Introduction
Pulmonary hypertension (PH) is a pathophysiological
syndrome caused by heterogenous diseases and diverse pathogenesis,
with the dominant feature of continuous increase of pulmonary
vascular resistance. Characterized by its clinical manifestations
of increased load behind right ventricle, decreased activity
endurance and even death out of heart failure, PH is a severe
chronic pulmonary circulatory illness which could contribute to
potential fatality. Provided that no effective treatment is
conducted, the prognosis for majority of PH patients will be
extremely poor, with ~15% mortality rate within 1 year on modern
therapy (1–4).
Hypoxic pulmonary hypertension (HPH) is a clinically
common type of PH, with frequent occurrence in various chronic
pulmonary ailments. With interaction of long-term anoxia, chronic
inflammation, a variety of active vascular substances and growth
factors, the structure of pulmonary vessels may undergo
transformation, resulting in vascular remodeling. On the other
hand, remodeling of pulmonary arterial smooth muscle cells (PASMCs)
is a key feature known to result from an imbalance between
apoptosis and cell proliferation (5).
As a member of the family of serine/threonine
protein kinases, protein kinase C (PKC) was first discovered by
Nishizuka in 1997 (6). So far, at
least 11 subtypes of PKC have been isolated and purified from
different species and genuses of tissue organs (7). According to dissimilarity of
molecular structure and sensitivity to activators, PKC subtypes
could be grouped into three categories involving the conventional
(cPKCα, βI, βII and γ), the novel (nPKCδ, ε, η and θ) as well as
the atypical (aPKCλ/ι and ζ). The classical PKCs are diacylglycerol
(DAG) and Ca2+-dependent enzymes; whereas, the novel
PKCs require DAG, but not calcium, for activation. The atypical are
not responsive to activation by DAG or calcium, but are activated
by other lipid-derived second messengers (8).
PKC may exist in almost all the tissue cells,
including PASMCs (9,10). In addition, PKC not only
participates in the process of cell growth, differentiation,
apoptosis and contraction of smooth muscle cells (SMCs), but also
serves a significant part in signal transduction pathways mediated
by hormones, neurotransmitters, growth factors and antigens
(11,12). PKC has been reported to act as a
signaling mediator in hypoxia-induced cell proliferation (13). Recently, PKCα has been demonstrated
to serve a vital role in proliferation of diverse cells, including
PASMCs (14–17). An study investigating ox pulmonary
arterial smooth muscle cells demonstrated that PKC initiates and
promotes PASMC proliferation (14). In a study of rat thoracic aortic
smooth muscle cells, however, Sasaguri et al (18) revealed that PKC activation inhibits
SMC proliferation. These studies suggested that PKC and its
mediated cell signaling pathways may occupy an important position
in SMC proliferation, but with inconsistent and conflicting
findings.
To investigate hypoxia-induced PASMC proliferation,
the present study aimed to establish an external model of hypoxic
pulmonary hypertension and to observe the change and underlying
molecular mechanism of PKCα expression in hypoxia-induced rat
PASMCs, as well as its impact upon PASMC proliferation. The present
study may further uncover the molecular mechanism of PH pulmonary
vascular remodeling, providing a theoretical basis for its
prevention and treatment.
Materials and methods
Animals and agents
A total of 20 adult rats (age, 8 weeks; weight, ~200
g) purchased from the Experimental Animal Center of Shanxi Medical
University (Taiyuan, China) were maintained in a temperature-(22°C)
and humidity (between 60 and 65%)-controlled room on a 12-h
light/dark cycle with free access to food and water for 1 week
prior to use. All procedures were approved by the Animal Management
Guidelines of the Ministry of Health of the People's Republic of
China, in accordance with the National Institutes of Health Guide
for the Care and Use of Laboratory Animals. Dulbecco's modified
Eagle's medium (DMEM) and 20% fetal bovine serum (FBS) were
obtained from Hyclone; GE Healthcare Life Sciences (Logan, UT,
USA). Monoclonal antibodies against ERK (cat. no. 9102) and
phosphorylated (p)-ERK (cat. no. 9101) were from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Polyclonal antibodies against
smoothlin (cat. no. sc-20481), PKCα (cat. no. sc-208) were from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Polyclonal
antibody against β-actin (cat. no. AP0060) was from Bioworld
Technology, Co., Ltd. (Nanjing, China). Polyclonal antibodies
against smooth muscle myosin heavy chain (cat. no. ab53219) were
obtained from Abcam (Cambridge, MA, USA).
Isolation and culture of PASMCs
Rat PASMCs were isolated and cultured in accordance
with previously described methods (19). Rats were anaesthetized by
intraperitoneal injection of pentobarbital sodium (Sinopharm
Chemical Reagent Co., Ltd., Beijing, China; 50 mg/kg body weight),
then the main trunk of pulmonary arteries and the right and left
branches were isolated under a dissecting light microscope (Olympus
Corporation, Tokyo, Japan). After connective tissues of arteries
were cleaned and vessels cut open longitudinally, luminal
endothelia were removed by gentle scraping with cotton swabs. The
isolated pulmonary arteries were dissected into small pieces of 1×1
mm, maintained in DMEM supplemented with 20% FBS and incubated in a
humidified atmosphere with 5% CO2 at 37°C. Culture
medium was changed twice per week and cells were harvested with
trypsin (0.25%; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) containing EDTA. Passages ranging from 4 to 6 were used for
all experiments, which were divided into three groups: Normoxia,
hypoxia and control. In the normoxia group, PASMCs were placed at
37°C in a humidified atmosphere containing 5% CO2. In
the hypoxia group, PASMCs were placed into three-gas chambers
containing 3% O2, 5% CO2 and 92%
N2 for 24, 48 and 72 h, respectively. In the control
group, cells were pre-treated with drugs (12-myristate 13-acetate,
safingol, PD98059 and U0126) and placed into three-gas chambers
containing 3% O2, 5% CO2 and 92%
N2 for 72 h. Prior to exposure to hypoxia or treatment,
cells were incubated in DMEM with free FBS for 24 h and then
exposed to hypoxia or treated in DMEM supplemented with 2% FBS.
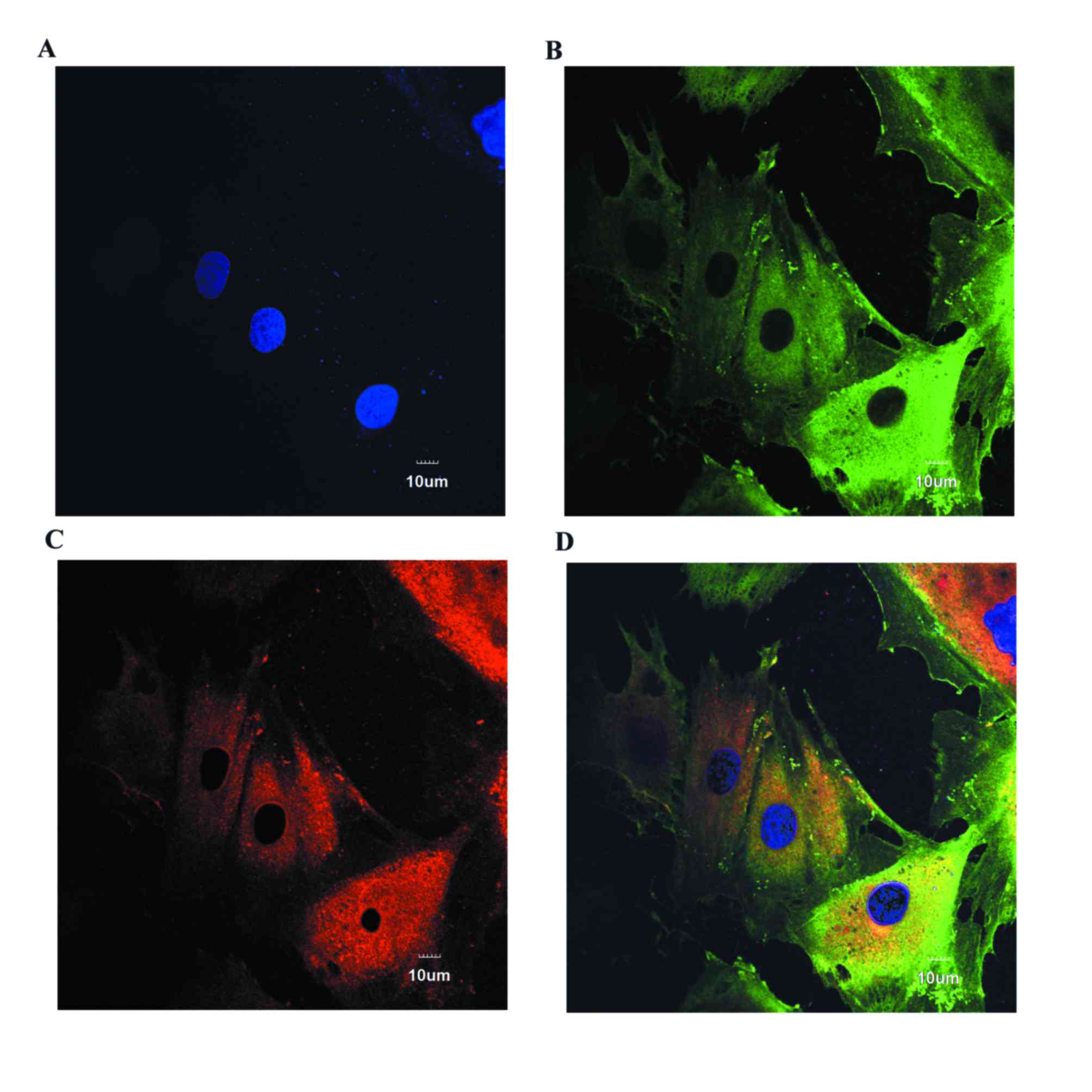
Immunofluorescence staining of
PASMCs
PASMCs were plated in glass chambers, fixed with 4%
paraformaldehyde for 10 min, and added to 0.2% permeable Triton
X-100 for 15 min. Following three washes in PBS, cells were blocked
with goat serum (Solarbio, Beijing, China) for 1 h, followed by
incubation with anti-smoothlin (1:100) and anti-smooth muscle heavy
chain (1:80) primary antibodies overnight at 4°C. Cells were then
washed with PBS three times, followed by incubation with
fluorescein isothiocyanate-conjugated secondary antibody (cat. no.
A16000; dilution, 1:500; Thermo Fisher Scientific, Inc.) and
tetramethylrhodamine-conjugated secondary antibody (cat. no.
A16040; dilution, 1:500; Thermo Fisher Scientific, Inc.) for 1 h in
the dark. Following three washes with PBS, nuclei were stained with
DAPI for 10 min and observed using confocal laser scanning
microscope. All the aforementioned procedures were performed at
room temperature.
Cell proliferation assay
PASMCs at a density of 5×103 cells were
plated into 96-well glass chamber plates. Cell proliferation in the
normoxia, hypoxia and control groups were measured. MTT (20 µl; 5
mg/ml) was added to each well and the slides were incubated in a
humidified incubator with 5% CO2 at 37°C for 4 h. At the
end of incubation, the supernatant was removed and dimethyl
sulfoxide (DMSO; 150 µl/well) was added to the plates to solubilize
for 10 min. The optical densities were determined using a
multi-well scanning spectrophotometer (Spectra Max Plus 384,
Molecular Devices, LLC, Sunnyvale, CA, USA) at 490 nm.
Western blot analysis
After various treatments, cells were washed three
times in cold PBS, harvested and scraped with cell lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China) for 2 h at
4°C. The lysates were then centrifuged at 13,000 × g for 20 min at
4°C, the supernatants collected and the total protein
concentrations were determined by the Bicinchoninic Acid Protein
Assay kit (Beyotime Institute of Biotechnology, Shanghai, China)
following the manufacturer's protocol. Equal amounts of proteins
(30 µg) were separated by 10% SDS-PAGE, and then transferred to
polyvinylidene difluoride membranes. After blocking with 5% bull
serum albumin (Solarbio) at room temperature for 1 h, membranes
were incubated overnight at 4°C with antibodies specific for rabbit
anti-p-ERK1/2 (1:1,000), rabbit anti-ERK (1:1,000), rabbit
anti-PKCα (1:1,000) and rabbit anti-β-actin (1:2,000).
Subsequently, the membranes were incubated with a goat anti-rabbit
peroxidase-conjugated IgG secondary antibody (cat. no. ZB-2301;
dilution, 1:5,000; ZSGB-BIO, Beijing, China) for 1 h at room
temperature. The expressed protein amount was determined through
p-ERK1/2/ERK1/2 for ERK1/2 and PKCα/β-actin for PKCα. Proteins were
visualized using enhanced chemiluminescence (Pierce; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Proteins were quantified using
a Gel Doc XR system (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
using Quantity One software (version 4.62; Bio-Rad Laboratories,
Inc.).
Drugs
The following drugs were used for the hypoxia
treated control cells. Prior to hypoxic treatment, cells were
treated with the PKCα promoter 12-myristate 13-acetate (PMA; 0.4
mM; Alomone Labs, Israel), the PKCα special inhibitor safingol (0.6
mM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), the
extracellular signal-regulated kinase kinase (MEK1/2) inhibitor
PD98059 (20 mM, Cell Signaling Technology, Inc.) or U0126 (5 mM,
Cell Signaling Technology, Inc.) and incubated in a humidified
atmosphere with 5% CO2 at 37°C for 30 min one time.
Statistical analysis
Data are expressed as mean ± standard deviation.
Comparisons between two groups was made with an unpaired two-tailed
Student's t-test. Comparisons between multiple groups was made with
a one-way analysis of variance followed by Dunnett or Tukey test.
SPSS software, version 20.0 (IBM Corp., Armonk, NY, USA) was used
for all statistical analysis. P<0.05 was considered to indicate
a statistically significant difference.
Results
Activation of hypoxia on PASMC
proliferation
Immunofluorescence staining determined that PASMCs
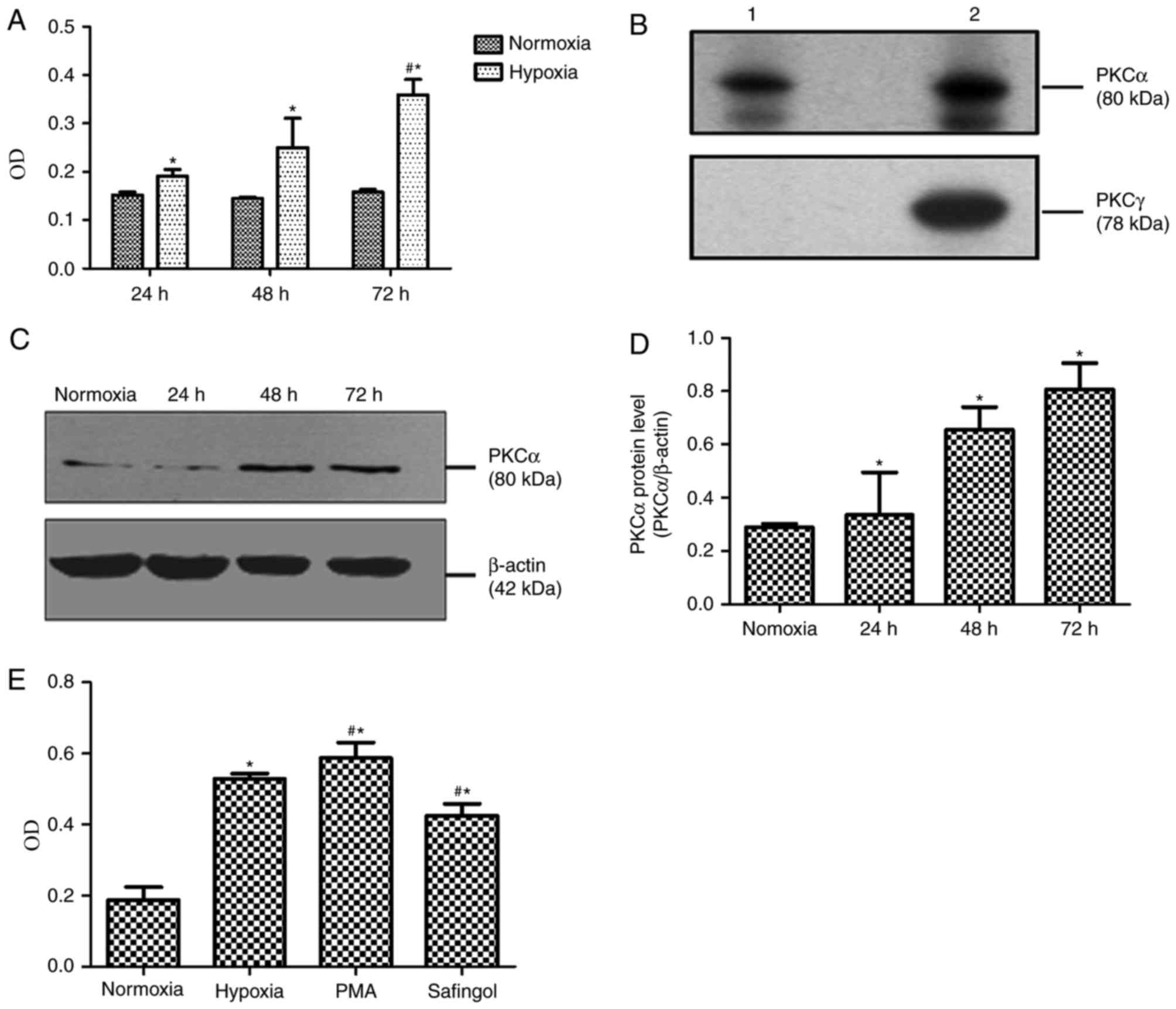
stain positively for smooth muscle markers (Fig. 1). MTT demonstrated that the optical
densities (ODs) of cells exposed to hypoxia with 3% O2
for 24, 48 and 72 h, respectively, were significantly increased,
compared with cells exposed to normoxia with 21% O2 for
24, 48 and 72 h (Fig. 2A). The
mean OD values of cells exposed to normoxia for 24, 48 and 72 h
were 0.152±0.006, 0.145±0.003 and 0.158±0.005, respectively;
however, the OD values of cells exposed to hypoxia for 24, 48 and
72 h were 0.191±0.014, 0.250±0.060 and 0.359±0.032, respectively
(Fig. 2A). Compared with cells
exposed to hypoxia for 24 h, MTT absorbance of PASMCs in hypoxic
exposure for 72 h was significantly increased (P<0.05; Fig. 2A).
Upregulation of PKCα in
hypoxia-induced PASMCs
The results of western blotting demonstrated that
PKCα (80 kD) was present in normal rat PASMCs (Fig. 2B). As presented in Fig. 2C and D, PKCα protein expression
levels in hypoxic group for 24, 48 and 72 h were 0.336±0.160,
0.656±0.085 and 0.808±0.098, respectively. On the other hand, PKCα
protein levels in cells exposed to normoxia for 72 h were
0.290±0.013. Compared with the 72-h normoxia group, PKCα protein
expression levels in PASMCs exposed to hypoxia were markedly
increased in a time-dependent manner (P<0.05).
PASMC proliferation induced by
PKCα
To examine the role of PKCα in hypoxia-induced
proliferative responses, greatly proliferated PASMCs in hypoxia for
72 h were selected, which were treated with a PKCα promoter (PMA)
and inhibitor (safingol). According to the MTT findings, the mean
OD values in the 72-h normoxia, 72-h hypoxia, hypoxic PMA control
and hypoxic safingol groups were demonstrated to be 0.187±0.037,
0.529±0.015, 0.587±0.044 and 0.426±0.033, respectively (Fig. 2E). In addition, the OD value in the
hypoxic PMA control group was significantly increased compared with
the hypoxia group (P<0.05), whereas the OD value in the hypoxic
safingol group was lower than that in hypoxic PMA control
(P<0.05; Fig. 2E).
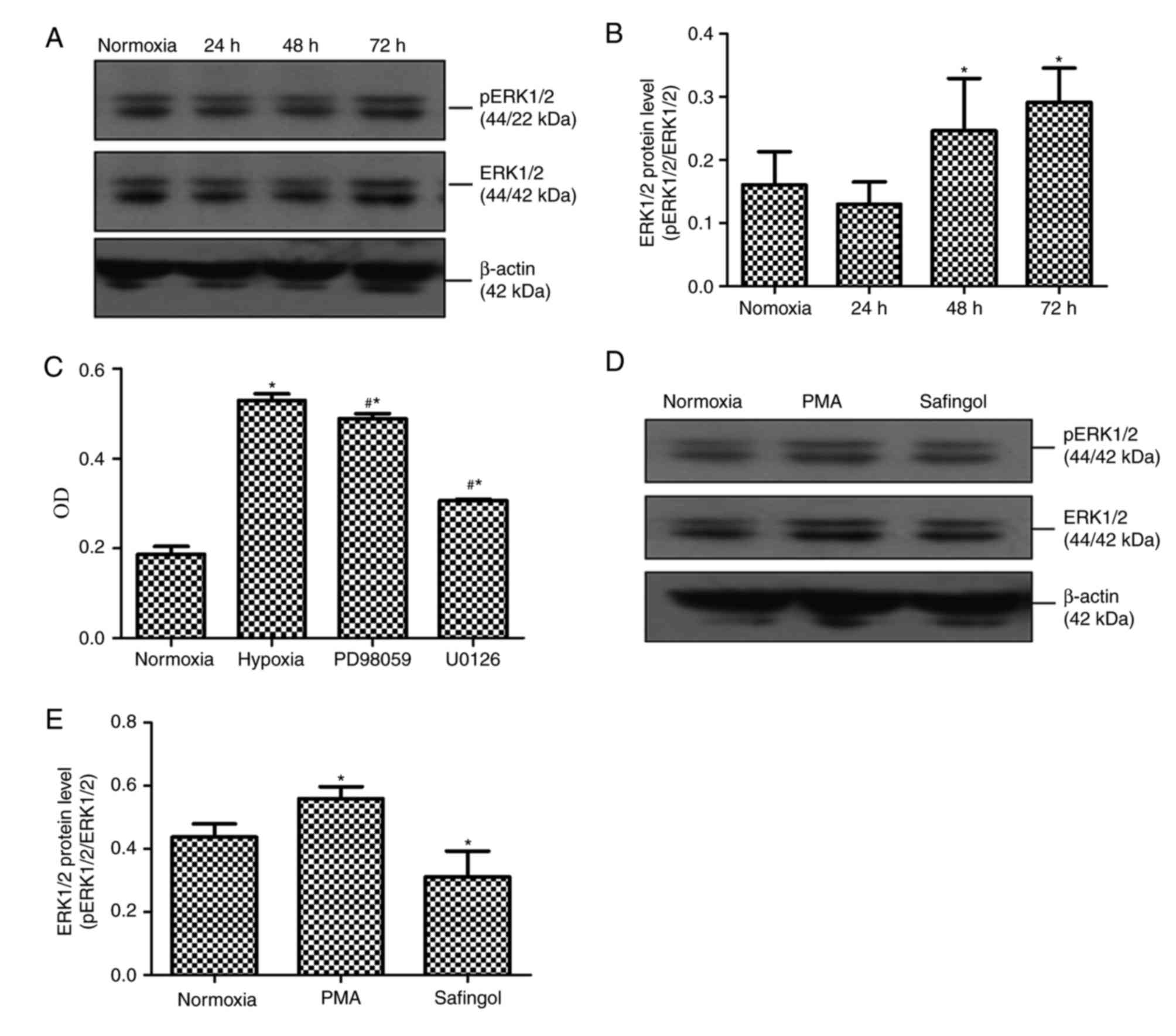
PASMC proliferation activated by
hypoxia through ERK1/2 phosphorylation
To determine the cellular pathway that may be
involved in the PASMC proliferation process, the present study
evaluated the possible modulation of ERK1/2 in signaling by western
blotting and MTT. According to the western blotting results,
p-ERK1/2 expression levels in cells exposed to hypoxia for 24, 48
and 72 h were 0.130±0.035, 0.246±0.083 and 0.291±0.054,
respectively; compared with cells exposed to normoxia for 72 h,
which was 0.160±0.053 (Fig. 3A and
B). Compared with the 72-h normoxia group, p-ERK1/2 expression
in PASMCs after exposure to hypoxia for 48 and 72 h was markedly
upregulated (P<0.05, Fig. 3A and
B). When PASMCs exposed to hypoxia for 72 h were pretreated
with the MEK1/2 inhibitor PD98059 or U0126, the mean OD values in
the normoxia 72 h, hypoxia 72 h, hypoxic PD98059 control and
hypoxic U0126 control groups were 0.187±0.017, 0.529±0.015,
0.489±0.011 and 0.306±0.004, respectively. Compared with the
hypoxia for 72 h group, the mean OD values in the hypoxic PD98059
control and hypoxic U0126 control groups were significantly
attenuated (P<0.05; Fig.
3C).
ERK1/2 phosphorylation stimulated by
PKCα
To investigate a potential association between PKCα
and ERK1/2, the present study tested the modulation of ERK1/2
expression through hypoxic PASMCs pretreated with a PKCα promoter
and inhibitor. Western blotting results indicated that p-ERK1/2
protein expression levels in the normoxia for 72 h, hypoxic PMA
control and hypoxic safingol control groups were demonstrated to be
0.437±0.042, 0.558±0.039 and 0.311±0.082, respectively. p-ERK1/2
protein expression levels in the presence of the PKCα promoter was
significantly increased compared with the other two hypoxic groups
(P<0.05). Compared with the other two groups, ERK1/2
phosphorylation in the presence of PKCα inhibitor was greatly
reduced (P<0.05, Fig. 3D and
E).
Discussion
The present study primarily demonstrated that PKCα
was upregulated in hypoxia-induced rat PASMC proliferation, and
through activation of ERK1/2 phosphorylation, upregulated PKCα
induced the proliferation of PASMCs, thus contributing to the
occurrence of pulmonary artery hypertension.
Chronic hypoxic pulmonary hypertension is a common
clinical problem, which has an association with increased vascular
tone, and imbalance between proliferation and apoptosis of PASMCs
(20,21). By MTT assay to determine PASMC
proliferation, the present study demonstrated that hypoxia may
induce PASMC proliferation, which is in consistent with findings by
Preston et al and Li et al (22,23).
As a type of protein kinase, PKC is expressed in
many tissue cells. To date, at least 11 subtypes of PKC have been
identified, among which each type has a different function. As a
classical subtype of PKC, PKCα is widely distributed and present in
all the tissues, and has a connection with cell apoptosis,
proliferation and migration (24).
Although PKCα has been reported to serve an important role in cell
proliferation, including in PASMCs, there is still a lack of
research on the association between PKCα subtypes and signal
transduction pathways in hypoxia-induced PASMC proliferation.
Using western blotting in the present study, PKCα
was demonstrated to be present in rat PASMCs. In comparison, the
presentation of various PKC subtypes including α, β and γ has been
validated in a study by Barman et al (25). Through further investigation, the
present study demonstrated that the expression level of PKCα in
hypoxia-induced PASMCs was increased. In addition, the PKCα special
inhibitor safingol suppressed hypoxia-induced PASMC proliferation,
and the PKCα promoter PMA significantly increased proliferation.
Therefore, it may be hypothesized that the promotion of PKCα
expression levels serve a vital role in hypoxia-induced PASMC
proliferation. Dempsey et al (26) demonstrated that hypoxia might
activate PKC in neonatal bovine PASMCs, and the activated PKC could
foster SMC proliferation through stimulating growth factors such as
insulin-like growth factor-1. In addition, PKC with PKCα in
particular has been demonstrated to serve a critical part in
hypoxia and mitogen-induced PASMC proliferation (27). Furthermore, hypoxia-activated PKC
in PASMCs has also been identified to be involved in
hypoxia-induced PASMC proliferation, thus participating in the
formation of hypoxic PH (28). The
findings resulting from the present study fit in with those
reported in the aforementioned investigations.
The mitogen-activated protein kinase (MAPK) family
is comprised of ERK1/2, c-Jun N-terminal kinase (JNK) and p38 MAPK.
MAPK has been reported to exhibit a distinct increase of activation
in chronic hypoxic pulmonary arteries (29–31).
In recent years, ERK1/2 has been identified to be activated by a
variety of extracellular stimuli, and is involved in
hypoxia-induced PASMC proliferation (20,22,23,32–34).
PD98059 and U0126 are universally used as MEK1/2 inhibitors, the
role of which is to inhibit mitochondrial metabolism, thus using up
p-ERK. With ERK1/2 being the only known MEK1/2 downstream substrate
(21), PD98059 and U0126 were used
to block ERK1/2. In the present study, PASMC proliferation was
markedly increased with exposure to hypoxia for 48 and 72 h, and
p-ERK1/2 expression was also significantly increased. However,
PD98059 and U0126 markedly weakened this proliferative response,
which suggested that hypoxia may activate ERK1/2, thus promoting
PASMC proliferation. Therefore, ERK1/2 may serve a vital part in
hypoxia-induced PASMC proliferation.
It has been reported that activated PKCα induces the
proliferation of capillary endothelial cells through the ERK1/2
signaling pathway (35). Another
investigation demonstrated that activated PKCα in human saphenous
vein SMCs strengthened the activity of MAPKs, which could
contribute to cell proliferation (36). However, relevant information
concerning the role and mechanism of PKCα in PH-induced PASMCs
proliferation is not yet available. The present study demonstrated
that PMA-induced PKCα activation upregulated the expression of
p-ERK1/2, thus increasing PASMC proliferation. On the other hand,
the PKCα inhibitor safingol may inhibit ERK1/2 phosphorylation,
suppressing PASMC proliferation. Therefore, increased expression of
PKCα may activate ERK1/2, leading to PASMC proliferation and PH
formation.
In conclusion, protein expression levels of PKCα in
rats were upregulated in hypoxia-induced rat PASMCs. Through
activating ERK1/2, PKCα served part in PASMC proliferation, which
adds to the understanding of its mechanism in PH formation and lays
a theoretical basis for prevention as well as treatment of HPH.
Whether PKCα could be considered as a target for PH treatment in
the future still requires further studies on the role of other
subtypes in PH formation. Limitations of the present study lie in
that early pathological variations of PH are primarily involved in
peripheral pulmonary artery. The present study would be of much
more significance if distal PASMCs could be obtained.
Acknowledgements
The present study work was supported by the Natural
Science Foundation of Shanxi Province of China (grant no.
201601D011110).
References
|
1
|
Hoette S, Jardim C and Souza Rd: Diagnosis
and treatment of pulmonary hypertension: An update. J Bras Pneumol.
36:795–811. 2010.(In English, Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shi Y, Wang C, Han S, Pang B, Zhang N,
Wang J and Li J: Determination of PKC isoform-specific protein
expression in pulmonary arteries of rats with chronic
hypoxia-induced pulmonary hypertension. Med Sci Monit.
18:BR69–BR75. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nie X, Shi Y, Yu W, Xu J, Hu X and Du Y:
Phosphorylation of PTEN increase in pathological right ventricular
hypertrophy in rats with chronic hypoxia induced pulmonary
hypertension. Chin Med J (Engl). 127:338–342. 2014.PubMed/NCBI
|
|
4
|
McLaughlin VV, Archer SL, Badesch DB,
Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH,
Rosenson RS, et al: ACCF/AHA 2009 expert consensus document on
pulmonary hypertension a report of the American College of
Cardiology Foundation Task Force on Expert Consensus Documents and
the American Heart Association developed in collaboration with the
American College of Chest Physicians; American Thoracic Society,
Inc.; and the pulmonary hypertension association. J Am Coll
Cardiol. 53:1573–1619. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang S, Wang Y, Jiang J, Wang R, Li L, Qiu
Z, Wu H and Zhu D: 15-HETE protects rat pulmonary arterial smooth
muscle cells from apoptosis via the PI3K/Akt pathway.
Prostaglandins Other Lipid Mediat. 91:51–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hurley JH, Newton AC, Parker PJ, Blumberg
PM and Nishizuka Y: Taxonomy and function of C1 protein kinase C
homology domains. Protein Sci. 6:477–480. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nitti M, Pronzato MA, Marinari UM and
Domenicotti C: PKC signaling in oxidative hepatic damage. Mol
Aspects Med. 29:36–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan HC, Fernandez-Hernando C and Lai JH:
Protein kinase C isoforms in atherosclerosis: Pro- or
anti-inflammatory? Biochem Pharmacol. 88:139–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haller H, Baur E, Quass P, Behrend M,
Lindschau C, Distler A and Luft FC: High glucose concentrations and
protein kinase C isoforms in vascular smooth muscle cells. Kidney
Int. 47:1057–1067. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Webb BL, Lindsay MA, Seybold J, Brand NJ,
Yacoub MH, Haddad EB, Barnes PJ, Adcock IM and Giembycz MA:
Identification of the protein kinase C isoenzymes in human lung and
airways smooth muscle at the protein and mRNA level. Biochem
Pharmacol. 54:199–205. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Newton AC: Protein kinase C: Structure,
function, and regulation. J Biol Chem. 270:28495–28498. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Throckmorton DC, Packer CS and Brophy CM:
Protein kinase C activation during Ca2+-independent
vascular smooth muscle contraction. J Surg Res. 78:48–53. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang H, Okamoto M, Panzhinskiy E, Zawada
WM and Das M: PKCδ/midkine pathway drives hypoxia-induced
proliferation and differentiation of human lung epithelial cells.
Am J Physiol Cell Physiol. 306:C648–C658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agbani EO, Coats P, Mills A and Wadsworth
RM: Peroxynitrite stimulates pulmonary artery endothelial and
smooth muscle cell proliferation: Involvement of ERK and PKC. Pulm
Pharmacol Ther. 24:100–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mandegar M, Fung YC, Huang W, Remillard
CV, Rubin LJ and Yuan JX: Cellular and molecular mechanisms of
pulmonary vascular remodeling: Role in the development of pulmonary
hypertension. Microvasc Res. 68:75–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamamoto M, Acevedo-Duncan M, Chalfant CE,
Patel NA, Watson JE and Cooper DR: The roles of protein kinase C
beta I and beta II in vascular smooth muscle cell proliferation.
Exp Cell Res. 240:349–358. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu BH, Yao ZX, Luo SJ, Jiang LM, Xiao JW,
Liu SC, Liu JB, Sun JM and Pei ZY: Effects of antisense
oligonucleotides of PKC-alpha on proliferation and apoptosis of
HepG2 in vitro. Hepatobiliary Pancreat Dis Int. 4:75–79.
2005.PubMed/NCBI
|
|
18
|
Sasaguri T, Kosaka C, Hirata M, Masuda J,
Shimokado K, Fujishima M and Ogata J: Protein kinase C-mediated
inhibition of vascular smooth muscle cell proliferation: The
isoforms that may mediate G1/S inhibition. Exp Cell Res.
208:311–320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pisarcik S, Maylor J, Lu W, Yun X, Undem
C, Sylvester JT, Semenza GL and Shimoda LA: Activation of
hypoxia-inducible factor-1 in pulmonary arterial smooth muscle
cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol.
304:L549–L561. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li XW, Hu CP, Wu WH, Zhang WF, Zou XZ and
Li YJ: Inhibitory effect of calcitonin gene-related peptide on
hypoxia-induced rat pulmonary artery smooth muscle cells
proliferation: Role of ERK1/2 and p27. Eur J Pharmacol.
679:117–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ripple MO, Kim N and Springett R: Acute
mitochondrial inhibition by mitogen-activated protein
kinase/extracellular signal-regulated kinase kinase (MEK) 1/2
inhibitors regulates proliferation. J Biol Chem. 288:2933–2940.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Preston IR, Hill NS, Warburton RR and
Fanburg BL: Role of 12-lipoxygenase in hypoxia-induced rat
pulmonary artery smooth muscle cell proliferation. Am J Physiol
Lung Cell Mol Physiol. 290:L367–L374. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li GW, Xing WJ, Bai SZ, Hao JH, Guo J, Li
HZ, Li HX, Zhang WH, Yang BF and Wu LY: The calcium-sensing
receptor mediates hypoxia-induced proliferation of rat pulmonary
artery smooth muscle cells through MEK1/ERK1,2 and PI3K pathways.
Basic Clin Pharm Toxicol. 108:185–193. 2011. View Article : Google Scholar
|
|
24
|
Wang P, Xu J, Hou Z, Wang F, Song Y, Wang
J, Zhu H and Jin H: miRNA-34a promotes proliferation of human
pulmonary artery smooth muscle cells by targeting PDGFRA. Cell
Prolif. 49:484–493. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barman SA, Zhu S and White RE: Protein
kinase C inhibits BKCa channel activity in pulmonary arterial
smooth muscle. Am J Physiol Lung Cell Mol Physiol. 286:L149–L155.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dempsey EC, Badesch DB, Dobyns EL and
Stenmark KR: Enhanced growth capacity of neonatal pulmonary artery
smooth muscle cells in vitro: Dependence on cell size, time from
birth, insulin-like growth factor I, and auto-activation of protein
kinase C. J Cell Physiol. 160:469–481. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dempsey EC, Frid MG, Aldashev AA, Das M
and Stenmark KR: Heterogeneity in the proliferative response of
bovine pulmonary artery smooth muscle cells to mitogens and
hypoxia: Importance of protein kinase C. Can J Physiol Pharmacol.
75:936–944. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang HP, Xu YJ, Zhang ZX, Ni W and Chen
SX: Effect of protein kinase C-nuclear factor-kappa B signal
transduction pathway on proliferation and expression of vascular
endothelial growth factor in human pulmonary artery smooth muscle
cells. Zhonghua Jie He He Hu Xi Za Zhi. 27:218–223. 2004.PubMed/NCBI
|
|
29
|
Jin N, Hatton N, Swartz DR, Xia Xl,
Harrington MA, Larsen SH and Rhoades RA: Hypoxia activates
jun-N-terminal kinase, extracellular signal-regulated protein
kinase, and p38 kinase in pulmonary arteries. Am J Respir Cell Mol
Biol. 23:593–601. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weerackody RP, Welsh DJ, Wadsworth RM and
Peacock AJ: Inhibition of p38 MAPK reverses hypoxia-induced
pulmonary artery endothelial dysfunction. Am J Physiol Heart Circ
Physiol. 296:H1312–H1320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Church AC, Martin DH, Wadsworth R, Bryson
G, Fisher AJ, Welsh DJ and Peacock AJ: The reversal of pulmonary
vascular remodeling through inhibition of p38 MAPK-alpha: A
potential novel anti-inflammatory strategy in pulmonary
hypertension. Am J Physiol Lung Cell Mol Physiol. 309:L333–L347.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun Y, Tian Y, Prabha M, Liu D, Chen S,
Zhang R, Liu X, Tang C, Tang X, Jin H and Du J: Effects of sulfur
dioxide on hypoxic pulmonary vascular structural remodeling. Lab
Invest. 90:68–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xia S, Tai X, Wang Y, An X, Qian G, Dong
J, Wang X, Sha B, Wang D, Murthi P, et al: Involvement of Gax gene
in hypoxia-induced pulmonary hypertension, proliferation, and
apoptosis of arterial smooth muscle cells. Am J Respir Cell Mol
Biol. 44:66–73. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang B, Shen M, Xu M, Liu LL, Luo Y, Xu
DQ, Wang YX, Liu ML, Liu Y, Dong HY, et al: Role of macrophage
migration inhibitory factor in the proliferation of smooth muscle
cell in pulmonary hypertension. Mediators Inflamm. 2012:8407372012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park MJ, Park IC, Lee HC, Woo SH, Lee JY,
Hong YJ, Rhee CH, Lee YS, Lee SH, Shim BS, et al: Protein kinase
C-alpha activation by phorbol ester induces secretion of gelatinase
B/MMP-9 through ERK 1/2 pathway in capillary endothelial cells. Int
J Oncol. 22:137–143. 2003.PubMed/NCBI
|
|
36
|
Okazaki J, Mawatari K, Liu B and Kent KC:
The effect of protein kinase C and its alpha subtype on human
vascular smooth muscle cell proliferation, migration and
fibronectin production. Surgery. 128:192–197. 2000. View Article : Google Scholar : PubMed/NCBI
|