 30 min. The
cells were then washed three times with pre-warmed PBS. NAC (4 µM)
was added and the cell culture plate was incubated at 37°C for the
required duration. The plates were immediately read using the
Flx800 plate reader (BioTek Instruments, Inc.). Red fluorescence
was measured at 550 nm (excitation) and 600 nm (emission). Green
fluorescence was measured at 485 nm (excitation) and 535 nm
(emission). The ratio of red fluorescence to green fluorescence was
determined, and mitochondrial depolarization was indicated by a
decrease in the red/green fluorescence ratio.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The untreated and treated H9c2 cells were rinsed
twice with PBS. The total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was synthesized
from 1 µg total RNA using reverse transcription reagents from
Applied Biosystems; Thermo Fisher Scientific, Inc., according to
the manufacturer's instructions. PCR amplifications were performed
using 10 µl SYBR-Green PCR Master mix, 2 µl cDNA sample (equivalent
to 100 ng) and 1 µl of 2 µM forward and reverse primers on the ABI
Prism 7000 Sequence Detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the following thermal cycling
conditions: 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C
for 30 sec. PCR amplifications were performed in duplicate wells.
The quantification was performed using the comparative
quantification cycle (2−∆∆Cq) method (14), using human GAPDH as an internal
control.
Statistical analysis
All experiments were repeated three times. For the
western blot analysis, one representative image is shown in
figures. SPSS software version 22.0 (IBM Corp., Armonk, NY, USA)
was used for statistical analysis. Values are presented as the mean
± standard deviation. Student's t-test was used for statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
NAC induces the apoptosis of H9c2
cells
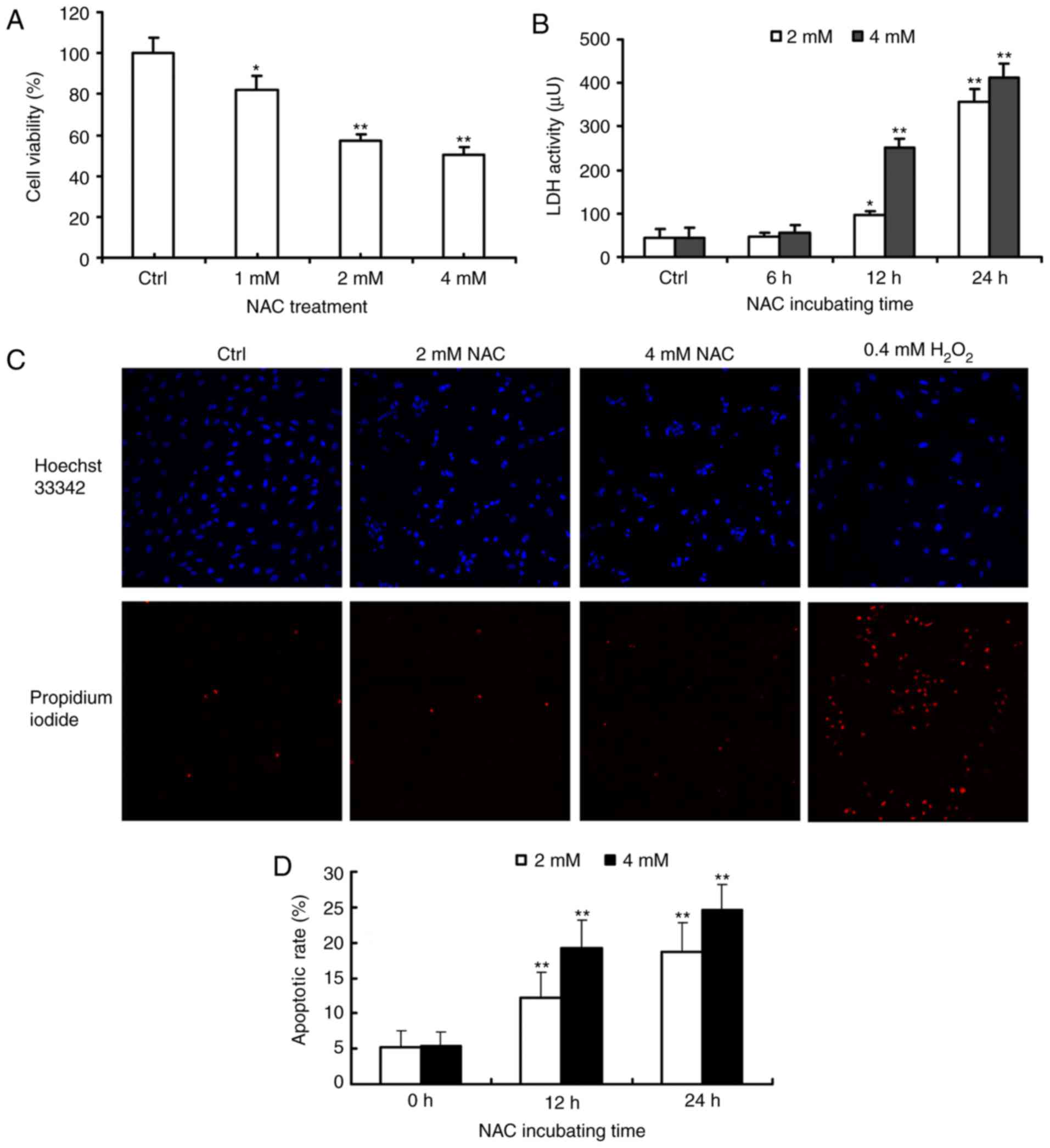
Firstly, the present study investigated the effects
of NAC treatment on the growth of H9c2 cells by using the
alamarBlue® assay. Compared with the control group, the
viability of H9c2 cells was significantly decreased in a
dose-dependent manner in response to 1, 2 and 4 µM of NAC for 24 h
(Fig. 1A). LDH release was also
measured in the supernatant of the NAC-treated H9c2 cells. NAC
treatment induced the release of LDH in a dose- and time-dependent
manner (Fig. 1B).
To determine whether the observed decrease in cell
viability was associated with apoptosis or necrosis, the nuclear
morphology and plasma membrane permeability of the NAC-treated H9c2
cells were examined using Hoechst 33342 (blue) and PI (red)
staining. In the H2O2-treated cells, the rate
of necrosis was markedly increased, whereas treatment with 2 or 4
µM NAC had effect on the rate of necrosis (Fig. 1C). The ability of NAC to induce
H9c2 cell apoptosis was quantified using Annexin V-FITC/PI double
staining and was calculated using Multicycle software. Following
treatment with various concentrations of NAC for 24 h, the
percentages of apoptotic cells were 18.6±4.1 and 24.5±3.7%,
respectively, which was significantly higher, compared with that in
the untreated control cells (5.4±1.8, P<0.01; Fig. 1D).
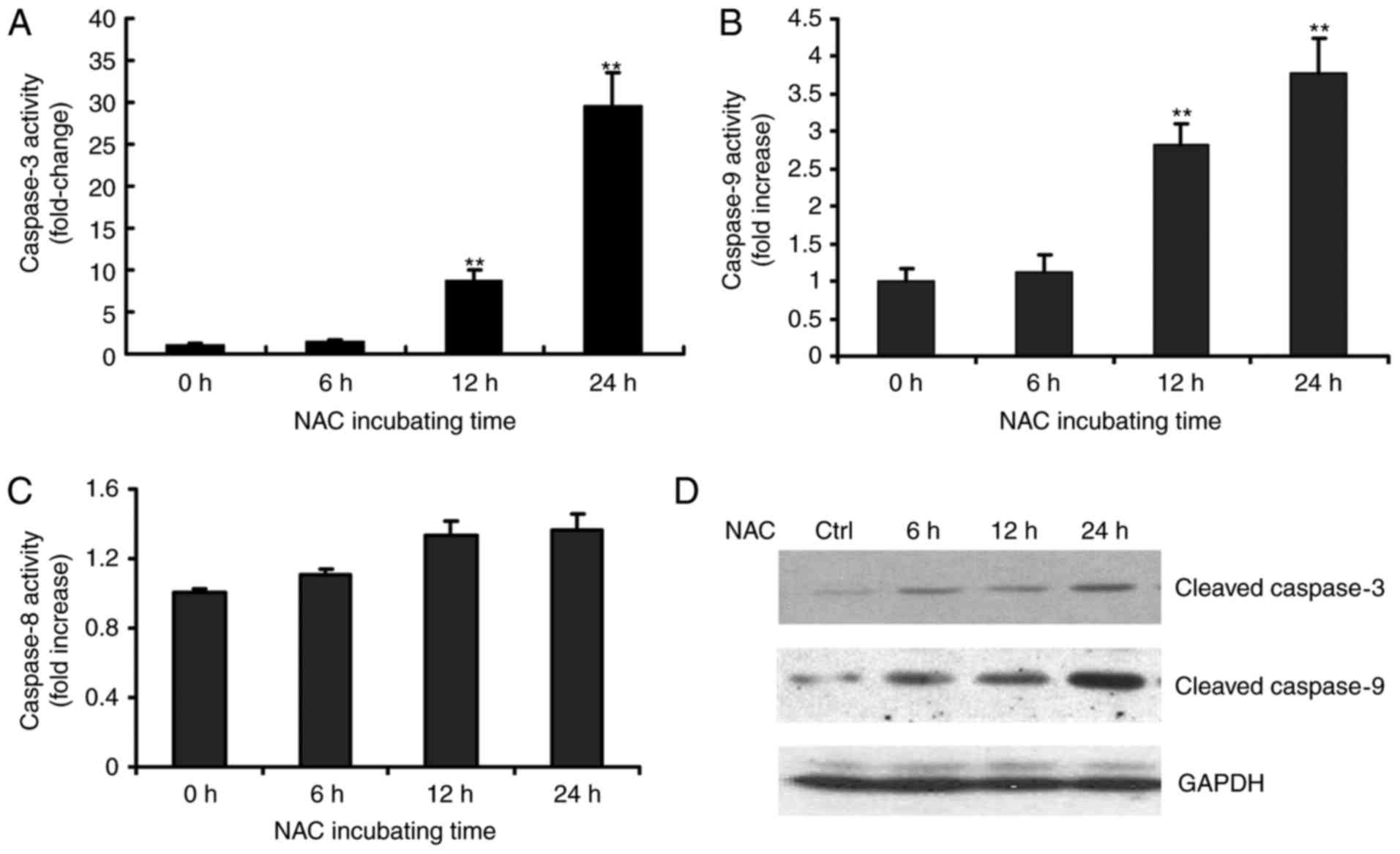
Activation of caspase-9 and −3, but
not caspase-8, is involved in NAC-induced apoptosis of H9c2
cells
The present study also investigated the possible
mechanisms underlying the NAC-induced apoptosis of H9c2 cells. As
caspases are known to be pivotal in mediating various apoptotic
signals, the present study measured the activity of initiator
caspases (caspase-8 and −9) and effector caspase (caspase-3) in the
NAC-treated H9c2 cells using fluorometrical assay kits. As shown in
Fig. 2, exposure of the H9c2 cells
to 4 µM of NAC led to increased enzymatic activities of caspase-3
and −9 in a time-dependent manner during the treatment period
(Fig. 2A and B). The increased
activities of caspases-3 and −9 were observed as early as 12 h. By
contrast, no significant change in the activity of caspase-8 was
observed (Fig. 2C). The
NAC-induced caspase activation was further confirmed by detecting
the cleavage of procaspase-9 and procaspase-3 following NAC
treatment in H9c2 cells (Fig.
2D).
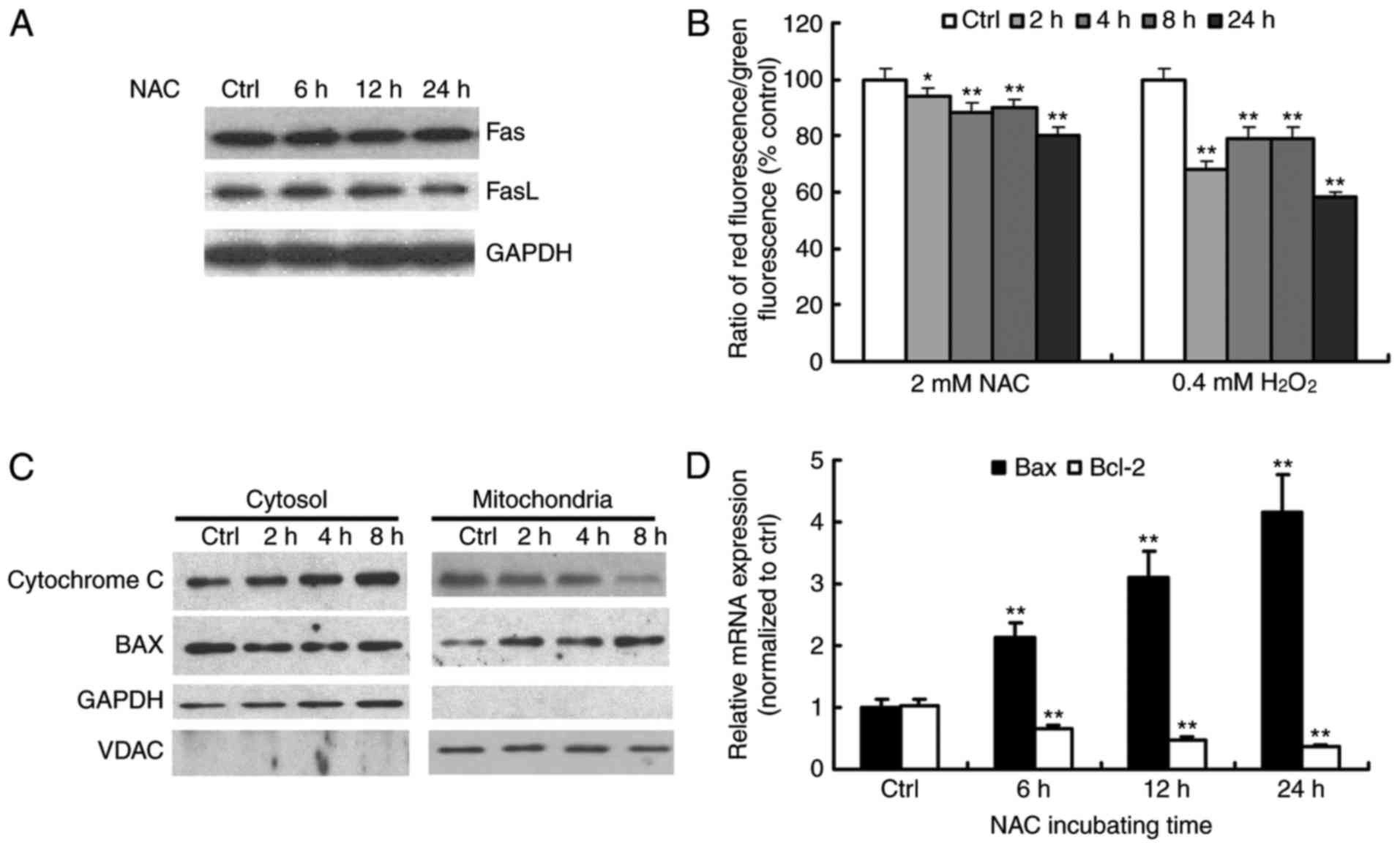
NAC induces apoptosis through
activation of the intrinsic mitochondrial signaling pathway in H9c2
cells
Apoptosis is usually induced via two main pathways
involving either the activation of death receptors (extrinsic
pathway) or the mitochondrial pathway (intrinsic pathway). The
death receptor pathway is usually triggered by the binding of death
receptors, including Fas or tumor necrosis factor receptor, by
their respective ligands, namely FasL and TNFRL, which recruit
initiator caspase-8 via the adaptor protein, FADD, leading to the
proteolytic activation of caspase-8 (15). Consistent with the results of
caspase-8 activity, NAC treatment did not affect the levels of Fas
or FasL (Fig. 3A). These results
suggested that NAC treatment did not activate the Fas-mediated
death receptor pathway in H9c2 cells.
Mitochondria are key in the regulation of apoptosis.
One of the major events in mitochondrial dysfunction is the loss of
Δψm and the subsequent release of cytochrome c (16). The present study investigated Δψm
using the dual-emission fluorescent dye, JC-1, which characterizes
the dissipation of Δψm by a significant shift of red to green
fluorescence. The treatment of cells with NAC enhanced the level of
green fluorescence in a time-dependent manner, which demonstrated
the loss of Δψm during NAC-induced apoptosis (Fig. 3B).
The release of cytochrome c from mitochondria
combines with apoptotic protease activating factor 1 and
procaspase-9 to form the apoptosome, which leads to the activation
of caspse-9 and −3. The Bcl-2 family members are known to be
critical in regulating the release of cytochrome c. Under
apoptotic stimuli, pro-apoptotic Bcl-2 members, including Bax and
BH3 interacting-domain death agonist, are activated, whereas
anti-apoptotic Bcl-2 and Bcl-extra large prevent this process
(17). The balance of pro- and
anti-apoptotic proteins is associated with the ultimate fate of
cells. To assess whether the mitochondrial pathway was involved in
NAC-induced apoptosis, the present study detected the levels of
cytochrome c and Bax in proteins extracts from cytosolic and
mitochondrial fractions of the NAC-treated cells. The release of
cytochrome c from the mitochondria to the cytosol was
observed as early as 2 h following treatment (Fig. 3C). Consistent with this, a
time-dependent increase in mitochondrial Bax and a concomitant
decrease in the cytosolic fraction were observed (Fig. 3C). The expression levels of Bax and
Bcl-2 were examined using RT-qPCR analysis. As shown in Fig. 3D, following exposure of the H9c2
cells to 4 µM NAC for different durations (0–24 h), The mRNA levels
of Bax increased, whereas the mRNA levels of Bcl-2 decreased
gradually with time. Therefore, NAC treatment increased the ratio
of Bax/Bcl-2, which favors the occurrence of apoptosis. These
results suggested that NAC-induced apoptosis occurred mainly
through the mitochondria-dependent pathway.
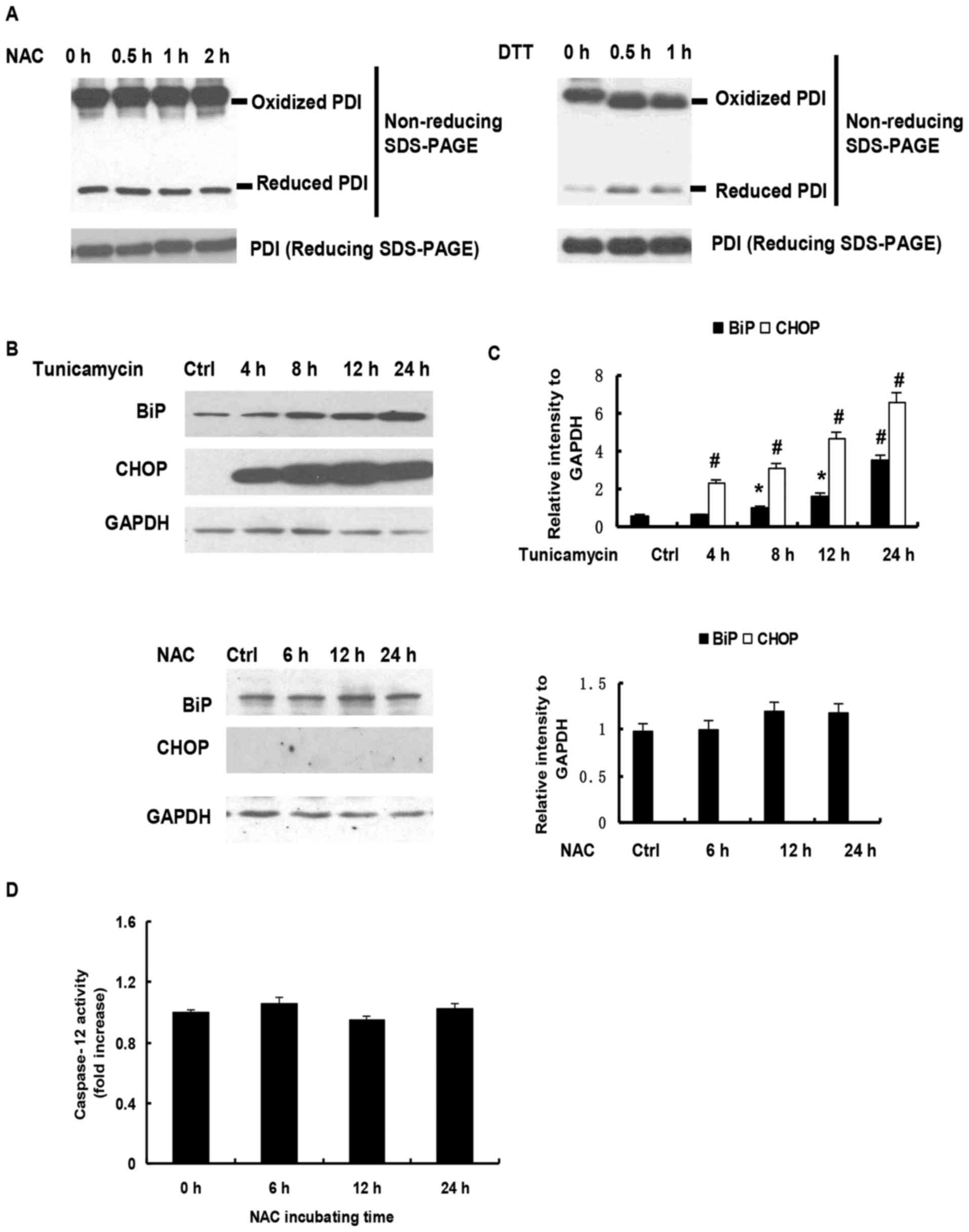
NAC induced apoptosis is independent
of ER stress
In the ER, nascent proteins are folded with the
assistance of ER chaperones. The highly oxidized redox milieu of
the ER matrix facilitates disulfide bond formation and the
maturation of secretory proteins. Exogenous reductants disrupt
disulfide bond formation and abrogate oxidative protein folding in
the ER, which triggers the ER stress response. Our previous study
demonstrated that the repletion of GSH by NAC in H9c2 cells caused
a reduction of the glutathione redox potential. To examine whether
NAC exposure induced H9c2 cell apoptosis through altering the
oxidized environment in ER and causing ER stress, the present study
analyzed the redox state of PDI. PDI is an essential chaperone,
which constitutes 2% of the protein in the ER and has long been
known to assist in the formation of disulfide bonds. PDI is found
predominantly in an oxidized form in vivo (18). As shown in Fig. 4A, NEM derivatization allowed the
identification of reduced PDI and oxidized PDI, which migrates at a
slower rate than the reduced form. Following reduction with DTT,
PDI shifted to the reduced isoform, whereas minimal change in
reduced PDI was observed on exposure to NAC for 2 h (Fig. 4A). Further analysis showed that the
levels of BiP and CHOP were significantly upregulated in the H9c2
cells treated with tunicamycin, a common agent to induce ER stress,
but not in NAC-treated H9c2 cells (Fig. 4B and C). Increasing evidence has
demonstrated that caspase-12 is key in ER stress-mediated apoptosis
(19). In the present study, no
change in caspase-12 activity was observed following NAC treatment
(Fig. 4D). Taken together, these
data indicated that exogenous GSH repletion by NAC-induced H9C2
cell apoptosis was independent of ER stress in H9c2 cells.
Discussion
As an antioxidant precursor to glutathione, the
clinical applications of NAC have broadened. NAC appears to be
promising in the treatment of several illnesses, including chronic
obstructive pulmonary disease and contrast-induced nephropathy
(20). There is also evidence to
support its use in Alzheimer's disease and psychiatric disorders,
particularly schizophrenia and bipolar disorder (21). The primary mechanisms underlying
the beneficial effects for these disorders may be that NAC
maintains the redox balance in the cell through augmentation of
intracellular glutathione levels and its scavenging activity of
free radicals (22). NAC is safe
and well tolerated when administered orally, but has documented
risks on intravenous administration.
Although NAC can inhibit
H2O2-mediated cell death, it enhances the
apoptosis induced by other stimuli, including hypoxia, ultraviolet,
imatinib, 5-fluorouracil and fisetin. NAC can also induce apoptosis
in specific cells. Tsai et al (23) reported that NAC induces apoptosis
in rat and human smooth muscle cells, and that the overexpression
of Bcl-2 suppressed the cell death induced by NAC. Consistent with
these results, the present study showed that NAC was cytotoxic
towards H9c2 cells through inducing apoptosis. The increased
apoptosis was not attributable to the extrinsic apoptosis pathway
due to the lack of activation of caspase-8, and no increase in the
levels of Fas and FasL in response to NAC. By contrast, NAC
appeared to increase the activities of caspase-9 and −3, and the
cleavage of procaspase-9 and −3. Bcl-2 family proteins are known to
be either pro-apoptotic or anti-apoptotic via regulating the
permeability of the mitochondrial outer membrane. Bcl-2 is
anti-apoptotic and the Bax protein is pro-apoptotic in initiating
apoptosis. The data obtained in the present study showed that NAC
resulted in the simultaneous upregulation of Bax and downregulation
of Bcl-2, loss of Δψm, and the subsequent release of cytochrome
c and translocation of Bax to mitochondria. These results
support the hypothesis that NAC-induced apoptosis in H9c2 cells is
mediated by the intrinsic mitochondrial pathway.
The mitochondrion is the most important organelle in
determining continued cell survival and cell death. GSH/GSSG ratios
of 20:1-40:1 and reduced milieu have been reported in the matrix of
mitochondria, whereas its inner membrane space is more oxidizing
(1). Our previous investigations
(12) revealed that the repletion
of GSH from NAC in H9c2 cells disrupted the reduced milieu of
mitochondria, which was confirmed by the redox states of
mitochondrial thrioredoxin 2 and roGFP. A previous study also
showed that GSH ethyl ester or NAC induce mitochondrial oxidation
via respiratory complex III (24).
Singh et al (25) reported
that NAC reductive stress impairs L6 myoblast mitochondrial
respiratory chain function, leading to mitochondrial ROS production
and the activation of mitochondrial biogenesis pathways. These
findings further clarify the possible mechanism by which the
mitochondria pathway is involved in NAC-induced apoptosis.
GSH is known to be present in the ER, and the redox
status of the ER is defined by the status of glutathione. This
compartment contains millimolar concentrations of GSH and GSSG, in
which the GSH/GSSG ratio ranges between 1:1 and 3:1 to achieve a
more oxidizing environment (26).
Previous findings have demonstrated that BiP and CHOP are
upregulated in HeLa cells following treatment with NAC, and that
the protein kinase R-like ER kinase-activating transcription factor
4 pathway is activated in NAC-treated cells, indicating that
NAC-induced apoptosis in HeLa cells is mediated by the ER stress
pathway (27). By contrast, the
present study found that NAC did not upregulate the expression of
BiP or CHOP in H9c2 cells. The fact that the redox status of PDI
did not alter following exposure to NAC was consistent with NAC
causing a decreased GSH/GSSG ratio due to a corresponding increase
in GSSG. This observation suggests that the mechanism of
NAC-induced apoptosis in H9c2 cells and HeLa cells is cell
type-specific.
To the best of our knowledge, the present study is
the first to show that NAC induced the apoptosis of H9c2 cells via
the mitochondria-dependent pathway but not via ER stress, and that
exogenous GSH from NAC did not alter the oxidized milieu of the
ER.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81270279 and 81471897) and
the Hunan Natural Science Foundation (grant no. 2013JJ1009).
References
|
1
|
Hansen JM, Go YM and Jones DP: Nuclear and
mitochondrial compartmentation of oxidative stress and redox
signaling. Annu Rev Pharmacol Toxicol. 46:215–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris C and Hansen JM: Oxidative stress,
thiols and redox profiles. Methods Mol Biol. 889:325–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schafer FQ and Buettner GR: Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple. Free Radic Biol Med.
30:1191–1212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trotter EW and Grant CM: Thioredoxins are
required for protection against a reductive stress in the yeast
Saccharomyces cerevisiae. Mol Microbiol. 46:869–878. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rajasekaran NS, Connell P, Christians ES,
Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM,
Leopold JA, et al: Human alpha B-crystallin mutation causes
oxido-reductive stress and protein aggregation cardiomyopathy in
mice. Cell. 130:427–439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rand JD and Grant CM: The thioredoxin
system protects ribosomes against stress-induced aggregation. Mol
Biol Cell. 17:387–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mayer M and Noble M: N-acetyl-L-cysteine
is a pluripotent protector against cell death and enhancer of
trophic factor-mediated cell survival in vitro. Proc Natl Acad Sci
USA. 91:7496–7500. 1994; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park SA, Choi KS, Bang JH, Huh K and Kim
SU: Cisplatin-induced apoptotic cell death in mouse hybrid neurons
is blocked by antioxidants through suppression of
cisplatin-mediated accumulation of p53 but not of Fas/Fas ligand. J
Neurochem. 75:946–953. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rakshit S, Bagchi J, Mandal L, Paul K,
Ganguly D, Bhattacharjee S, Ghosh M, Biswas N, Chaudhuri U and
Bandyopadhyay S: N-acetyl cysteine enhances imatinib-induced
apoptosis of Bcr-Abl+ cells by endothelial nitric oxide
synthase-mediated production of nitric oxide. Apoptosis.
14:298–308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu MS, Lien GS, Shen SC, Yang LY and Chen
YC: N-acetyl-L-cysteine enhances fisetin-induced cytotoxicity via
induction of ROS-independent apoptosis in human colonic cancer
cells. Mol Carcinog. 53 Suppl 1:E119–E129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qanungo S, Wang M and Nieminen AL:
N-Acetyl-L-cysteine enhances apoptosis through inhibition of
nuclear factor-kappaB in hypoxic murine embryonic fibroblasts. J
Biol Chem. 279:50455–50464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Limphong P, Pieper J, Liu Q,
Rodesch CK, Christians E and Benjamin IJ: Glutathione-dependent
reductive stress triggers mitochondrial oxidation and cytotoxicity.
FASEB. 26:1442–1451. 2012. View Article : Google Scholar
|
|
13
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuznetsov AV, Margreiter R, Amberger A,
Saks V and Grimm M: Changes in mitochondrial redox state, membrane
potential and calcium precede mitochondrial dysfunction in
doxorubicin-induced cell death. Biochim Biophys Acta.
1813:1144–1152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sharpe JC, Arnoult D and Youle RJ: Control
of mitochondrial permeability by Bcl-2 family members. Biochim
Biophys Acta. 1644:107–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mezghrani A, Fassio A, Benham A, Simmen T,
Braakman I and Sitia R: Manipulation of oxidative protein folding
and PDI redox state in mammalian cells. EMBO J. 20:6288–6296. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An endoplasmic reticulum stress-specific
caspase cascade in apoptosis. Cytochrome c-independent activation
of caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dodd S, Dean O, Copolov DL, Malhi GS and
Berk M: N-acetylcysteine for antioxidant therapy: Pharmacology and
clinical utility. Expert Opin Biol Ther. 8:1955–1962. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dean O, Giorlando F and Berk M:
N-acetylcysteine in psychiatry: Current therapeutic evidence and
potential mechanisms of action. J Psychiatry Neurosci. 36:78–86.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aruoma OI, Halliwell B, Hoey BM and Butler
J: The antioxidant action of N-acetylcysteine: Its reaction with
hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous
acid. Free Radic Biol Med. 6:593–597. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsai JC, Jain M, Hsieh CM, Lee WS,
Yoshizumi M, Patterson C, Perrella MA, Cooke C, Wang H, Haber E, et
al: Induction of apoptosis by pyrrolidinedithiocarbamate and
N-acetylcysteine in vascular smooth muscle cells. J Biol Chem.
271:3667–3670. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kolossov VL, Beaudoin JN, Ponnuraj N,
DiLiberto SJ, Hanafin WP, Kenis PJ and Gaskins HR: Thiol-based
antioxidants elicit mitochondrial oxidation via respiratory complex
III. Am J Physiol Cell Physiol. 309:C81–C91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singh F, Charles AL, Schlagowski AI,
Bouitbir J, Bonifacio A, Piquard F, Krähenbühl S, Geny B and Zoll
J: Reductive stress impairs myoblasts mitochondrial function and
triggers mitochondrial hormesis. Biochim Biophys Acta.
1853:1574–1585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hwang C, Sinskey AJ and Lodish HF:
Oxidized redox state of glutathione in the endoplasmic reticulum.
Science. 257:1496–1502. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan D, Xu Y, Yang M, Wang H, Wang X and
Shen Z: N-acetyl cysteine and penicillamine induce apoptosis via
the ER stress response-signaling pathway. Mol Carcinog. 49:68–74.
2010.PubMed/NCBI
|