Introduction
Liver fibrosis is the common consequence of chronic
liver diseases and may progress to cirrhosis and hepatocellular
carcinoma (HCC). It is characterized by accumulation of
extracellular matrix (ECM) components, including collagen,
fibronectin and laminin (1). The
mechanisms underlying liver fibrosis are still largely unclear.
Hepatic stellate cells (HSCs) are the predominant cell type in the
development of liver fibrosis. The activation and
transdifferentiation of HSCs are pivotal events in liver fibrosis
(2,3). Following liver injury, quiescent HSCs
are exposed to different inflammatory cytokines, including
transforming growth factor (TGF)-β1, and then undergo a process of
activation to a myofibroblastic phenotype, finally resulting in the
excess production of ECM components (4). Thus, suppression of HSCs activation
is the main approach for the treatment of liver fibrosis.
Follistatin-like 1 (Fstl1), also known as TSC-36, is
a secreted glycoprotein that belongs to the follistatin and SPARC
(secreted protein, acidic and rich in cysteine) families.
Increasing evidences have reported that Fstl1servescritical roles
in angiopoiesis, immunomodulation, embryonic development and
tumorigenesis (5–8). For example, overexpression of FSTL1
significantly inhibited cell proliferation and invasion in ovarian
and endometrial cancers (9). More
recently, it was reported that Fstl1 is induced in response to lung
injury; and blockage of Fstl1 with a neutralizing antibody
attenuated bleomycin-induced lung fibrosis in vivo (10). However, the role of Fstl1 in liver
fibrosis remains undefined. Therefore, the aim of the present study
was to investigate the role of Fstl1 in liver fibrosis. The results
demonstrated that knockdown of Fstl1 inhibited activation of HSCs
through the TGF-β1/Smad3 signaling pathway.
Materials and methods
Specimen collection
Liver samples were collected by trans-parietal
puncture from 11 healthy individuals and 11 patients with liver
fibrosis. Written informed consent was obtained from all patients,
and the study was approved by the Medical Ethics Committee of First
Teaching Hospital of Tianjin University of Traditional Chinese
Medicine (Tianjin, China).
Cell culture
Primary HSCs were isolated as described previously
(11). Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% (v/v)
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
L-glutamine (4 mmol/l; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), penicillin (100 IU/ml; Sigma-Aldrich; Merck KGaA), and
streptomycin (100 µg/ml; Sigma-Aldrich; Merck KGaA) at 37°C in a
humidified 5% CO2 atmosphere.
RNA interference and cell
transfection
Small-interfering RNA targeting Fstl1 (si-Fstl1) and
non-targeting control siRNA (scramble) were synthesized by
Guangzhou Ribo Bio Co., Ltd. (Guangzhou, China). HSCs at a density
of 5×104 cells/well were seeded in each cell of a
24-well micro-plate, grown for 24 h to reach 30–50% confluence, and
then transfected with si-Fstl1 or scramble using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of quiescent HSCs and activated HSCs, as
well as normal and hepatic fibrosis tissues, was extracted and
purified using an RNeasy Mini kit according to the instructions of
the manufacturer (Qiagen, Inc., Valencia, CA, USA). Up to 5 µg of
the total RNA was reverse-transcribed into cDNA using M-MLV reverse
transcriptase (Sigma-Aldrich; Merck KGaA). RT-qPCR was performed
with the Applied Biosystems 7900HT Fast Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using a SYBR
Green Real-Time PCR Master Mix kit (Takara Biotechnology Co., Ltd.,
Dalian, China). The following primers were used: Fstl1,
5′-CGAGGTGGAGTTGACGAGAAAC-3′ (sense),
5′-AGGACTGGATCATCATGACGTTCT-3′ (antisense); and β-actin
5′-CCGTGAAAAGATGACCCAGATC-3′ (sense), 5′-CACAGCCTGGATGGCTACGT-3′
(antisense). The relative levels of transcript were determined by
using the2−∆∆Cq method (12) and normalized by β-actin.
Western blotting
Total protein was extracted from hepatic fibrosis
tissues or HSCs using RIPA Cell Lysis buffer (Takara Biotechnology
Co., Ltd.). Lysates were sonicated for 5 sec on ice and centrifuged
at 6,000 × g for 5 min at 4°C. Supernatants were collected and the
protein concentration was detected using a Bio-Rad Protein Assay
kit II (cat. no. 500-0002; Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The cell lysates (30 µg/lane) were subjected to SDS-PAGE
and subsequently transferred to a polyvinylidene difluoride
membrane. Then, nonspecific binding was blocked by incubating with
5% non-fat milk in TBS containing 0.1% Tween-20 at room temperature
for 1 h. The blots were incubated with primary antibodies:
anti-Fstl1 (1:1,000; cat. no. sc-80408), anti-collagen I (1:2,000;
cat. no. sc-59772), anti-α-SMA (1:2,000; cat. no. sc-130616),
anti-Smad3 (1:2,000; cat. no. sc-101154), anti-p-Smad3 (1:2,000;
cat. no. sc-11769) or anti-GAPDH (1:5,000; cat. no. sc-400163; all
from Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at 4°C
overnight. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (1:2,000;
cat. no. sc-2789; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. Following washing, the blots were visualized using an
enhanced chemiluminescence detection system (GE Healthcare Life
Sciences, Chalfont, UK). Protein expression was analyzed using
BandScan software version 5.0 (Glyko; BioMarin Pharmaceutical,
Inc., San Rafael, CA, USA). All experiments were repeated ≥3
times.
Cell proliferation assay
Cell proliferation was determined using the Cell
Counting kit-8 assay. Briefly, infected HSCs were plated at a
density of 1×104 cells/well in a 96-well culture plate
and treated with or without TGF-β1 for 24 h. Then, 10 µl CCK-8
reagent was added (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) to each well. Following 1 h of incubation at 37°C, the
absorbance was measured with a microplate reader (Bio-Rad
Laboratories, Inc.) at a wavelength of 490 nm.
Statistical analysis
All statistical analyses were performed using the
SPSS software (version, 13.0; SPSS, Inc., Chicago, IL, USA).
Results were presented as mean ± standard deviation for three
experiments. Statistical comparisons were performed using one-way
analysis of variance followed by the Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
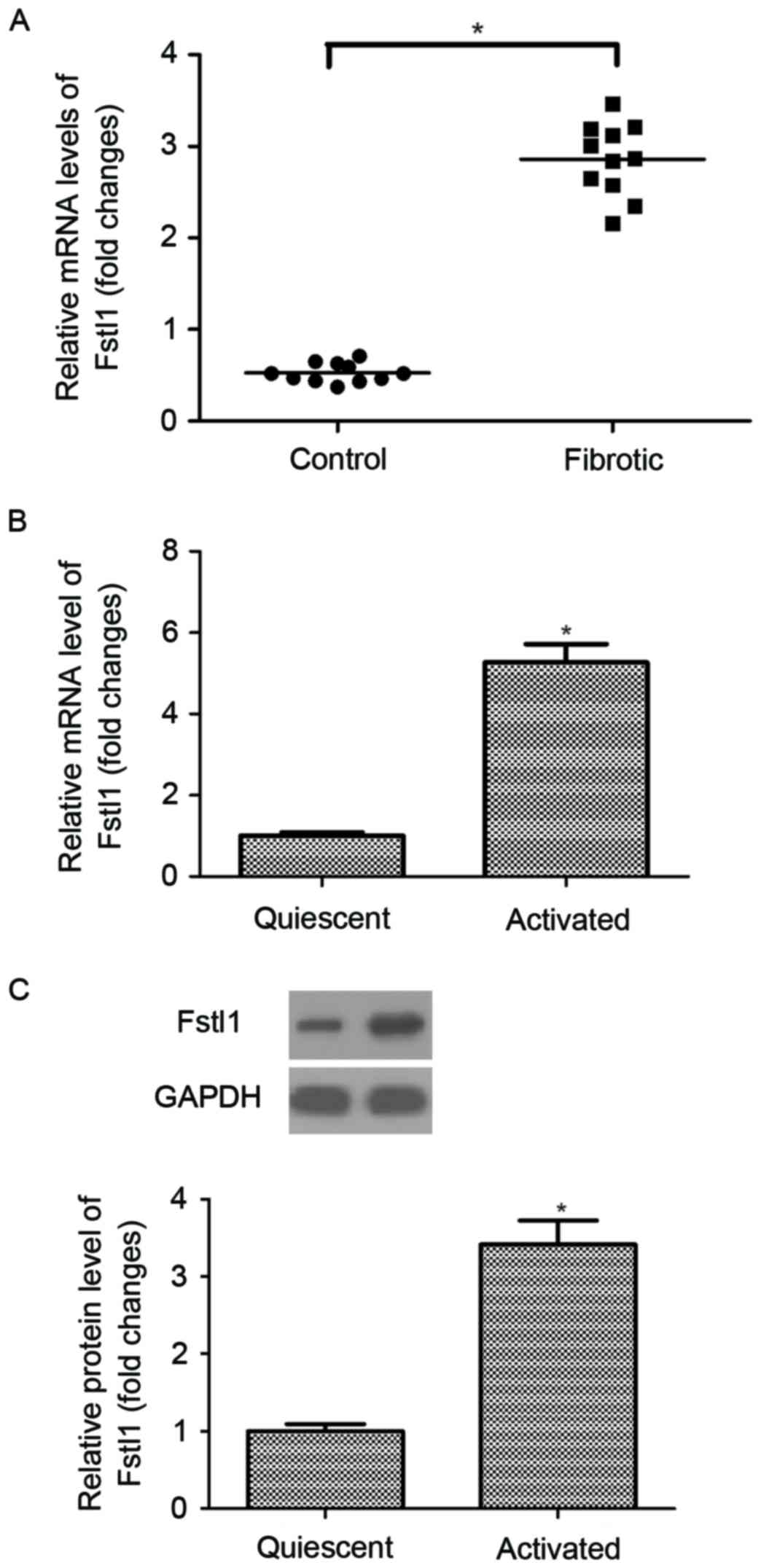
Fstl1 is highly expressed in hepatic
fibrosis tissues and activated HSCs
The authors initially measured the expression of
Fstl1 in human hepatic fibrosis tissues. As indicated in Fig. 1A, the mRNA expression levels of
Fstl1 were significantly upregulated in human hepatic fibrosis
tissues, as compared with the normal liver tissues. Moreover, the
authors observed that the expression levels of Fstl1 at both mRNA
and protein were higher in activated HSCs than that of in the
quiescent cells (Fig. 1B and
C).
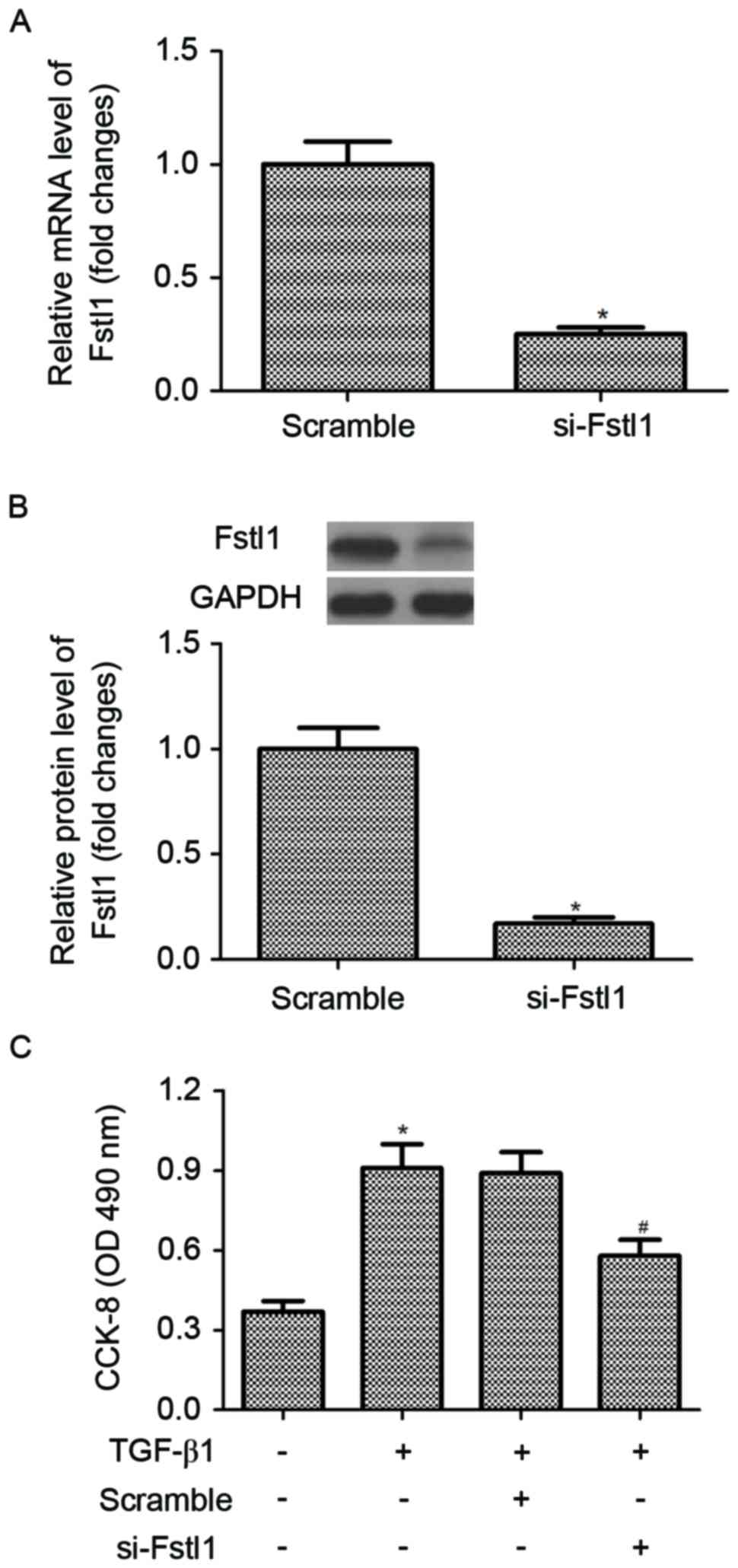
Knockdown of Fstl1 inhibits the
proliferation of activated HSC
In order to investigate the effect of Fstl1 on HSC
proliferation, HSCs were transfected with si-Fstl1 or scramble,
respectively. The efficiency of transfection was confirmed by
RT-qPCR and western blotting. The results of RT-qPCR analysis
indicated that the mRNA expression of Fstl1 was obviously decreased
in HSCs transfected with si-Fstl1 (Fig. 2A). Similarly, knockdown of Fstl1
greatly downregulated the protein expression level of Fstl1 in HSCs
(Fig. 2B). Then, the authors
performed the CCK-8 assay to detect the effect of Fstl1 on HSC
proliferation. As indicated in Fig.
2C, TGF-β1 treatment markedly promoted the proliferation of
HSCs, compared with the control group. However, knockdown of Fstl1
significantly inhibited HSC proliferation in TGF-β1-induced
HSCs.
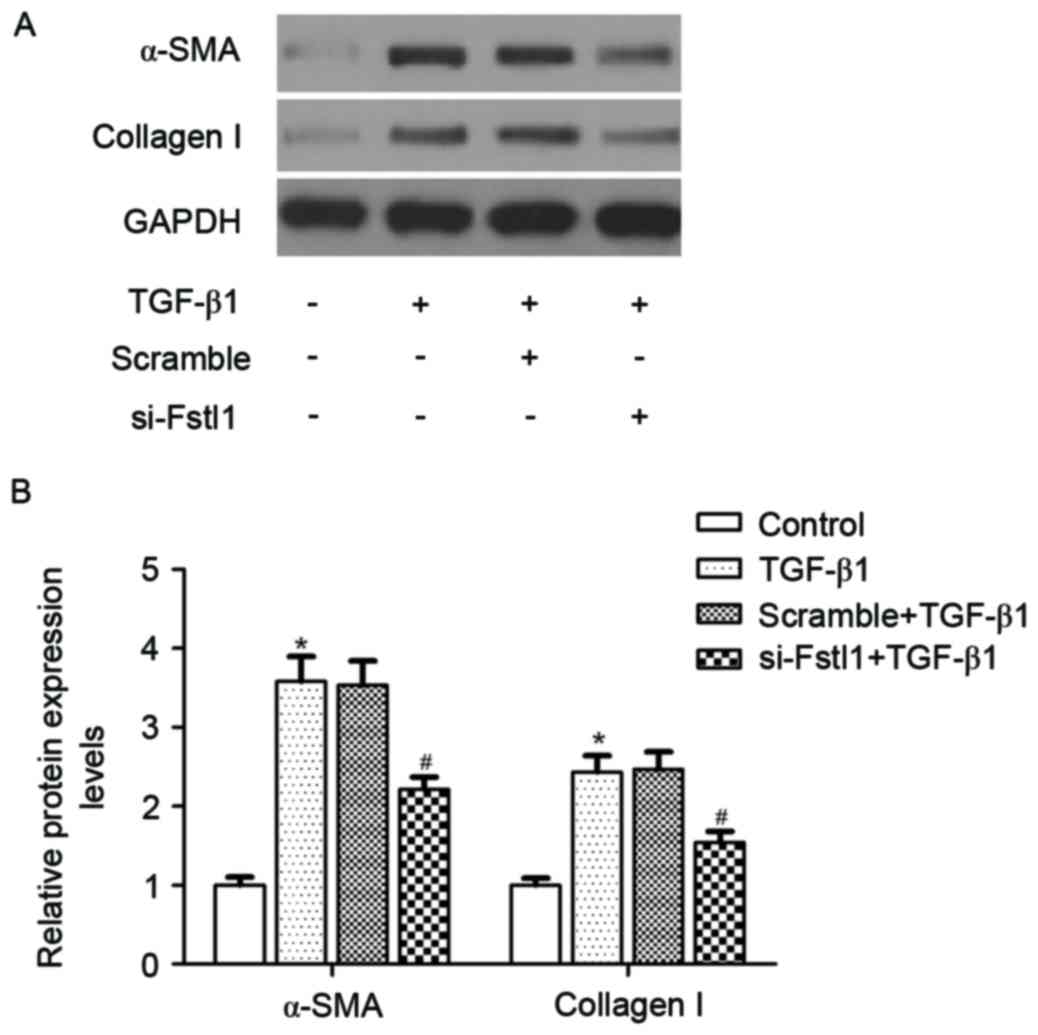
Knockdown of Fstl1 inhibits the
expression levels of α-SMA and collagen I in HSCs
The authors next investigated whether si-Fstl1
attenuated ECM expression in HSCs. The results of western blotting
analysis demonstrated that TGF-β1 obviously induced the protein
expression levels of α-SMA and type I collagen, as compared with
the control group. Meanwhile, knockdown of Fstl1 significantly
blunted TGF-β1-induced α-SMA and type I collagen expression in HSCs
(Fig. 3).
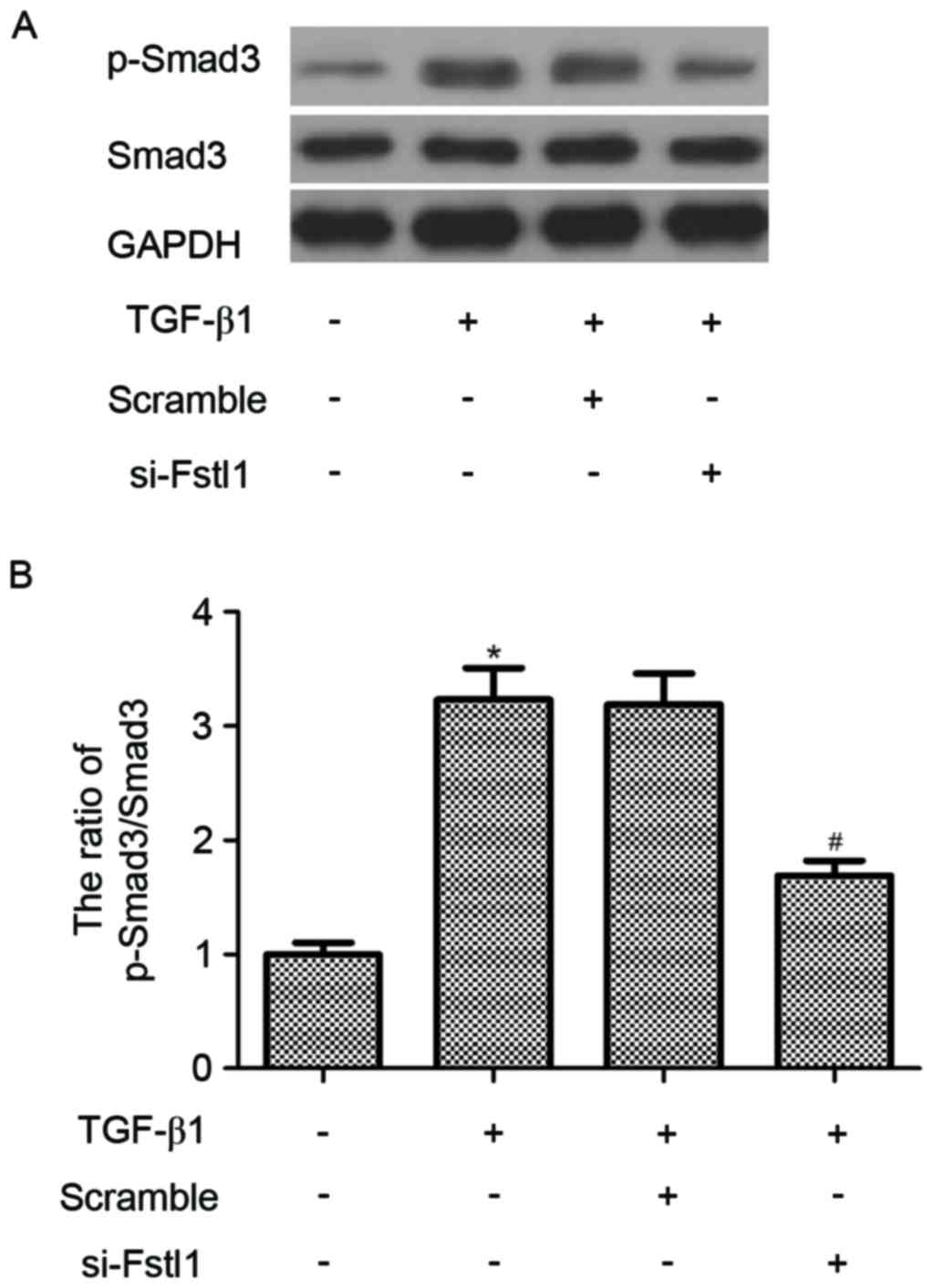
Fstl1 is involved in the regulation of
TGF-β1-mediated Smad3 signaling pathway in HSCs
To determine the molecular mechanism of Fstl1
regulates HSCs activation described above, the effect of Fstl1 on
the activation of Smad3 signaling pathway in HSCs was examined.
Western blotting indicated that TGF-β1 led to a marked increase in
Smad3 phosphorylation; however, knockdown of Fstl1 remarkably
decreased the phosphorylation level of Smad2/3 in TGF-β1-induced
HSCs (Fig. 4).
Discussion
In the current study, the results indicated that
Fstl1 was highly expressed in human hepatic fibrosis tissues and
HSCs. Furthermore, knockdown of Fstl1effectively suppressed HSC
proliferation and the protein expression levels of α-SMA and
collagen I in TGF-β1-treated HSCs. Mechanistically, knockdown of
Fstl1remarkably decreased the phosphorylation level of Smad3 in
TGF-β1-induced HSCs.
Prior studies have documented that Fstl1 contributes
to the fibrogenesis. Maruyama et al (13) confirmed that the expression of
Fstl1 was upregulated in ischemic portions of the myocardium, and
the reparative fibrotic response conferred by Fstl1 is the
consequence of its early activation of cardiac fibroblasts that
leads to myofibroblast accumulation in the infarct area. Murphy
et al (14) reported that
the protein expression level of Fstl1 was greatly increased in the
lungs of bleomycin-treated mice and in the lungs of idiopathic
pulmonary fibrosis patients. In agreement with the results above,
herein, the authors observed that Fstl1 is overexpressed in human
liver fibrotic tissues comparing with the normal liver tissues, and
the expression of Fstl1 was higher in activated HSCs than that of
quiescent HSCs. These findings suggested that Fstl1 servesa
pro-fibrotic role in liver fibrosis.
HSC activation is the primary feature during the
progression of liver fibrosis; these activated HSCs increase
proliferation and migration, and acquire contractility and
pro-inflammatory properties (15).
The induction of HSCs proliferation is stimulated by a variety of
cytokines (16–18). TGF-β1 is the most potent stimulus
for HSC-mediated fibrogenesis (19). Herein, the authors observed that
TGF-β1 treatment markedly promoted the proliferation of HSCs.
However, knockdown of Fstl1 significantly inhibited HSC
proliferation.
Liver fibrosis is characterized by an exacerbated
accumulation of deposition of ECM proteins (20). There is extensive evidence
demonstrating that TGF-β1 upregulated expression of α-SMA and
collagen I in HSCs (21–23). Recently, it was reported that the
expression of α-SMA, type I collagen and fibronectin in fibroblasts
isolated from bleomycin-treated
Fstl1+/− lungs was markedly decreased
(10). Similarly, in the present
study, knockdown of Fstl1 effectively suppressed the protein
expression levels of α-SMA and collagen I in TGF-β1-treated HSCs.
These data suggested that knockdown of Fstl1 inhibited HSC
activation by downregulating the protein expression levels of α-SMA
and collagen I in TGF-β1-treated HSCs.
The TGF-β1/Smad signaling pathway serves an
important role in the development of liver fibrosis (24–26).
During fibrogenesis, TGF-β1 exerts its biological functions via a
heteromeric receptor complex of type II and type I receptor
serine-threonine kinases. Subsequently, Smad2/3 is phosphorylated
and binds with Smad4 to form multimeric complexes, then activated
R-Smads translocate to the nucleus and induce the expression of
target genes, including ECM proteins (27). It has been reported that knockdown
of Smad3 significantly reduced TGF-β1-induced collagen I production
in HSCs (28). In the present
study, the authors indicated that TGF-β1 led to a marked increase
in Smad3 phosphorylation; however, knockdown of Fstl1 remarkably
decreased the phosphorylation level of Smad3 in TGF-β1-induced
HSCs. These data suggested that Fstl1 silencing inhibits HSCs
activation through suppressing the TGF-β1/Smad3 signaling
pathway.
In conclusion, it has been demonstrated that
Fstl1servesan important role in liver fibrosis and target deletion
of Fstl1 attenuated HSCs activation through suppressing the
TGF-β1/Smad3 signaling pathway. Therefore, Fstl1 may be a potential
therapeutic target for the treatment of liver fibrosis.
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bataller R and Brenner DA: Hepatic
stellate cells as a target for the treatment of liver fibrosis.
Semin Liver Dis. 21:437–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reeves HL and Friedman SL: Activation of
hepatic stellate cells-a key issue in liver fibrosis. Front Biosci.
7:d808–d826. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ouchi N, Oshima Y, Ohashi K, Higuchi A,
Ikegami C, Izumiya Y and Walsh K: Follistatin-like 1, a secreted
muscle protein, promotes endothelial cell function and
revascularization in ischemic tissue through a nitric-oxide
synthase-dependent mechanism. J Biol Chem. 283:32802–32811. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kudo-Saito C: FSTL1 promotes bone
metastasis by causing immune dysfunction. Oncoimmunology.
2:e265282013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adams D, Larman B and Oxburgh L:
Developmental expression of mouse Follistatin-like 1 (Fstl1):
Dynamic regulation during organogenesis of the kidney and lung.
Gene Expr Patterns. 7:491–500. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kudo-Saito C, Fuwa T, Murakami K and
Kawakami Y: Targeting FSTL1 prevents tumor bone metastasis and
consequent immune dysfunction. Cancer Res. 73:6185–6193. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chan QK, Ngan HY, Ip PP, Liu VW, Xue WC
and Cheung AN: Tumor suppressor effect of follistatin-like 1 in
ovarian and endometrial carcinogenesis: A differential expression
and functional analysis. Carcinogenesis. 30:114–121. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y,
Li X, Dong S, Liu X, Li X, et al: Blocking follistatin-like 1
attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med.
212:235–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang W, Yang M, Song L, Shen K, Wang H,
Gao X, Li M, Niu W and Qin X: Isolation and culture of hepatic
stellate cells from mouse liver. Acta Biochim Biophys Sin
(Shanghai). 46:291–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maruyama S, Nakamura K, Papanicolaou KN,
Sano S, Shimizu I, Asaumi Y, van den Hoff MJ, Ouchi N, Recchia FA
and Walsh K: Follistatin-like 1 promotes cardiac fibroblast
activation and protects the heart from rupture. EMBO Mol Med.
8:949–966. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Murphy N, Gaynor KU, Rowan SC, Walsh SM,
Fabre A, Boylan J, Keane MP and McLoughlin P: Altered expression of
bone morphogenetic protein accessory proteins in murine and human
pulmonary fibrosis. Am J Pathol. 186:600–615. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Svegliati-Baroni G, Ridolfi F, Hannivoort
R, Saccomanno S, Homan M, De Minicis S, Jansen PL, Candelaresi C,
Benedetti A and Moshage H: Bile acids induce hepatic stellate cell
proliferation via activation of the epidermal growth factor
receptor. Gastroenterology. 128:1042–1055. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan F, Peltekian KM and Peterson TC:
Effect of interferon-alpha, ribavirin, pentoxifylline, and
interleukin-18 antibody on hepatitis C sera-stimulated hepatic
stellate cell proliferation. J Interferon Cytokine Res. 28:643–651.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bendia E, Benedetti A, Baroni GS,
Candelaresi C, Macarri G, Trozzi L and Di Sario A: Effect of
cyanidin 3-O-beta-glucopyranoside on hepatic stellate cell
proliferation and collagen synthesis induced by oxidative stress.
Dig Liver Dis. 37:342–348. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li J, Si HF, Lü X, Guo A and Jiang H:
Suppressive effects of leflunomide on leptin-induced TIMP-1
production involves on hepatic stellate cell proliferation and
apoptosis. Eur J Pharmacol. 580:63–69. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He Y, Huang C, Sun X, Long XR, Lv XW and
Li J: MicroRNA-146a modulates TGF-beta1-induced hepatic stellate
cell proliferation by targeting SMAD4. Cell Signal. 24:1923–1930.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wiercinska E, Wickert L, Denecke B, Said
HM, Hamzavi J, Gressner AM, Thorikay M, ten Dijke P, Mertens PR,
Breitkopf K and Dooley S: Id1 is a critical mediator in
TGF-beta-induced transdifferentiation of rat hepatic stellate
cells. Hepatology. 43:1032–1041. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He P, Yu ZJ, Sun CY, Jiao SJ and Jiang HQ:
Knockdown of eIF3a attenuates the pro-fibrogenic response of
hepatic stellate cells induced by TGF-β1. Cell Mol Biol
(Noisy-le-grand). 62:107–111. 2016.PubMed/NCBI
|
|
23
|
Ma HH, Yao JL, Li G, Yao CL, Chen XJ and
Yang SJ: Effects of c-myb antisense RNA on TGF-beta1 and beta1-I
collagen expression in cultured hepatic stellate cells. World J
Gastroenterol. 10:3662–3665. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo JY, Zhang LT, Ming-Xia LU, Yan XM,
Xiao P and Medicine DO: Smurf 2 impact on TGF-β/Smad pathway in
liver fibrosis. Med Recapitulate. 17:125–131. 2015.
|
|
25
|
Zhou Y, Tong X, Ren S, Wang X, Chen J, Mu
Y, Sun M, Chen G, Zhang H and Liu P: Synergistic anti-liver
fibrosis actions of total astragalus saponins and glycyrrhizic acid
via TGF-β1/Smads signaling pathway modulation. J Ethnopharmacol.
190:83–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang JW and Wook K: The role of Pin1 in
liver fibrosis: Regulation of Tgf-β1 expression and Smad2/3
phosphorylation. Toxicol Lett. 229 Suppl:S2402014. View Article : Google Scholar
|
|
27
|
Breitkopf K, Weng H and Dooley S:
TGF-β/Smad-signaling in liver cells: Target genes and inhibitors of
two parallel pathways. Signal Transduction. 6:329–337. 2006.
View Article : Google Scholar
|
|
28
|
Zhang L, Liu C, Meng XM, Huang C, Xu F and
Li J: Smad2 protects against TGF-β1/Smad3-mediated collagen
synthesis in human hepatic stellate cells during hepatic fibrosis.
Mol Cell Biochem. 400:17–28. 2015. View Article : Google Scholar : PubMed/NCBI
|