Introduction
Metabolic syndrome consists of a group of complex
metabolic disorders associated with the metabolism of
carbohydrates, lipids and proteins. According to the definition of
the International Diabetes Federation, patients who meet at least
three of the following criteria are diagnosed with metabolic
syndrome: i) Abdominal obesity; ii) hypertension, hyperlipemia,
hypercholesterolemia; and iii) reduced high-density lipoprotein
cholesterol levels (1). In 2005,
>25% the total world population was diagnosed with metabolic
syndrome (2). In 2012, >1/3 of
the adult population and half of the population above 60 years of
age have been diagnosed with metabolic syndrome in the United
States (3). The negative impact of
metabolic syndrome is primarily due to its influence on other
diseases. In patients with metabolic syndrome, the risk of diabetes
mellitus is increased 5-fold, the risk of cardiovascular diseases
3-fold, the risk of cardiovascular death 2-fold and the overall
risk of death is increased 1.5-fold (4,5).
Glucocorticoids are involved in the regulation of
carbohydrate, lipid, protein, water and salt metabolism and have
important physiological functions (6–8).
Previous studies have confirmed that excessive glucocorticoids may
induce insulin resistance, inhibit glucose absorption and reduce
insulin release, thus increasing the possibility developing
metabolic syndrome (6). In
patients with metabolic syndrome, the glucocorticoid levels in
blood circulation are normal; however, the glucocorticoid levels in
local tissues are increased. 11β-hydroxysteroid dehydrogenase type
1 (11β-HSD1) is a metabolic reductase and dehydrogenase that
converts glucocorticoids between active cortisol and inactive
cortisone (7). Therefore, 11β-HSD1
is a local amplifier of glucocorticoid functions (9). Alberts et al (8) confirmed that the 11β-HSD1-specific
inhibitor BTV2733 reduced the levels of fasting blood glucose,
insulin, the concentrations of blood cholesterol, free fatty acids
and triglycerides in mice. Additionally, 11β-HSD1 knockout mice
exhibited weakened insulin resistance and gluconeogenesis reaction,
enhanced glucose tolerance and improved lipid distribution
(10). Masuzaki et al
(11) and Paterson et al
(12) already confirmed that the
tissue-specific overexpression of 11β-HSD1 in the liver and fat
induced metabolic syndrome characteristics in mice. However, these
two transgenic models could not reflect the overall effect of the
systemic expression of the 11β-HSD1 gene in patients with metabolic
syndrome. Therefore, the present study established transgenic mice
that systemically expressed the 11β-HSD1 gene and fed these mice
with a high-fat diet to induce metabolic syndrome. The current
findings revealed that transgenic mice exhibited metabolic syndrome
characteristics and pathological features in key tissues.
Materials and methods
Experimental animals
The porcine 11β-HSD1 gene was ligated into the T
vector and then ligated into the pcDNA3.1 plasmid (donated by Dr Li
Li, Institute of Zoology, Chinese Academy of Sciences) using
double-enzyme digestion. The digestion sites were NheI and
PmeI. The constructed vector plasmid was injected into 10
C57BL/6L mice (Institute of Laboratory Animal Science, Chinese
Academy of Medical Sciences; Beijing, China) via pronuclear
microinjection to obtain transgenic mice. This was conducted at the
Institute of Laboratory Animal Science, Chinese Academy of Medical
Sciences. Matched C57 mice were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. (Beijing, China). The
offspring of transgenic founders were divided into the transgenic
group and the negative control group and each group consisted of 5
mice. High-fat diets were purchased from Beijing Biopike
Biotechnology Co., Ltd. (Beijing, China) with fat content of 58%.
All mice received humane care according to the criteria outlined in
the ‘Guide for the Care and Use of Laboratory Animals, Institute of
Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing,
China.’ The procedures were approved by the Institutional Animal
Care and Use Committee of Chinese Academy of Agricultural Sciences.
The mice were housed under a 12-h light/dark cycle with access to
food and water ad libitum.
Following the termination of high-fat diet feeding,
the mice in the two groups were anaesthetized by intraperitoneal
injection of sodium pentobarbital (Sigma-Aldrich, Shanghai, China)
at a dose of 50 mg/kg body weight and subsequently decapitated.
Blood samples were collected and pancreatic, liver, lipid, kidney
and muscle tissues were immediately removed, rinsed with cold
physiological saline, frozen in liquid nitrogen and stored at
−80°C. Pieces of liver about ~10 mm diameter were fixed in 4%
paraformaldehyde at 4°C for histopathological studies.
Polymerase chain reaction (PCR)
detection
At 2 weeks of age, the tail tips of the mice were
cut to extract genomic DNA using the phenol-chloroform method
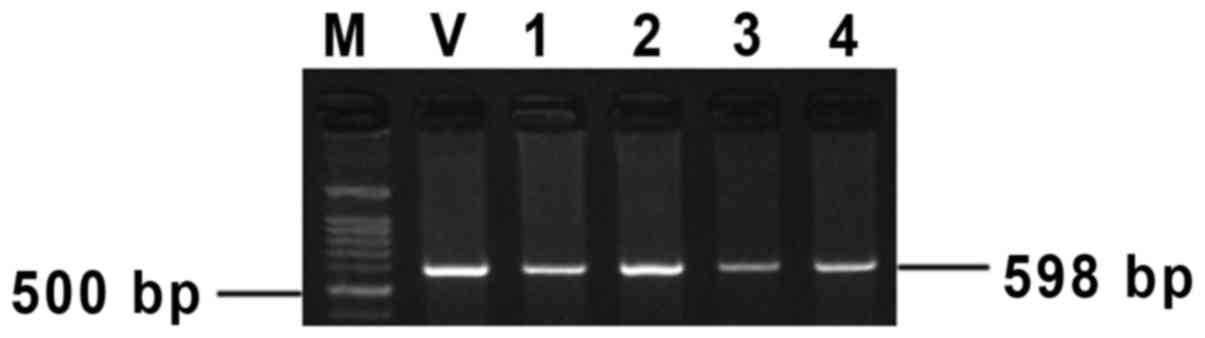
(13). The PCR product was 598 bp
long for the following primers: F 5′-CCCATAGTAACGCCAATA-3′ and R,
5′-CTACTGCTATTCCGCAAA-3′. The reaction was conducted in a total
volume of 20 µl and performed for 35 cycles under the following
conditions: Pre-denaturation at 95°C for 5 min, denaturation at
95°C for 30 sec, annealing at 58°C for 30 sec and extension at 72°C
for 30 sec and a final extension at 72°C for 5 min. All reagents
were obtained from Takara Biotechnology Co., Ltd. (Dalian, China).
Following PCR, 1% agarose gel electrophoresis (Biowest Regular
Agarose G-10; Biowest, Hong Kong, China). was performed.
Body weight, glucose and insulin
tolerance test
During the high-fat diet feeding period, the body
weight of the mice was measured once every other week. At the end
of the experiment, a glucose tolerance test was performed. Before
the test, the mice were fasted for 12 h and an intraperitoneal
injection of 20% glucose at a dose of 2 g/kg body weight was then
administered. Blood samples (2 µl) were collected from the tail
tips prior to injection and at 15, 30, 60, 90 and 120 min after
injection to quantify the blood glucose levels. For the insulin
tolerance test, the mice were fasted for 6 h prior to an
intraperitoneal injection of insulin at a dose of 1 IU/kg body
weight. Blood samples (2 µl) were collected from tail tips prior to
the injection and 15, 30, 60 and 90 min after the injection to
measure the blood glucose levels. Glucose was purchased from
Mallinckrodt Pharmaceuticals Ltd. (Staines-upon-Thames, UK) and
insulin (Novolin) was purchased from Novo Nordisk (Bagsværd,
Denmark). A Onetouch Ultra blood glucose meter and blood glucose
test strips were obtained from LifeScan, Inc. (Milpitas, CA,
USA).
Serological quantification
Biochemical parameters were analyzed using a AU480
automatic biochemistry analyzer (Olympus Corporation, Tokyo,
Japan). Assay kits were purchased from InTec Products, Inc.
(Xiamen, China). Alanine aminotransferase (ALT), aspartate
aminotransferase (AST) and uric acid were analyzed using the
dehydrogenase enzyme method, triglyceride and creatinine were
analyzed using the oxidase enzyme method, high-density
lipoprotein-cholesterol and low-density lipoprotein-cholesterol
were analyzed using direct method and total cholesterol was
analyzed using the enzyme method as previously described (14).
Western blotting
Total protein was extracted from tissues using a
tissue total protein extraction reagent kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and the total protein
concentration was quantified using an enzyme-linked immunosorbent
assay (ELISA) using a microplate reader (Spectra Max M5, Molecular
Devices, LLC, Sunnyvale, CA, USA). Subsequently, the proteins were
denatured and stored at −20°C. For each sample, 30 µg of protein
was separated using SDS-PAGE and the proteins were then transferred
onto a nitrocellulose (NC) membrane. The NC membrane was blocked in
5% non-fat milk. To quantitatively detect the 11β-HSD1 protein, a
primary 11β-HSD1 antibody (cat. no. ab83522; 1:1,000; Abcam,
Cambridge, UK) was used at room temperature for 2 h and a
horseradish peroxidase-labeled secondary goat anti-rabbit antibody
(cat. no. ab6721; 1:5,000; Abcam) was added at room temperature for
40 min. To assess endoplasmic reticulum stress, a DNA damage
inducible transcript 3 primary antibody (DDIT3; cat. no. ab11419;
1:1,000; Abcam) was used for incubation at room temperature for 2 h
and a secondary goat anti-mouse antibody (cat. no. ab6789; 1:5,000;
Abcam) were added for incubation at room temperature for 40 min.
Following washing, Super Signal West Pico Chemiluminescent
substrate (Thermo Fisher Scientific, Inc.) was used to detect the
immunoblots.
Hematoxylin and eosin (HE)
staining
Tissues were sectioned at a thickness of 4 µm, and
the sectioned slides were warmed at 60°C for 1 h, deparaffinized
using xylene, rehydrated, stained with hematoxylin at room
temperature for 2 min, washed, differentiated, blued, stained with
eosin at room temperature for 1 min, and washed with water. The
stained sections were dehydrated in 70, 80, 90, 95 and 100% ethanol
(2 min for each concentration), followed by clearing in xylene 1
and xylene 2 (5 min each), the sections were subsequently mounted
in resin.
Statistical analysis
The differences of data between the two study groups
were statistically analyzed using SPSS version 18.0 (SPSS, Inc.,
Chicago, IL, USA). One-way analysis of variance and the least
significant difference post-hoc test were used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference. Data are presented as the mean ± standard
deviation.
Results
Detection of positive transgenic
mice
The founders of 11β-HSD1 transgenic mice and their
offspring were detected using PCR. The positive F0 individuals had
the band of 598 bp (Fig. 1).
Significant increase in obesity of
transgenic mice
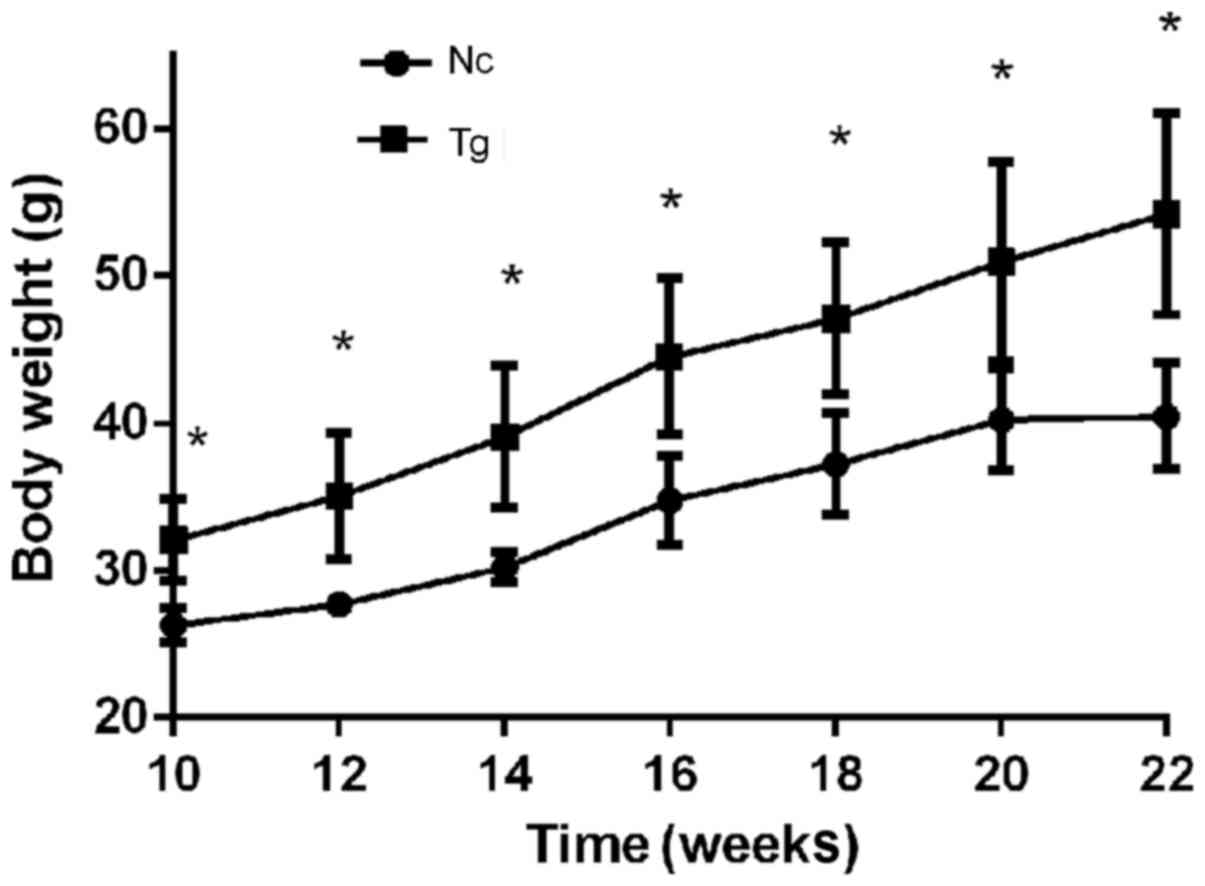
The average body weight of transgenic mice was
32.07±2.43 g prior to feeding with high-fat diet and the negative
control group weight was 26.29±1.09 g (P<0.05). From week 2 of
high-fat diet induction until the end of the induction experiment,
transgenic mice were significantly more obese compared with control
mice. In addition, the body weights of transgenic mice consistently
increased, whereas the body weights of control mice plateaued at
week 20 and subsequently decelerated (Fig. 2). Following induction, the average
body weight of the transgenic group was 1.56-fold higher compared
with the control group, 54.22±5.90 g and 40.46±3.23 g
(P<0.05).
Biochemical parameters
Concentration of biochemical parameters was
increased, indicating metabolic syndrome and damage to key organs
in the transgenic group. In the transgenic group, the values of the
triglyceride, total cholesterol, and low-density lipoprotein
cholesterol significantly increased and were 2.13, 1.68, and
2.08-fold higher compared with the control group (Table I). These values were indicative of
metabolic syndrome in the transgenic group. The values of ALT and
AST, which reflect liver function, significantly increased and were
2.94 and 1.43-fold higher compared with the control group,
respectively (Table I). The levels
of uric acid and creatinine, which reflect kidney function,
significantly increased and were 1.48 and 2.25-fold higher compared
with the control group values, respectively (Table I). These findings revealed that
transgenic mice exhibited characteristics of metabolic syndrome and
that important organs involved in metabolic syndrome were
damaged.
 | Table I.Biochemical parameters of the Tg and
Nc groups. |
Table I.
Biochemical parameters of the Tg and
Nc groups.
| Biochemical
parameters | Tg | Nc | P-value |
|---|
| ALT, IU/l |
211.33±13.47 |
70.4±30.71 | <0.01 |
| AST, IU/l |
254.67±23.80 |
178±8.88 | 0.04 |
| Triglyceride,
µmol/l |
0.81±0.06 |
0.38±0.17 | <0.01 |
| Total cholesterol,
µmol/l |
5.57±0.64 |
3.31±0.85 | 0.02 |
| HDL-C, µmol/l |
2.23±0.13 |
1.72±0.40 | 0.04 |
| LDL-C, µmol/l |
0.59±0.09 |
0.28±0.05 | 0.03 |
| Uric acid,
µmol/l |
269.7±10.50 |
182.72±18.27 | <0.01 |
| Creatinine,
µmol/l |
10.33±1.25 |
4.6±1.85 | <0.01 |
Severely impaired glucose tolerance in
transgenic mice
Fasting glucose tolerance is an important indicator
that comprehensively reflects insulin release and insulin
resistance in an organism. Following high-fat diet induction, the
intraperitoneal glucose tolerance was quantified in the transgenic
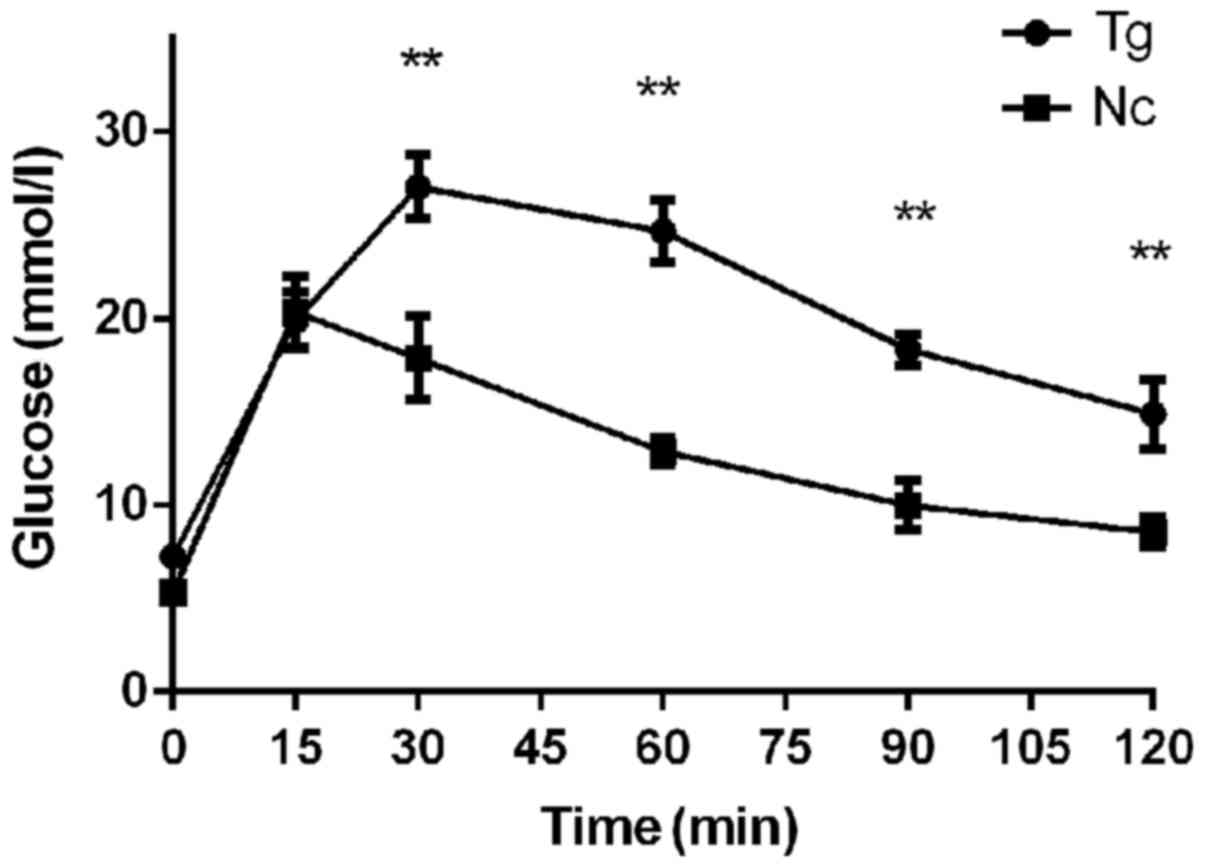
and the control groups (Fig. 3).
The peak glucose of the control group appeared at 15 min after
injection, whereas the peak value of the transgenic group appeared
at 30 min. The peak glucose of the control group and the transgenic
group were 20.35±1.65 mmol/l and 27.03±1.49 mmol/l, respectively
(P<0.01). Following the peak, the blood glucose levels in these
two groups began to decrease. At 4 detection points between 30 and
120 min, the blood glucose levels in the transgenic group were all
significantly higher compared with the in the control group. After
2 h, the blood glucose level in the transgenic group remained at
14.9 mmol/l, whereas the blood glucose level in the control group
decreased to 8.58 mmol/l. Therefore, after 12 weeks of high-fat
diet feeding, the transgenic group exhibited severely impaired
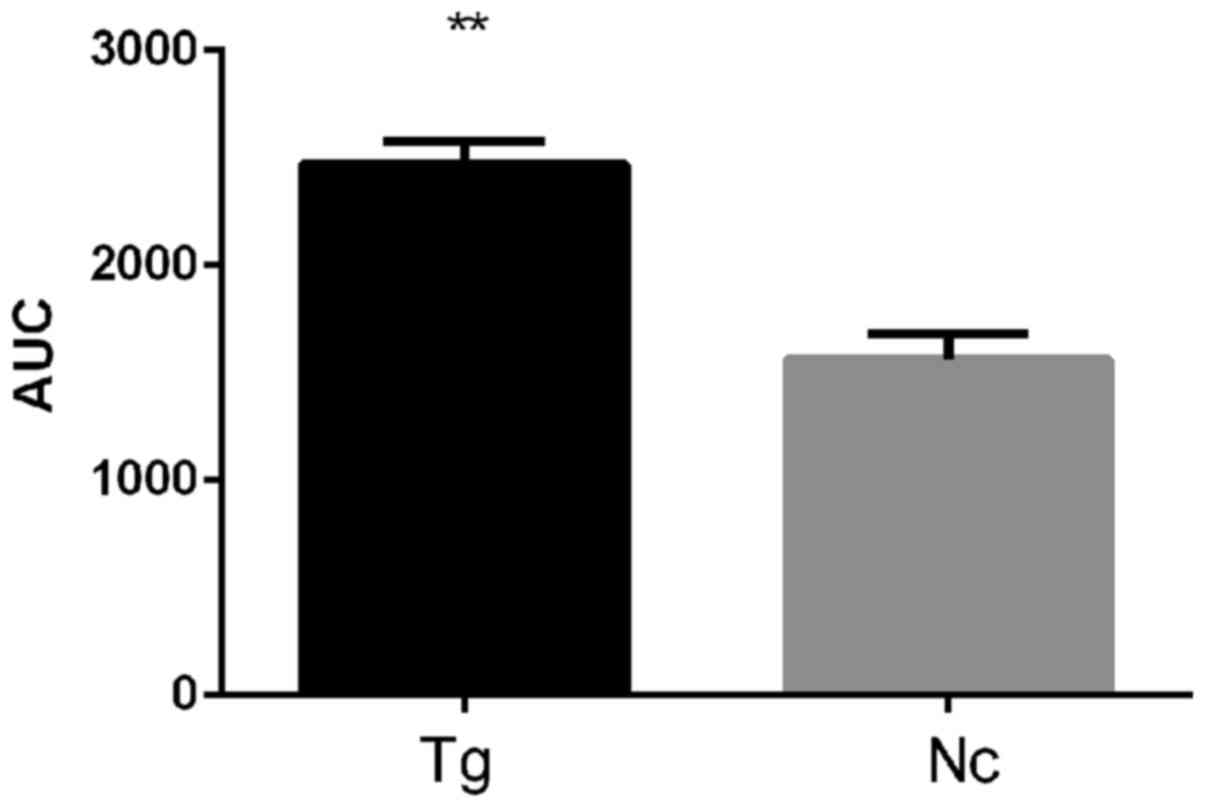
glucose tolerance. This finding is supported by the area under the
glucose tolerance curve (Fig. 4),
which was 1.58-fold higher for the transgenic group compared with
the control group (P<0.01).
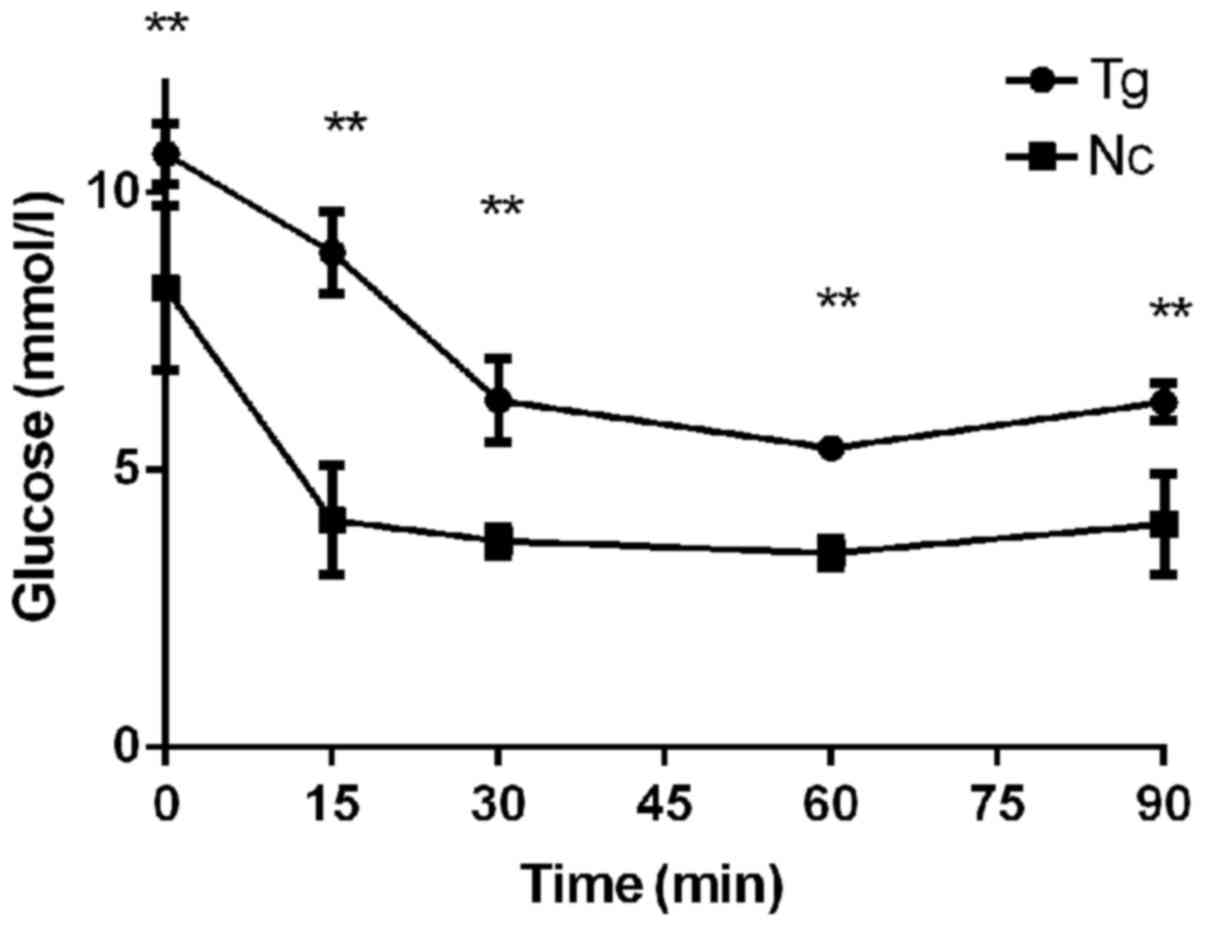
Significant insulin resistance in the
transgenic group
Prior to feeding with a high-fat diet, the 6 h
fasting glucose value was 10.7 mmol/l in the transgenic group and
8.28 mmol/l in the control group (P<0.01). Following an insulin
injection, the glucose of these two groups rapidly reduced and
reached a minimum after 30 min at 6.25 mmol/l in the transgenic
group and 3.73 mmol/l in the control group a significant difference
was identified between these values. After 90 min, the blood
glucose levels began to increase again and reached 6.23 and 4.03
mmol/l in the transgenic group and the control group, respectively,
the former was 1.55 times higher than the latter. These findings
revealed that the transgenic group exhibited reduced insulin
sensitivity and severe insulin resistance (Fig. 5).
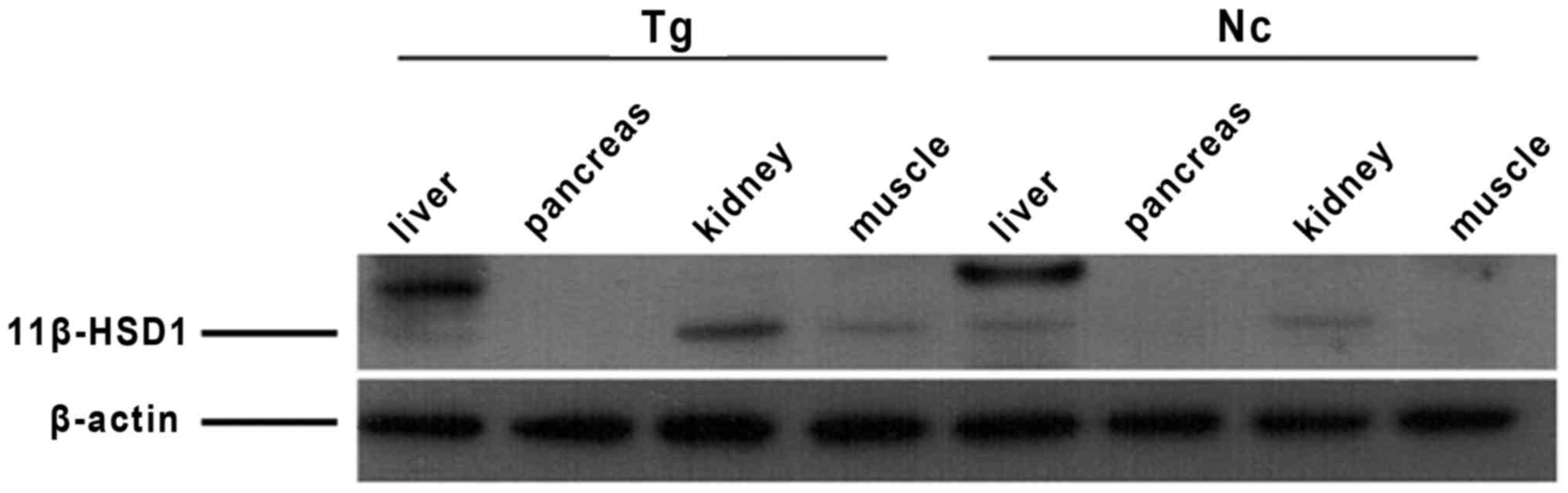
Tissue expression profiles of 11β-HSD1
in transgenic mice
The expression of 11β-HSD1 was the highest in the
liver, followed by the kidney and muscle. The pancreases did not
express evident quantity of 11β-HSD1 protein. The sizes of 11β-HSD1
protein differed by tissue, and expression levels in the positive
individuals were higher compared with the negative individuals
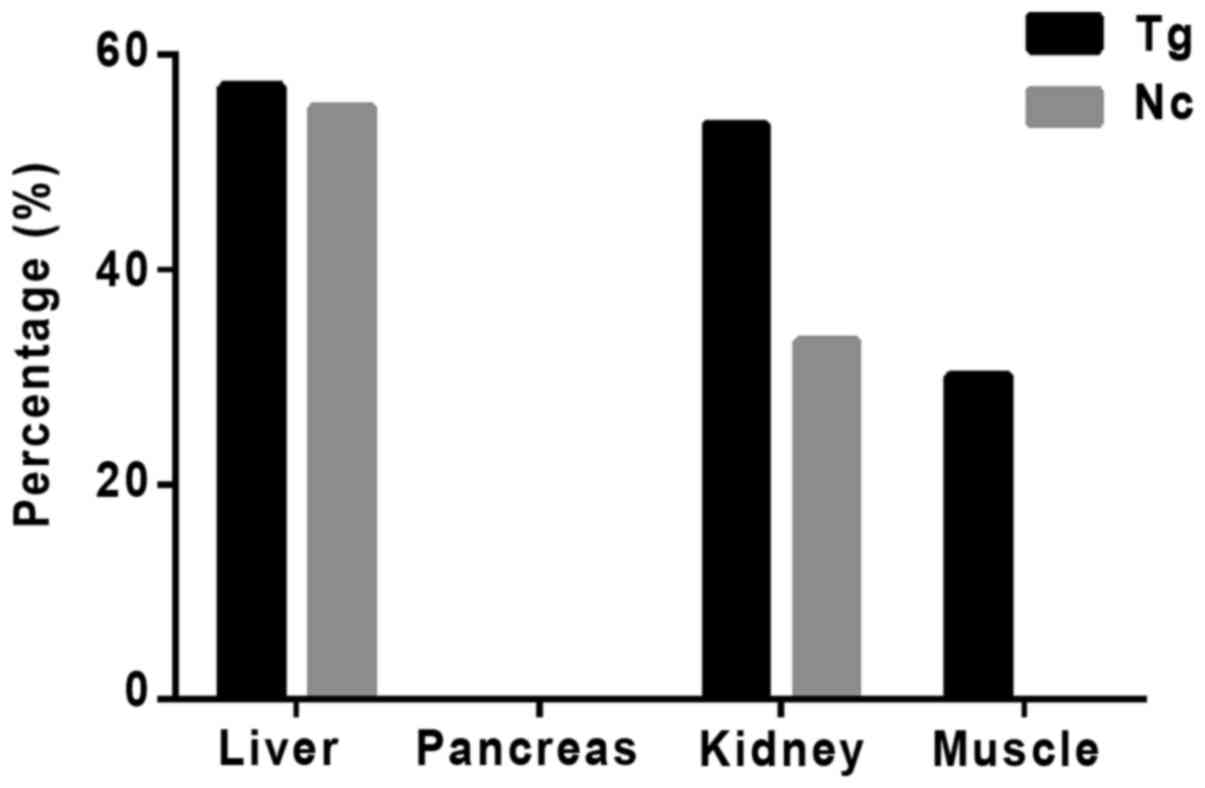
(Fig. 6). The percentage of
11β-HSD1 protein levels increased in transgenic mouse compared with
the control group is presented in Fig.
7.
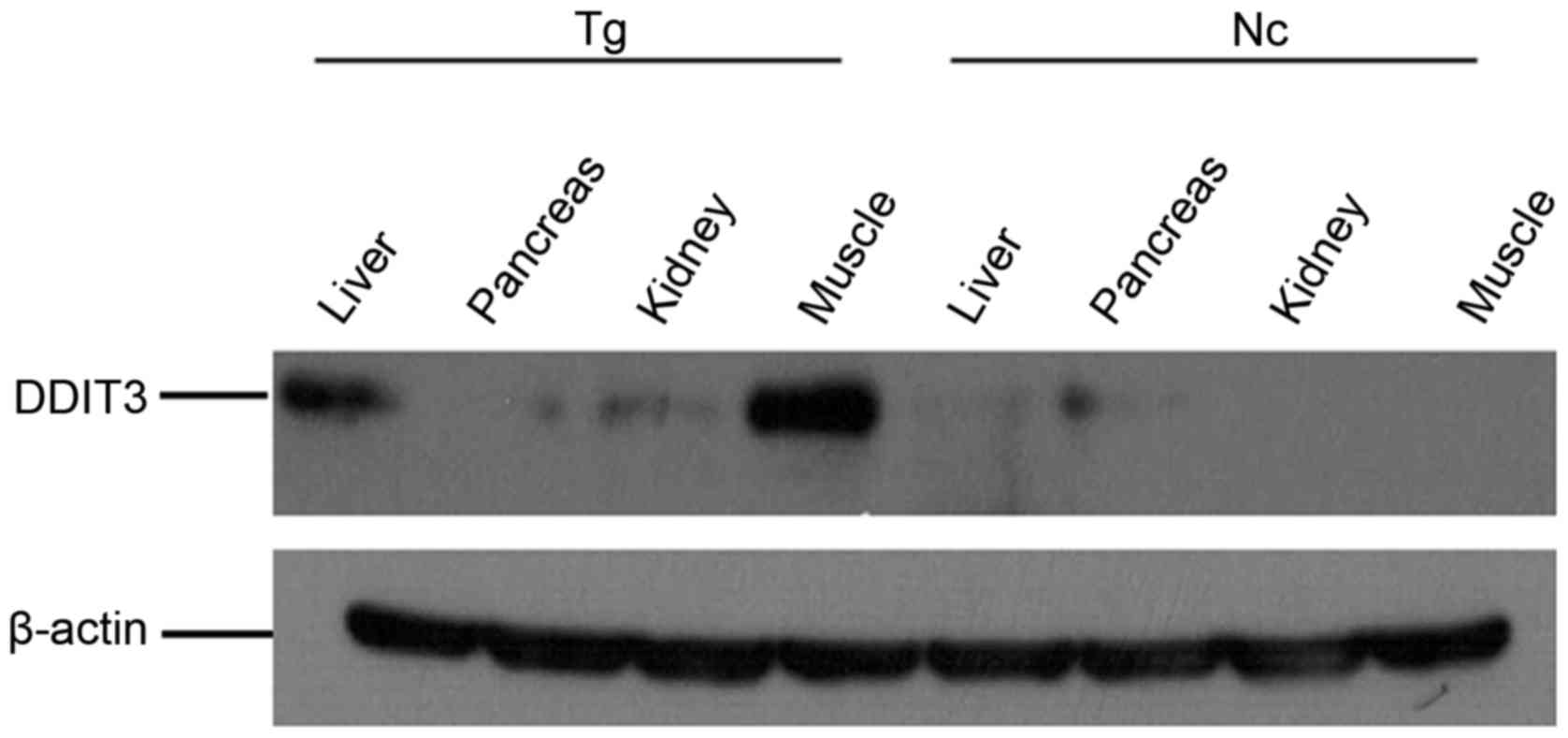
Severe endoplasmic reticulum stress
associated with the expression of 11β-HSD1 in the transgenic
group
Endoplasmic reticulum stress is central to the
development of metabolic syndrome induced by insulin resistance
(15). Therefore, the present
study assessed endoplasmic reticulum stress in the mice. The muscle
and liver in the transgenic group exhibited severe endoplasmic
reticulum stress due to high DDIT3 expression levels, and the
kidney also revealed some endoplasmic reticulum stress (Fig. 8).
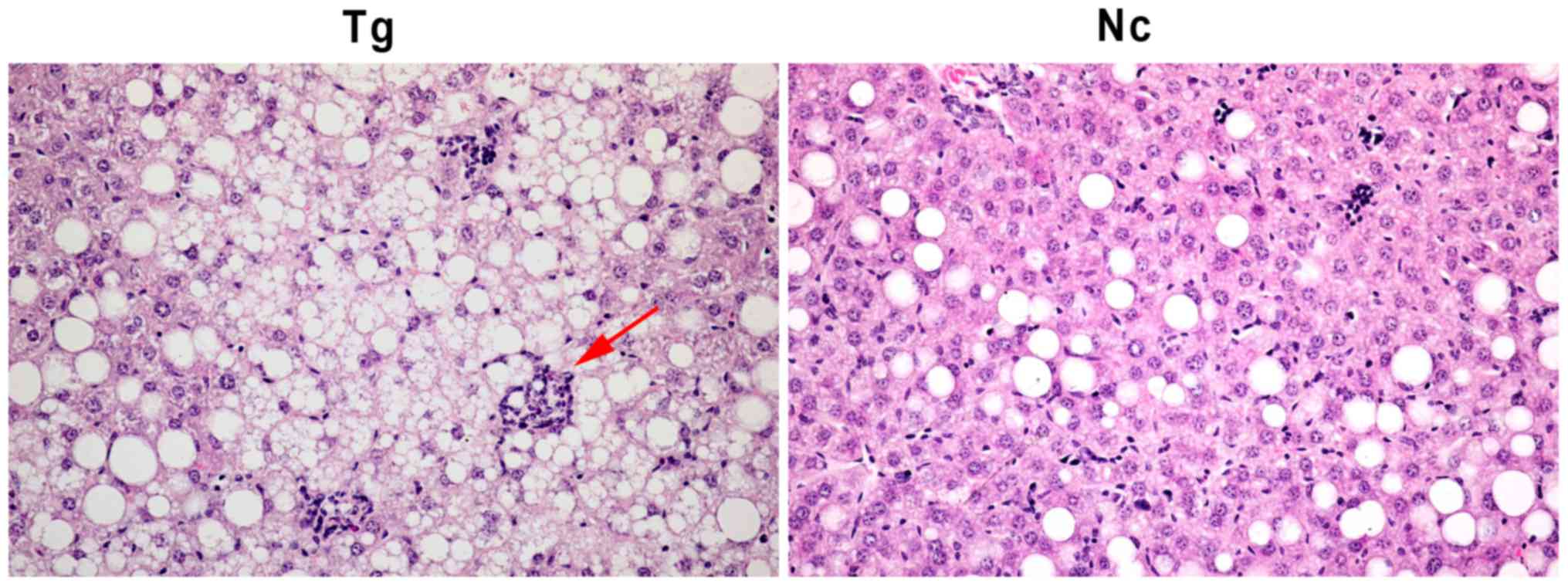
Hepatic lipidosis and hepatocyte
injury in transgenic mice
The liver is the largest detoxification organ in the
human body and impaired liver detoxification due to hepatic
lipidosis and impaired liver function are important elements in the
development of metabolic syndrome, with almost two thirds of
patients with type 2 diabetic having hepatic steatosis (16). Therefore, the mice liver tissues in
these two groups were stained with. The it was determined that
transgenic mice suffered from severe hepatic lipidosis, with lipid
droplets being diffused throughout the liver and liver cells
exhibiting necrosis and lysis. The sections exhibited inflammatory
foci and infiltration of inflammatory cells. Conversely, the
control group exhibited only mild hepatic lipidosis, hepatocytes
were intact and inflammatory cell infiltration was absent (Fig. 9).
Discussion
Glucocorticoids are closely associated with
metabolic syndrome, primarily by promoting gluconeogenesis,
reducing insulin sensitivity in peripheral tissues, inhibiting
insulin secretion and promoting islet β cell apoptosis (6). 11β-HSD1 is a metabolic enzyme of
glucocorticoids it may activate glucocorticoids and amplify their
function locally (9). The current
study established transgenic mice that systemically overexpress the
11β-HSD1 gene in order to simulate the development of metabolic
syndrome. After 12 weeks of high-fat diet, mice that overexpressed
the 11β-HSD1 gene exhibited obesity and significantly impaired
glucose tolerance and insulin resistance. Additionally, the levels
of triglycerides, total cholesterol, and high- and low-density
lipoprotein cholesterol significantly increased in the transgenic
mice, indicating that mice overexpressing 11β-HSD1 exhibited
characteristics of metabolic syndrome. In addition, serology
measurements indicated that liver and kidney function in transgenic
mice was impaired. Masuzaki et al (11) determined that the specific
overexpression of 11β-HSD1 in adipose tissues may lead to increased
corticosteroid levels in adipose tissues. Paterson et al
(12) established transgenic mice
that specifically overexpressed 11β-HSD1 in the liver. These mice
exhibited mild insulin resistance, hepatic lipidosis and
dyslipidemia, which was accompanied by increased hepatic lipid
synthesis/efflux, elevated liver X receptor and peroxisome
proliferator-activated receptor mRNA expression levels, impaired
hepatic lipid clearance and significant transgenic dose-dependent
hypertension combined with increased expression levels of hepatic
vascular angiotensinogen (12).
The findings of the present study were consistent with these
previous findings.
The current study revealed that the endoplasmic
reticulum stress was associated with the expression of 11β-HSD1 in
the tissues of transgenic mice. Additionally, Kim et al
(17) quantified the expression of
the endoplasmic reticulum stress-associated marker genes X-box
binding protein 1, activating transcription factor 4 (ATF4), ATF6
and DDIT3 (also termed CHOP) to confirm that the 11β-HSD1 inhibitor
carbenoxolone attenuated tunicamycin-induced endoplasmic reticulum
stress and neuronal apoptosis in the hypothalamus (17). These findings indicated that
11β-HSD1 directly induced endoplasmic reticulum stress in
hypothalamic neurons and the subsequent apoptosis, which also
indirectly confirmed the association between 11β-HSD1 and
endoplasmic reticulum stress (17). Endoplasmic reticulum stress induced
by overexpression of 11β-HSD1 activates unfolded protein response,
which activates c-Jun N-terminal kinase (JNK) via
inositol-requiring enzyme-1, activated JNK may induce the serine
phosphorylation of insulin receptor substrates and inhibits the
normal tyrosine phosphorylation of insulin receptor substrate,
which in turn inhibit the transduction of insulin, ultimately
leading to insulin resistance (15).
The present study assessed endoplasmic reticulum
stress in important metabolic syndrome-associated organs. The
current findings revealed that the liver and muscle tissues of mice
overexpressing 11β-HSD1 has severe endoplasmic reticulum stress and
that the kidney also exhibited some endoplasmic reticulum stress.
The liver is the largest detoxification organ in the human body.
Impaired liver detoxification due to hepatic lipidosis and impaired
liver function is an important element in the development of
metabolic syndrome (18). It is
possible that that the transgenic mice in the present study
overexpressing 11β-HSD1 suffered from liver lesions due to their
metabolic syndrome phenotype.
The quantification of liver function-associated
indicators in mice revealed that the level of ALT, the most
sensitive indicator of liver function, significantly increased.
Additionally, the level of AST also significantly increased and was
higher than the ALT level, indicating that the mouse liver
parenchyma in the transgenic group was severely injured and that
liver function was severely impaired. These findings were
consistent with the significant endoplasmic reticulum stress
observed in the liver. Histological evaluation identified severe
fat deposition, lipid droplets that had diffused throughout the
liver, necrotic and lysed hepatocytes, many inflammatory foci and
significant inflammatory cell infiltration in the livers of
transgenic mice. However, endoplasmic reticulum stress-induced
hepatocyte apoptosis may be directly caused by high 11β-HSD1
expression in the liver or indirectly caused by 11β-HSD1-induced
lipid deposition in the liver and this mechanism requires further
investigation.
In conclusion, the present study successfully
established transgenic mice that systemically overexpressed
11β-HSD1. These mice exhibited obvious characteristics of metabolic
syndrome, such as severe hepatic lipidosis, hepatocyte necrosis,
impaired liver function, and endoplasmic reticulum stress in
muscle, liver and kidney. These findings indicate that the systemic
overexpression of 11β-HSD1 in mice produces pathological changes
that approximate to the clinical symptoms of human metabolic
syndrome. Therefore, these mice may be used as a model for future
studies of metabolic syndrome.
Acknowledgements
The current study was supported by The National
Natural Science Foundation of China (grant no. 31372276), The
National Basic Research Program of China (grant no. 2015CB943100),
National Science and Technology Major Project (grant no.
2016ZX08006-001), The State Key Laboratory of Animal Nutrition
(grant no. 2004DA125184G1602) and The Agricultural Science and
Technology Innovation Program (grant nos. ASTIP-IAS05 and
ASTIP-IAS-TS-4).
References
|
1
|
Alberti KG, Eckel RH, Grundy SM, Zimmet
PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith
SC Jr, et al: Harmonizing the metabolic syndrome: A joint interim
statement of the International Diabetes Federation Task Force on
Epidemiology and Prevention; National Heart, Lung and Blood
Institute; American Heart Association; World Heart Federation;
International Atherosclerosis Society; and International
Association for the Study of Obesity. Circulation. 120:1640–1645.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas GN, Ho SY, Janus ED, Lam KS, Hedley
AJ and Lam TH: Hong Kong Cardiovascular Risk Factor Prevalence
Study Steering Committee: The US National Cholesterol Education
Programme Adult Treatment Panel III (NCEP ATP III) prevalence of
the metabolic syndrome in a Chinese population. Diabetes Res Clin
Pract. 67:251–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aguilar M, Bhuket T, Torres S, Liu B and
Wong RJ: Prevalence of the metabolic syndrome in the United States,
2003–2012. Jama. 313:1973–1974. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sookoian S and Pirola CJ: Metabolic
syndrome: From the genetics to the pathophysiology. Curr Hypertens
Rep. 13:149–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eckle RH, Grundy SM and Zimmet PZ: The
metabolism syndrome. Lancet. 365:1415–1428. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee MJ, Pramyothin P, Karastergiou K and
Fried SK: Deconstructing the roles of glucocorticoids in adipose
tissue biology and the development of central obesity. Biochim
Biophys Acta. 1842:473–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seckl JR and Walker BR: Minireview:
11beta-hydroxysteroid dehydrogenase type 1 a tissue-specific
amplifier of glucocorticoid action. Endocrinology. 142:1371–1376.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alberts P, Nilsson C, Selen G, Engblom LO,
Edling NH, Norling S, Klingström G, Larsson C, Forsgren M, Ashkzari
M, et al: Selective inhibition of 11 beta-hydroxysteroid
dehydrogenase type 1 improves hepatic insulin sensitivity in
hyperglycemic mice strains. Endocrinology. 144:4755–4762. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andrew R, Westerbacka J, Wahren J,
Yki-Järvinen H and Walker BR: The contribution of visceral adipose
tissue to splanchnic cortisol production in healthy humans.
Diabetes. 54:1364–1370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Liu J, Zhang A, Cheng P, Zhang X,
Lv S, Wu L, Yu J, Di W, Zha J, et al: BVT.2733, a selective
11beta-hydroxysteroid dehydrogenase type 1 inhibitor, attenuates
obesity and inflammation in diet-induced obese mice. PLoS One.
7:e400562012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Masuzaki H, Paterson J, Shinyama H, Morton
NM, Mullins JJ, Seckl JR and Flier JS: A transgenic model of
visceral obesity and the metabolic syndrome. Science.
294:2166–2170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paterson JM, Morton NM, Fievet C, Kenyon
CJ, Holmes MC, Staels B, Seckl JR and Mullins JJ: Metabolic
syndrome without obesity: Hepatic overexpression of
11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc
Natl Acad Sci USA. 101:7088–7093. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tebbe CC and Vahjen W: Interference of
humic acids and DNA extracted directly from soil in detection and
transformation of recombinant DNA from bacteria and a yeast. Appl
Environ Microbiol. 59:2657–2665. 1993.PubMed/NCBI
|
|
14
|
Li L, Zhao Z, Xia J, Xin L, Chen Y, Yang S
and Li K: A Long-Term High-Fat/High-Sucrose Diet Promotes Kidney
Lipid Deposition and Causes Apoptosis and Glomerular Hypertrophy in
Bama Minipigs. PLoS One. 10:e01428842015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Targher G, Bertolini L, Padovani R,
Rodella S, Tessari R, Zenari L, Day C and Arcaro G: Prevalence of
nonalcoholic fatty liver disease and its association with
cardiovascular disease among type 2 diabetic patients. Diabetes
Care. 30:1212–1218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim J, Jung EJ, Moon SS and Seo M:
Protective effect of carbenoxolone on ER stress-induced cell death
in hypothalamic neurons. Biochem Biophys Res Commun. 468:793–799.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nannipieri M, Gonzales C, Baldi S, Posadas
R, Williams K, Haffner SM, Stern MP and Ferrannini E: Mexico City
diabetes study: Liver enzymes, the metabolic syndrome, and incident
diabetes: The Mexico City diabetes study. Diabetes Care.
28:1757–1762. 2005. View Article : Google Scholar : PubMed/NCBI
|