Introduction
Focal segmental glomerulosclerosis (FSGS) is a
description of a histological lesion, rather than a disease;
characterized by focal and segmental glomerular sclerosis and
podocyte foot-process effacement and its clinical manifestations
include proteinuria and progressive renal failure. Current
treatments for FSGS frequently fail to achieve remission (1,2).
Therefore, unravelling the pathogenesis of FSGS is of primary
concern for the development of targeted therapy.
The etiology of FSGS has been identified as diverse.
FSGS may occur following immunologically-mediated injury, genetic
factors, circulating permeability factor/s, and hemodynamic
adaptations resulting in glomerular hypertrophy and direct podocyte
injury also lead to FSGS (3). The
most common clinical manifestation of FSGS is proteinuria, which
may range from subnephrotic to nephrotic levels. However, a number
of the patients with proven-FSGS present with hematuria (4).
The renal glomerular filtering apparatus consists of
three major components: The fenestrated endothelial cell layer, the
glomerular basement membrane (GBM) and the epithelial podocyte
layer. Injury to any layer may result in red blood cells or protein
escaping into the urine through a defect in the glomerular
filtration barrier. It has been demonstrated that podocyte damage
serves a central role in the pathogenesis of FSGS (5,6). A
number of genes have also been demonstrated to be mutated in FSGS
(7–24) (Table
I), and most of the encoded proteins are localized in
podocytes, whereas others are expressed in other tissues and cell
types including in GBM (20–22,25–26).
According to statistical analysis, 1/3-1/2 of children with
isolated, persistent hematuria have a familial history (27–28).
It has been demonstrated by previous studies that long-term
persistent microscopic hematuria (MH) may lead to chronic kidney
disease (CKD) (29–32). A total of ~14–50% of familial cases
progress to end-stage renal disease (ESRD) on long-term follow-ups
(33).
 | Table I.Selected list of 18 genes associated
with FSGS syndrome. |
Table I.
Selected list of 18 genes associated
with FSGS syndrome.
| Author, year | Gene | Locus | Inheritance | Protein | Phenotype | (Refs.) |
|---|
|
| SD-associated and
adaptor protein: |
|
|
|
| Santin, 2009 | NPHS1 | 19q13.1 | AR | Nephrin | CNS/NS, FSGS | (7) |
| Tonna, 2008 | NPHS2 | 1q25-q31 | AR | Podocin | CNS, NS-childhood
and adult onset, FSGS | (8) |
| Gigante, 2009 | CD2AP | 6p12.3 | AD | CD2 associated
protein | Early-onset NS, HIV
nephropathy, FSGS | (9) |
| Hinkes, 2006 | PLCE1 | 10q23.33 | AR | Phospholipase C ε
1 | Early-onset NS,
DMS | (10) |
| Santin, 2009 | TRPC6 | 11q22.1 | AD | Transient receptor
potential cation channel subfamily C member 6, | Adult onset NS,
FSGS | (11) |
|
| Nuclear
proteins: |
|
|
|
| Hall, 2013 | WT1 | 11p13 | Sporadic, AD | Wilms' tumor 1 | Adult onset NS,
Denys-Drash and Frasier Syndromes, DMS, FSGS | (12) |
| Boyer, 2013 | LMX1B | 9q34.1 | AR | LIM homeobox
transcription factor 1, β | Nail-Patella
Syndrome/NS only, | (13) |
| Boerkoel, 2002 | SMARCAL1 | 2q34-q36 | AR | SWI/SNF related,
matrix associated, actin dependent regulator of chromatin,
subfamily a-like 1 | Schimke
immuno-osseous dysplasia (syndromic immune complex nephritis) | (14) |
|
| Actin cytoskeleton
and signaling |
|
|
|
| Choi, 2008 | ACTN4 | 19q13 | AD | α-actinin 4 | Adult onset NS,
FSGS | (15) |
| Gbadegesin,
2011 | INF2 | 14q32 | AD | Inverted formin
2 | Familial/sporadic
NS; Charcot-Marie-Tooth, FSGS | (16) |
| Lowik, 2005 | Mitochondrial |
|
| MTTL1 | mtDNA | Maternal
inheritance | tRNA-LEU | MELAS syndrome; NS
± deafness and diabetes | (17) |
| Diomedi-Camassei,
2007 | COQ2 | 4q21.23 | AR | Coenzyme Q2
4-hydroxybenzoate polyprenyltransferase | Mitochondrial
disease/isolated nephropathy | (18) |
| Lopez, 2006 | PDSS2 GBM | 6q21 | AR | Prenyl diphosphate
synthase subunit 2 | Leigh syndrome,
FSGS or collapsing glomerulopathy | (19) |
| Matejas, 2010 | LAMB2 | 3p21 | AR | Laminin β 2 | Pierson syndrome:
CNS with ocular abnormalities, isolated early-onset NS, DMS
FSGS | (20) |
| Hatei, 2005 | LAMB3 | 1q32 | AR | Laminin β 3 | Epidermolysis
bullosa, junctional, non-herlitz type, somatic mosaic revertant,
Junctional epidermolysis bullosa gravis of Herlitz | (21) |
| Kambham, 2000 | ITGB4 | 17q25 | AR | β4-integrin | NEP syndrome-NS,
epidermolysis bullosa and pulmonary disease | (22) |
|
| Other-Metabolic or
lysosomal |
|
|
|
| Berkovic, 2008 | SCARB2
(lysosomal) | 4q21.1 | AR | Scavenger receptor
class B member 2 | Nephrotic syndrome,
nephrotic syndrome with C1q deposits, progressive myoclonic
epilepsy (Action myoclonus renal failure syndrome ± hearing
loss) | (23) |
| Serebrinsky,
2015 | GLA | Xq22.1 | XLR | α-galactosidase
A | Andeson-Fabry
disease | (24) |
Alport's Syndrome (AS) and thin basement membrane
neuropathy (TBMN), which occur most frequently in glomerular MH,
result from defects in type IV collagen. The type IV collagen
α3α4α5 chain is a major component of the GBM and a heterotrimer
that is encoded by three genes: Collagen type IV α 3 chain
(COL4A3), COL4A4 and COL4A5 (34).
During the last three decades, six genes (COL4A3, COL4A4, COL4A5,
complement factor H related 5, myosin heavy chain 9 and fibronectin
1) have so far been identified in familial microscopic hematuria of
glomerular origin (34). In
addition to AS and TBMN, familial FSGS may also be a factor
resulting in familial glomerular microscopic hematuria (GMH)
(4).
It has been demonstrated by previous studies that
long-term persistent MH may lead to renal injury regardless of
TBMN, AS or other disease presenting with hematuria (29–32).
Therefore, pediatric nephrologists need to be aware that children
with familial hematuria and a family history of CKD have a high
probability of developing proteinuria and progressing to renal
failure in adult life. Especially at early stages when MH appears
as an isolated warning sign, it is worth having a step-wise
algorithm for deeper investigations of the etiology and
pathogenesis of the disease.
Advances in DNA analysis technology may facilitate
greater use of molecular diagnostics, which reduce the need to use
invasive methods including renal biopsy (4). Importantly, molecular diagnostics may
be performed at an early stage of disease, frequently providing a
broader set of therapeutic options and an increased window of
opportunity to ameliorate disease progression (35).
Recently the implementation of high-throughput
sequencing technologies including mass array technology and whole
exome sequencing (WES) make it possible to test multiple genes
simultaneously in a single experiment faster and more efficiently
(36). The performance of the
next-generation sequencing may help to identify novel genes or
novel unreported mutations and discover co-segregating genetic
regions. However, the appropriate application and combination of
sequencing methods with conventional gene-discovery strategies
should be considered for each patient and research project
(36). Only then may they be of
use in making a diagnosis in a more precise way.
The present study reports on a family in which
affected individuals exhibited familial hematuria and the siblings
had biopsy-proven FSGS and normal GBM. Renal biopsies demonstrated
non-specific pathological alterations and failed to exhibit
glomerular or basement membrane defects consistent with an
inherited glomerulopathy, and therefore a possible underlying
genetic cause for a unifying and definitive diagnosis was pursued.
The present study hypothesized that FSGS in the siblings resulted
from a defect in the 18 genes [nephrin (NPHS1), podocin (NPHS2),
CD2 associated protein (CD2AP), phospholipase C ε
(PLCE1), actinin α 4 (ACTN4), transient receptor
potential cation channel subfamily C member 6, (TRPC6),
inverted formin, FH2 and WH2 domain containing (INF2),
Wilms tumor 1 (WT1), LIM homeobox transcription factor 1β
(βLMX1B), laminin subunit β (LAMB) 2, LAMB3,
galactosidase α, integrin subunit β 4, scavenger receptor
class B member 2 (SCARB2), coenzyme Q2 (COQ2),
decaprenyl diphosphate synthase subunit 2 (PDSS2),
mitochondrially encoded tRNA leucine 1 (UUA/G; TRNL1)
and SWI/SNF related, matrix associated, actin dependent
regulator of chromatin, subfamily a like 1]. Under this
assumption, the siblings were identified as possessing a homozygous
mutation for TRNL1 (m. 3290T>C), which may segregate with
disease using matrix-assisted laser desorption/ionization
time-of-flight (MALDI-TOF) mass spectrometry technology. WES on the
siblings was performed to identify the existence of other genes or
mutations that co-segregate with familial hematuria or FSGS when
mutated. The results demonstrated that the two sisters carried a
single heterozygous mutation c. A4195T (p. M1399L) in the COL4A4
gene, which may serve a role in the pathology of FSGS and act as a
modifier to TRNL1.
To the best of the authors' knowledge, this is the
first report of a family with familial hematuria and proven-FSGS
with a mutation in the TRNL1 gene, and with a mutation in the
COL4A4 gene that co-segregated with disease. In addition, this may
be the first study to use mass array technology and WES
simultaneously in the identification of disease genes.
Materials and methods
Clinical data and DNA preparation
Clinical data and historical renal biopsies were
reviewed where available. Following informed consent being
obtained, DNA was obtained from the siblings (1~14 years old) and
their father (33–34 years old; data not available from their
mother) obtained from the Second Xiangya Hospital during the period
March/2014-March/2015. The research was approved by the Ethics
Commission of the Second Xiangya Hospital (Changsa, China). DNA was
isolated from peripheral leukocytes using the DNA purification kit
(Tiangen Biotech Co., Ltd., Beijing, China) according to the
manufacturer's protocol.
Single nucleotide polymorphism (SNP)
analysis using MALDI-TOF technology
The genes and SNPs were selected on the basis of
currently available literature (22). The 15 genes were selected following
searching databases including PubMed, the Online Mendelian
Inheritance in Man (OMIM; www.ncbi.nlm.nih.gov/omim) and the Human Gene Mutation
Database (nihlibrary.nih.gov/about-us/news/categories/3051).
Database ClinVar and OMIM were searched for clinically relevant
mutations or SNPs from the 18 genes. The database search identified
179 candidates. Certain candidates either lacked complete
information or were not compatible for MassArray technology. A
total of 138 mutations were included in the assay.
Polymerase chain reaction (PCR)
The assay was designed using MassARRAY®
software (version 4.0; Sequenom, San Diego, CA, USA). The 138
mutations were assigned to six multiplex assays. PCR primers were
designed using Mass ARRAY® Assay Design 4.0 Software
(Sequenom) (Table II). PCR was
first performed using the following protocol: 4 min at 95°C for
activation of Faststart taq DNA polymerase (Roche Diagnostics,
Basel, Switzerland, cat. no. 12032937001) and 30 sec at 95°C, 30
sec at 56°C, 1 min at 72°C for 45 cycles, followed by 5 min at
72°C. The PCR products were subjected to shrimp alkaline
phosphatase (SAP) reaction for the degradation of residual dNTPs.
The SAP reaction was performed as follows: 40 min at 37°C and 5 min
at 85°C. Following this, extension reaction was performed by the
following protocol: 30 sec at 94°C, 40 cycles for 5 sec at 94°C,
from 5 sec at 52°C to 5 sec at 80°C for 5 cycles, and finally 3 min
at 72°C. Then, the products were desalted using resin. The final
products were analyzed by MALDI-TOF mass spectrometry (Mass
ARRAY® Typer 4.0.5 Software, Sequenom) to identify the
mass. SNP genotyping was performed on
SEQUENOM®MassARRAY® platform using the
iPLEX™ assay (Sequenom) (37).
| Table II.Primer combinations for the 138
mutations used in the multiplex assays |
Table II.
Primer combinations for the 138
mutations used in the multiplex assays
| WELL | SNP_ID | 5′ PCR primer | 3′ PCR primer | UEP_SEQ |
|---|
| W1 | rs121912602 |
ACGTTGGATGGTCTTGAGGATCAGAACCAC |
ACGTTGGATGATGAGATGGCAACCCGAAAG |
AACCACTCCGGCTTC |
| W1 | rs121918233 |
ACGTTGGATGGGAAAACAACCTGGTTCAGC |
ACGTTGGATGTTGCTTAGGAACCTGGCTTC |
GCTGCCAAACCAATG |
| W1 | rs28935493 |
ACGTTGGATGGTTTATCATAGCTACAGCCC |
ACGTTGGATGAGGGAGACAACTTTGAAGTG |
AAGCCTGAGAGAGGT |
| W1 | rs121912489 |
ACGTTGGATGCCCAGGTACTTGATATTTCC |
ACGTTGGATGTGTCCGTACCAGCTTCAGAT |
AGCCTGGTATCTCCTA |
| W1 | rs121912462 |
ACGTTGGATGAGATGTAGCTCTCATCTCCC |
ACGTTGGATGTCCAACCAGGCTCACACACA |
ccTCCCTCTCTGCAGAC |
| W1 | rs121918231 |
ACGTTGGATGAGTTGAAATGTCTCCAGCGG |
ACGTTGGATGACATGAAGCTAAGGTATCTG |
ccCCAGCGGCTATTGGA |
| W1 | rs80356680 |
ACGTTGGATGACCTGCTTGTTGGGAGGAC |
ACGTTGGATGTACTGGGTGCAGTAGGTCTC |
GGAGGACCCGGTTTCTC |
| W1 | rs121434393 |
ACGTTGGATGTCTGCGTTCAACTCACCTTC |
ACGTTGGATGAAGATATGTACTGCAGGCCC |
TCACTTCATCACTCTCCT |
| W1 | rs121912490 |
ACGTTGGATGATCTGGCAATGTGAGTGGAG |
ACGTTGGATGTTGGGTCACGGTAGAAGAAG |
TGTGAGTGGAGGTGTGTG |
| W1 | rs28941777 |
ACGTTGGATGCTTCTCTGTCCATTTAGGTG |
ACGTTGGATGTATGAGTCCTGGTGTGGGTC |
CCATTTAGGTGTGAAACCA |
| W1 | rs119473037 |
ACGTTGGATGCTGCTGGAGGAAGCAGTCAT |
ACGTTGGATGGCAATCACCACTATCTTGCG |
tgacGCAGTCATGCTGCGG |
| W1 | rs28935485 |
ACGTTGGATGTCCTCCTCTCTTGTTTGAAT |
ACGTTGGATGCAGAGGGCCATCTGAGTTA |
ggtggGGCCTCAGCTGGAAT |
| W1 | rs118203956 |
ACGTTGGATGATGGAAACAAGGCACTGGAG |
ACGTTGGATGAATCTGGTCACAGCAAACAC |
cccgGGAGAGCTTTCCTCCCT |
| W1 | rs121909119 |
ACGTTGGATGGAGATATCGGGCCTGAAAAC |
ACGTTGGATGTGTGACTCACACAGTTGACG |
AAGATTTCATCTTTGTAGCCC |
| W1 | rs267606710 |
ACGTTGGATGGTTTTTAGGAAAGAACTGG |
ACGTTGGATGCCTCTAGATTACTTCTCATTG |
AGGAAAGAACTGGAAAAACTG |
| W1 | rs28939695 |
ACGTTGGATGTTCCTCTCCCCGCCAGATCC |
ACGTTGGATGAAACACACCAGCCTCACCC |
gactGCCCAGAAACTGTGGATT |
| W1 | rs267606955 |
ACGTTGGATGCAATTGTCAGGTTGGTGTAG |
ACGTTGGATGTTGATGTCTATGCAGTGCCC |
ggggaTGACTCGGAGCCAGATC |
| W1 | rs28935490 |
ACGTTGGATGCCTGATTGATGGCAATTACG |
ACGTTGGATGACATCAGCCCTCAAGCCAAA |
ccctATGGCAATTACGTCCTTAT |
| W1 | rs104894841 |
ACGTTGGATGCAGGAATCATCAATGTCAGC |
ACGTTGGATGTATCTGTTTTCACAGCCCA |
ATCATCAATGTCAGCAAAATTTC |
| W1 | rs121912466 |
ACGTTGGATGGATAAAGAGGGGCACCCGTA |
ACGTTGGATGACCTCTGACCACCTCCGAAC |
cccCCCTCCTGGAACTCCACCATG |
| W1 | rs121912461 |
ACGTTGGATGTGCTGTCCTCCACTCTGGC |
ACGTTGGATGACTCCATGTCCGATGATCTG |
atcccCTCTGGCCCTGCCGTACCC |
| W1 | rs121907904 |
ACGTTGGATGTGCTGTGCATCTGTAAGTGG |
ACGTTGGATGTCTGAGACCAGTGAGAAACG |
GACAGCTTAAAATATCTCTTATTG |
| W1 | rs199474665 |
ACGTTGGATGGCCCGGTAATCGCATAAAAC |
ACGTTGGATGGTTGGCCATGGGTATGTTGT |
caaaaTTTACAGTCAGAGGTTCAA |
| W1 | rs118203955 |
ACGTTGGATGAAGACCAGTGACTCCATGAC |
ACGTTGGATGATCCACAAATCTCTTCCAAG |
cccaTCAGCTCCTGTAGTCTTACAT |
| W1 | rs74315344 |
ACGTTGGATGCCTTTGCCCTCTTGTTCTCC |
ACGTTGGATGAGCTCTGAGGATGGAGAGGA |
aaacGCCCTCTTGTTCTCCTTGTGC |
| W1 | rs121912603 |
ACGTTGGATGGCTTTCTTTATCCTCTTTGGG |
ACGTTGGATGAACATTCTGGAAGACAGACC |
TGGGATTTCTTCATTATATTCAAACT |
| W1 | rs121912482 |
ACGTTGGATGGGGATTCCAGCAACTCAAAG |
ACGTTGGATGCCCATAGTTCCATGGACAAG |
aagaAACTCAAAGTCAAAAAATTCAA |
| W1 | rs121912488 |
ACGTTGGATGTTGTCTCCCAACGTGTGTAG |
ACGTTGGATGCAGACCTGTTGAAGATCACC |
ggtagGTAGACGAGTCAGGTTCACCC |
| W1 | rs121912485 |
ACGTTGGATGCAATGTCCTGCTTGGTCTGG |
ACGTTGGATGTACTGTTCTGCAGAAGATG |
ggTGGGGAGCCTGGCTGCAATGGCCT |
| W1 | rs80356681 |
ACGTTGGATGACCCTCCCAGCGCCTACTAT |
ACGTTGGATGTCAGCATGGCCGTGACAGAA |
cccaaCAGCGCCTACTATGCTGTGTCC |
| W1 | rs121912605 |
ACGTTGGATGGTTGAAGCCATTGATCGCAG |
ACGTTGGATGTCGTTGCTGAGGCAATGAAC |
gtccACTCTGACCTGCCAATCATCATAT |
| W1 | rs74315347 |
ACGTTGGATGAAGGAGCCCAAGAATCAAGC |
ACGTTGGATGAGCAAATGGCATCTATCTCC |
ctcagCCAAACTTTTTTCTGCCTAGATC |
| W1 | rs267606954 |
ACGTTGGATGTACAGAATCTCCTGACTAAC |
ACGTTGGATGGATGATGTAGAAGAAGACGC |
catAATCTCCTGACTAACAAAGTGGATC |
| W1 | rs121918232 |
ACGTTGGATGTTCTCCATTTCAAAGGAGAG |
ACGTTGGATGTGGCACTGGGTGTTCTTCTG |
tggacAACGTTTAATATACCTGTAGTAA |
| W1 | rs74315343 |
ACGTTGGATGGGTTGTACAAGAGTATGAAAG |
ACGTTGGATGGAGTGTTTTTTTACCAGGGC | cCAAGAGTATGAAAGAGTA
ATTATATTC |
| W2 | rs119473038 |
ACGTTGGATGAAGAGGGTGATCCTGTTGTC |
ACGTTGGATGAGAAAGTTGGCTTGACTGCG |
ACACCAGCCATGTCC |
| W2 | rs119473035 |
ACGTTGGATGCAGAGCTGAGAAGTTATTGG |
ACGTTGGATGTTTGGGATGGGCCTTGCTTG |
TTGGCAGAACAGCAT |
| W2 | rs121434390 |
ACGTTGGATGGTTAACGTTGAGTGAGTGGC |
ACGTTGGATGGGAACGCTTTTTGGATGCAG |
aATCTTCCGCACCACT |
| W2 | rs119473033 |
ACGTTGGATGAGCTCATTTCTCCCCAACAG |
ACGTTGGATGAATTGGTCTCAGAAAGCCCG |
ACAGGCCCCTGATTCAA |
| W2 | rs104894835 |
ACGTTGGATGTTGCACATGAAGCGCTCCCA |
ACGTTGGATGCCTCGTTTCCTGGGACATC |
ctaGTCCTTGCCAATCCA |
| W2 | rs74315346 |
ACGTTGGATGATCTCCAGAGTTTGGAGACG |
ACGTTGGATGCTCCTCCTCTCTTTTAGGTC |
TCAACCTTGTGGTAGGTA |
| W2 | rs121912601 |
ACGTTGGATGTTCAGGCATTACCTTTCAGC |
ACGTTGGATGGTCTTTGGGAGCAAGAAGTG |
TTACCTTTCAGCATCATTC |
| W2 | rs121907909 |
ACGTTGGATGCTTCTCTGTCCATTTAGGTG |
ACGTTGGATGTATGAGTCCTGGTGTGGGTC |
CCAGTGTAAAACTTGTCAG |
| W2 | rs2071225 |
ACGTTGGATGGATGTAGTTCTGGGTTCCTC |
ACGTTGGATGTTAAAAGCCCAGGTTACCCG |
gaatCTGCATTGTCACGGT |
| W2 | rs121907908 |
ACGTTGGATGAGAGGATGGGCGTTGTGTG |
ACGTTGGATGAGCCACACTGAGCCTTTTTC |
gggGTTGTGTGGTTATCGC |
| W2 | rs61747728 |
ACGTTGGATGAACCACTATGAAGCGTCTCC |
ACGTTGGATGCTAAGTACCTTTGCATCTTG |
taatCGTCTCCTAGCACATC |
| W2 | rs2717-192515 |
ACGTTGGATGTTCTGGGCTCACTATCTCAC |
ACGTTGGATGGCAGAAGCATTGTGTACTCC |
gAAGGGCCACATATAAAGAG |
| W2 | rs104894837 |
ACGTTGGATGATGTCGTAGTATCCAAAAC |
ACGTTGGATGAGCAAAGGACTGAAGCTAGG |
CGTAGTATCCAAAACTCCCAG |
| W2 | rs2717-192520 |
ACGTTGGATGCTGGTCCAGCAACATCAACA |
ACGTTGGATGGCTGACATTGATGATTCCTG |
CTGGTTAAAAGATGTCCAGTC |
| W2 | rs121912492 |
ACGTTGGATGTGCAAACACAACACACGTGG |
ACGTTGGATGGCAGGTCACGATAGAAATCC |
ccaccACACACGTGGCCTCAAC |
| W2 | rs28935197 |
ACGTTGGATGGAATCATCAATGTCAGCAAAA |
ACGTTGGATGGAAAGTAAACAGAAGAGTC |
gaACTGTCGGATTTCTGTATAA |
| W2 | rs121909118 |
ACGTTGGATGCTTACATCCTAACAGGTCAG |
ACGTTGGATGTCTGCAGGAACTTTATACCG |
TTTCAGTGACTATGAGAGTGTA |
| W2 | rs28935492 |
ACGTTGGATGCTCTTATTTACCTGTCTAAGC |
ACGTTGGATGATTGCCATCAATCAGGACCC |
catcTACCTGTCTAAGCTGGTAC |
| W2 | rs267607207 |
ACGTTGGATGATCGGACAGAGGCACTGATG |
ACGTTGGATGAGAGAGCTTGCCAAGTGCC |
ggggaTCAGAAGGAGGACTTCAA |
| W2 | rs28942089 |
ACGTTGGATGCCAGCAATGAGAAGTGAACC |
ACGTTGGATGTTCAGACCAGCTCAAAAGAC |
ctAAGTGAACCTACAAACCTGTAT |
| W2 | rs267606919 |
ACGTTGGATGGACGCAGGAGGAGGTGTCT |
ACGTTGGATGGGTACCTCTGAGTGAGGGAA |
actCAGGAGGAGGTGTCTTATTCC |
| W2 | rs199474663 |
ACGTTGGATGTTGTTAAGATGGCAGAGCCC |
ACGTTGGATGAGAGGAATTGAACCTCTGAC |
ttggtAGAGCCCGGTAATCGCATA |
| W2 | rs267607183 |
ACGTTGGATGGCAGGTGCTTACCGATAAAC |
ACGTTGGATGCAACGCCGTCATCTTGGG |
ccttTGCTTACCGATAAACTCGTTC |
| W2 | rs121909486 |
ACGTTGGATGAGACACTGGCAGCTGAGAC |
ACGTTGGATGGGTGGCCTCTTACCTTTGC |
gcggGGTCCAGGTCTGGTTTCAGAA |
| W2 | rs121912487 |
ACGTTGGATGCTCTGTAGGTCCAACT TAAC |
ACGTTGGATGGAGAAGTAACCACA CTGACC |
cccctATTCCAGCAACTCAAA GTCAA |
| W2 | rs104894834 |
ACGTTGGATGCCCAAAGAGATT CAGAAGGC |
ACGTTGGATGACATAATTAGCTAGC TGGCG |
aagtCAGACTTCAGGCAGACCCT CAG |
| W2 | rs121912467 |
ACGTTGGATGTTCACCGTGTAG CGGTAGG |
ACGTTGGATGTTGGGCCCATGAAGA AAGTG |
gggccGGTTCTCAATAAGCAGC ATCC |
| W2 | rs2717-192517 |
ACGTTGGATGCCCTCTGTCCATTCATTCTT |
ACGTTGGATGGGAGACATGGTAACAAGTCA |
ccTTAACCTGTTTAATTTTCTTCTCAG |
| W2 | rs121907911 |
ACGTTGGATGAGTTCACTGGCACAGCCGGA |
ACGTTGGATGTTAGGAAACATCCTGGCCTG |
gacgGGCACAGCCGGAGCCTGTCGCTA |
| W2 | rs121907911 |
ACGTTGGATGAGTTCACTGGCACAGCCGGA |
ACGTTGGATGTTAGGAAACATCCTGGCCTG |
gacgGGCACAGCCGGAGCCTGTCGCTA |
| W2 | rs121434394 |
ACGTTGGATGGGAAATTAAGCAGGACATCTC |
ACGTTGGATGAAGTTCTGCTAGGTCTTCTG |
ctcccTTAAGCAGGACATCTCAAGTCTC |
| W2 | rs28935488 |
ACGTTGGATGGTTTCCTCCTCTCTTGTTTG |
ACGTTGGATGAGGGCCATCTGAGTTACTTG |
gcATTATTTCATTCTTTTTCTCAGTTAG |
| W2 | rs121912465 |
ACGTTGGATGAGCAAACCGCTGCAAGAAGG |
ACGTTGGATGAGTAGGCGCAGTCCTTATCC |
ccgtTGCAAGAAGGCCCCAGTGAAGAGC |
| W2 | rs80356682 |
ACGTTGGATGCCGGATCCTAGATGCAAAGA |
ACGTTGGATGACCTCCTGCTCTGTGACTG |
ctcgGCAAAGAGTAAGATTGAGCAGATC |
| W3 | rs104894836 |
ACGTTGGATGTGATGCAGGAATCTGGCTCT |
ACGTTGGATGTGCACTGGGAGCGCTTCAT |
CTGGCTCTTCCTGGC |
| W3 | rs104894849 |
ACGTTGGATGTTACAGGCCACTCCTTTACC |
ACGTTGGATGGGGCTGTAGCTATGATAAAC |
AGCAACTGCGATGGT |
| W3 | rs267607208 |
ACGTTGGATGTTATGTCTGTCAGGCTCAGG |
ACGTTGGATGGGAGGACTTCAACAGCAAAC |
GGGTATGGGCAGAGA |
| W3 | rs28935196 |
ACGTTGGATGTGGGAGTTTTGGATACTACG |
ACGTTGGATGCTGTCACAGTAACAACCATC |
CCAGACCTTTGCTGAC |
| W3 | rs104894847 |
ACGTTGGATGAGAACTACATCTGGGCTGCG |
ACGTTGGATGTGCTCTAGCCCCAGGGATGT |
cTGCGCTTCGCTTCCTG |
| W3 | rs121907905 |
ACGTTGGATGTCTTCCCCAAGGTGAGAAAC |
ACGTTGGATGTGTCTTTTGAGCTGGTCTG |
AGTGTGACTTCAAGGAC |
| W3 | rs104894833 |
ACGTTGGATGAGACCATGAGCTCTGCCATC |
ACGTTGGATGTGGGAGGTACCTAAGTGTTC |
TCTCCATGAAGAGCTTCT |
| W3 | rs104894838 |
ACGTTGGATGTCTCATACAGGTTATAAGC |
ACGTTGGATGAAGGGCCACATATAAAGAGG |
AAGCATTGTGTACTCCTG |
| W3 | rs121909489 |
ACGTTGGATGTTCCTGATGCGAGTCAACGA |
ACGTTGGATGAAGTAGCAGCTGGTGGTGAG |
TGGCACGAGGAGTGTTTG |
| W3 | rs121909491 |
ACGTTGGATGTTGAAGGCTCTTCGCTGCTG |
ACGTTGGATGAGTCAGAGCAAGGGCAGCG |
gGCGTGGTGAGGATGGTC |
| W3 | rs137853042 |
ACGTTGGATGACTGGCTCTCCTCATATTCG |
ACGTTGGATGCACCTTCATCCTGGAAGGTC |
aTCATATTCGTTCCTGACTC |
| W3 | rs80338755 |
ACGTTGGATGTCTCCATGACCACGATGCTC |
ACGTTGGATGTTCTGGCCAGATGTTCAGGG |
ctaaCTCCGCCTGGGTGTTG |
| W3 | rs199474668 |
ACGTTGGATGTTGAACCTCTGACTGTAAAG |
ACGTTGGATGGTATTATACCCACACCCACC |
tgTTATGCGATTACCGGGCT |
| W3 | rs267606918 |
ACGTTGGATGGCGCTTACCAGTGCATTGTG |
ACGTTGGATGCCCACATCTGACAACAAGAC |
AGTGCATTGTGGACAATGGG |
| W3 | rs119473034 |
ACGTTGGATGCTTGCCTCTTACAGAGGAGC |
ACGTTGGATGATAACTTCTCAGCTCTGCGG |
AGGAAAAAGATTGAAGAGAAT |
| W3 | rs121912491 |
ACGTTGGATGCAGACCTGTTGAAGATCACC |
ACGTTGGATGTTGTCTCCCAACGTGTGTAG |
ccccTGAAGATCACCAACCTAC |
| W3 | rs2717-192513 |
ACGTTGGATGCAGTCCTCTGAATGAACAAG |
ACGTTGGATGAGACTTCAGGCAGACCCTCA |
cctcCATAATTAGCTAGCTGGC |
| W3 | rs121434392 |
ACGTTGGATGAGACAATGACAGGTAAGCCG |
ACGTTGGATGAGAAACAGAAGCATGACTCG |
TAGGCATTAATCCTAGATCTGG |
| W3 | rs199474660 |
ACGTTGGATGACAGTCAGAGGTTCAATTCC |
ACGTTGGATGTGGGTACAATGAGGAGTAGG |
AGAGGTTCAATTCCTCTTCTTAA |
| W3 | rs267606879 |
ACGTTGGATGTACCGATAAACTCGTTCCGC |
ACGTTGGATGTGATCAACGCCGTCATCTTG |
ggaaaTCGTTCCGCAGCTGGGTG |
| W3 | rs2717-192524 |
ACGTTGGATGTATTTACCTGTCTAAGCTGG |
ACGTTGGATGTCAAGCCAAAGCTCTCCTTC |
ccctTCCTGATTGATGGCAATTAC |
| W3 | rs121918230 |
ACGTTGGATGCTGTGATCCATCTGTTTGCC |
ACGTTGGATGAATTTCTTTACCTGATGGGC |
CTGGAGTTATGTGGACACTAATAT |
| W3 | rs104894842 |
ACGTTGGATGGGGCCACTTATCACTAGTTG |
ACGTTGGATGCAGGCTAAGCCTGAGAGAG |
gaAGGGAGACAACTTTGAAGTGTG |
| W3 | rs121909488 |
ACGTTGGATGAGGTGGTACACGCACTCCAG |
ACGTTGGATGTGTGCATCCGCAGGCTCTTC |
cccgtTGGGGGCGATCTTCTCCATG |
| W3 | rs121912486 |
ACGTTGGATGGAGAAGTAACCACACTGACC |
ACGTTGGATGCTCTGTAGGTCCAACTTAAC |
gttgTGACTTTGAGTTGCTGGAATC |
| W3 | rs104894844 |
ACGTTGGATGCTTCATCACACAGCTCCTCC |
ACGTTGGATGATGTGACTTCTTAACCTTG |
ggtggAAAGGAAGCTAGGGTTCTAT |
| W3 | rs119473036 |
ACGTTGGATGAGTACTTAAAGACTTTGCC |
ACGTTGGATGTGACCTTCTTAGCAAGTTGG |
ggacAGACTTTGCCAAGTTTCTTACA |
| W3 | rs267606953 |
ACGTTGGATGGAATGACATCCTGTGCAGTG |
ACGTTGGATGCCAAGTGCATGATGTTTCTC |
gacgaGGGGTGCTTTGATGACTGTTC |
| W3 | rs74315345 |
ACGTTGGATGATCTGACGCCCCTTAGTTAC |
ACGTTGGATGTGGTGGCGCTGTTGGAGAG |
tatCAGATAGTGGTGCTGAATCCGTAC |
| W4 | rs121909490 |
ACGTTGGATGAAGCGACCCCGGACCATCCT |
ACGTTGGATGACCTCGAAGGAGGCCTTGAA |
CTCACCACGCAGCAG |
| W4 | rs104894848 |
ACGTTGGATGGCTTCATGTGCAACCTTGAC |
ACGTTGGATGACACATGGAAAAGCAAAGGG |
CCAGATTCCTGCATCA |
| W4 | rs199474659 |
ACGTTGGATGTTGTTAAGATGGCAGAGCCC |
ACGTTGGATGAGAGGAATTGAACCTCTGAC |
tAGAGCCCGGTAATCG |
| W4 | rs121912468 |
ACGTTGGATGTGGAGGAGACGACATTGAAG |
ACGTTGGATGACAGCACTCTTCCTGTATTC |
GACATTGAAGGCCAGA |
| W4 | rs267607071 |
ACGTTGGATGTCTCCGACAGTTGGAACTGC |
ACGTTGGATGACATCCGCATCGATGGCTC |
tCTGGCACAGGTCCTCC |
| W4 | rs142059681 |
ACGTTGGATGCGATAACCACACAACGCCC |
ACGTTGGATGACCTGAATGCCTCTGAAGAC |
CACACAACGCCCATCCTC |
| W4 | rs121907907 |
ACGTTGGATGACCAGTGTGACTTCAAGGAC |
ACGTTGGATGGAAGTGAACCTACAAACCTG |
AGACCAGCTCAAAAGACA |
| W4 | rs121912484 |
ACGTTGGATGCCACGGTCCTCCAGCCCAG |
ACGTTGGATGATGCGGACCTCCGGGAGCAG |
cccaCAGCCCAGGCCCTGA |
| W4 | rs121912463 |
ACGTTGGATGACAGCTTGGGCCTGTCCAAC |
ACGTTGGATGTCTCATCTCCCTTCCTTGTC |
tAACCAGGCTCACACACAC |
| W4 | rs104894851 |
ACGTTGGATGTATCTGTTTTCACAGCCCA |
ACGTTGGATGCAGGAATCATCAATGTCAGC |
ATACAGAAATCCGACAGTA |
| W4 | rs74315348 |
ACGTTGGATGCTGAAGCGCAAAGACAAGCC |
ACGTTGGATGACGAGCAGGCCTTCCTAAAG |
gaAAAGACAAGCCAAAGTG |
| W4 | rs121908415 |
ACGTTGGATGCCCTTGTTGTTCACTTGCAG |
ACGTTGGATGAAAGGCATGGTAGAAGCTGG |
ggaaGGCCCGGCCCGACGAG |
| W4 | rs121912604 |
ACGTTGGATGTTCTGTGGGACAAGAACTGC |
ACGTTGGATGATCCATGCTGTCCAGATCTC |
caccAGAACTGCCCCATGTAT |
| W4 | rs121434391 |
ACGTTGGATGAGAAGCAAAGCATCCCCAAC |
ACGTTGGATGGAATGCCCTACAGTTGGCAG |
TCTGTAATTTCCAGATGCTCA |
| W4 | rs28935487 |
ACGTTGGATGTTGAACAAGGAGGGCTCAAG |
ACGTTGGATGCCAGGAGAGAATTGTTGATG |
GAGGGCTCAAGTTTTTACCATA |
| W4 | rs104894852 |
ACGTTGGATGCTTCAAGGTTAAGAAGTCAC |
ACGTTGGATGCTGCATTGTATTTTCTAGCTG |
ctTTAAGAAGTCACATAAATCCC |
| W4 | rs104894827 |
ACGTTGGATGTAGCCTGGGCTGTAGCTATG |
ACGTTGGATGTTACAGGCCACTCCTTTACC |
ggttGGCTGTAGCTATGATAAAC |
| W5 | rs2717-192514 |
ACGTTGGATGGAGGAACCCAGAACTACATC |
ACGTTGGATGAGGGATGTCCCAGGAAACGA |
CTGCGCGCTTGCGCT |
| W5 | rs267606880 |
ACGTTGGATGCGGAGTAGTTGACCACAGAG |
ACGTTGGATGACGGAGGCCAACCTGGAGA |
ACAGAGGGCATCTGG |
| W5 | rs2717-192511 |
ACGTTGGATGCTAGCTTCCTTTTCACAGGG |
ACGTTGGATGAGTTGCTTCCCTGGGTAAAG |
ACAGGGAGGAGCTGTG |
| W5 | rs2717-192516 |
ACGTTGGATGGCAGAAGCATTGTGTACTCC |
ACGTTGGATGCAGTTCTATTGGATTCTGGG |
CCCTTTCAAAAGGTGAG |
| W5 | rs121909487 |
ACGTTGGATGTTGAAGGCTCTTCGCTGCTG |
ACGTTGGATGAGTCAGAGCAAGGGCAGCG |
GAGGATGGTCCGGGGTC |
| W5 | rs28935495 |
ACGTTGGATGAGGGCCATCTGAGTTACTTG |
ACGTTGGATGGTTTCCTCCTCTCTTGTTTG |
ataCAGCTGAGGCCAAAG |
| W5 | rs2717-192521 |
ACGTTGGATGATAGGAAACAAGCCTACCGC |
ACGTTGGATGGTTGATGTTGCTGGACCAGG |
GGGTCTTGAACAAGGAGG |
| W5 | rs104894845 |
ACGTTGGATGAGCAAAGGACTGAAGCTAGG |
ACGTTGGATGTCGTAGTATCCAAAACTCCC |
ATGTTGGAAATAAAACCTGC |
| W5 | rs28941778 |
ACGTTGGATGTATGAGTCCTGGTGTGGGTC |
ACGTTGGATGCTTCTCTGTCCATTTAGGTG |
gtTGTGGGTCTTCAGGTGGT |
| W5 | rs121908417 |
ACGTTGGATGCTCCTGAAAAGGCATGGTAG |
ACGTTGGATGCTTGTTGTTCACTTGCAGAC |
gggcGGCATGGTAGAAGCTGG |
| W5 | rs121907910 |
ACGTTGGATGGAAGTGAACCTACAAACCTG |
ACGTTGGATGACCAGTGTGACTTCAAGGAC |
ccctTTTTGAGCTGGTCTGAAC |
| W5 | rs121434395 |
ACGTTGGATGAAGTTCTGCTAGGTCTTCTG |
ACGTTGGATGAGCAGGACATCTCAAGTCTC |
TGAGATTTTTCTTCAAGGAGTT |
| W5 | rs104894843 |
ACGTTGGATGGTTTATCATAGCTACAGCCC |
ACGTTGGATGAGGGAGACAACTTTGAAGTG |
caaaCTAAGCCTGAGAGAGGTC |
| W5 | rs2717-367202 |
ACGTTGGATGGCTGACATTGATGATTCCTG |
ACGTTGGATGCTGGTCCAGCAACATCAACA |
gTTCCTGGAAAAGTATAAAGAGT |
| W5 | rs199474658 |
ACGTTGGATGGCCCGGTAATCGCATAAAAC |
ACGTTGGATGGTTGGCCATGGGTATGTTGT |
aGGTAATCGCATAAAACTTAAAAC |
| W5 | rs104894828 |
ACGTTGGATGTGAAGGAGAGCTTTGGCTTG |
ACGTTGGATGCAAGTAACTCAGATGGCCCT |
gatgTGGCTTGAGGGCTGATGTGT |
| W6 | rs121912483 |
ACGTTGGATGACTGGCACCGAACATCCTG |
ACGTTGGATGACCTGGCGAGTGTACCAGTA |
CATCCTGCCAGCTCT |
| W6 | rs2717-192510 |
ACGTTGGATGTTACAGGCCACTCCTTTACC |
ACGTTGGATGGGGCTGTAGCTATGATAAAC |
CCAGGGAAGCAACTG |
| W6 | rs121907902 |
ACGTTGGATGTATGAGTCCTGGTGTGGGTC |
ACGTTGGATGCTTCTCTGTCCATTTAGGTG |
TGGGTCTTCAGGTGG |
| W6 | rs121909492 |
ACGTTGGATGTTCTGAAACCAGACCTGGAC |
ACGTTGGATGACCTGTTCCCCTCTCTCTGA |
GCTGCCAGTGTCTCTC |
| W6 | rs151195362 |
ACGTTGGATGTCAAGCCAAAGCTCTCCTTC |
ACGTTGGATGTATTTACCTGTCTAAGCTGG |
CAGGACCCCTTGGGCAA |
| W6 | rs2717-192522 |
ACGTTGGATGCGCAGCCCAGATGTAGTTCT |
ACGTTGGATGTTAAAAGCCCAGGTTACCCG |
acGTAGTTCTGGGTTCCTC |
| W6 | rs2717-192519 |
ACGTTGGATGGCTGACATTGATGATTCCTG |
ACGTTGGATGCTGGTCCAGCAACATCAACA |
TTGATGATTCCTGGAAAAG |
| W6 | rs199474661 |
ACGTTGGATGAGAGGAATTGAACCTCTGAC |
ACGTTGGATGACAGGGTTTGTTAAGATGGC |
GTAAAGTTTTAAGTTTTATGCGA |
WES analysis
WES was performed on the two siblings. A total of 6
µg sample DNA was prepared. First, the qualified DNA samples were
randomly fragmented to generate 200–300 bp DNA fragments. The
extracted DNA was amplified in a ligation-mediated (LM)-PCR, as
described earlier. The NimbleGen human exome array (SeqCap EZ Human
Exome Library; version 2.0; NimbleGen, Roche Diagnostics cat. no.
06465684001 or 06465692001) was used to capture the exons of the
human genome. High-throughput sequencing was performed on a
Hiseq2000 platform (Illumina), and the sequence of each library was
generated as 90 bp paired-end reads. The raw image files were
processed by Illumina base calling Software (Illumina Inc. San
Diego, CA, USA, version 1.7; HCS1.5.15.1, RTA1.13.48, OLB 1.9.4).
The obtained sequences were aligned to the reference genome [human
genome build37 (hg19)] using Burrows-Wheeler Aligner (BWA;
bio-bwa.sourceforge.net/; version:
0.5.9-r16). Single-nucleotide polymorphisms (SNPs) were detected by
SOAPsnp (http://soap.genomics.org.cn/soapsnp.html; version
1.05) and small insertions/deletions (indels) were detected by
SAMtools (version: 0.1.18; www.htslib.org/). Called SNP variants and indels were
annotated and classified using ANNOVAR (www.ncbi.nlm.nih.gov/pmc/articles/PMC2938201/).
Variants were filtered using data from dbSNP 142 and the 1000
Genomes Project.
Pathological diagnosis
The renal tissue was fixed in 10% neutral buffered
formalin and stored at room temperature or 4°C. The fixed tissue
was embedded in paraffin and 2-µm sections were cut. HE, PAS,
PASM+Masson and Masson staining were performed at 37°C as described
previously (22).
In silico analyses of the effect on
protein structure and function
Selected bioinformatics tools were used to assess
the effect of sequence variants on the structure and function of
the receptor. A total of two indirect in silico predictors,
PolyPhen2 (Polymorphism Phenotyping version 2; genetics.bwh.harvard.edu/pph2/index.shtml) and
SIFT (sift.jcvi.org), were used to evaluate
the possibly damaging effects of single amino-acid substitutions on
the expression of the proteins of these genes. To identify
potential pathogenic mutations, additional analysis focused on the
variants that are listed in OMIM as being associated with FSGS
(even if frequent) and also any other indels and nonsense variants.
For the missense variants, the high risk variants were determined
by a minor allele frequency (MAF) of 1% or unknown (using 1,000
Genomes population data; www.1000genomes.org/node/506).
Results
Quality control
Mass array technology assigns a quality code for
each genotyping call. Codes A, B, C, D and I stand for
conservative, moderate, aggressive, low possibility and bad
spectrum, respectively. The lower the order of the code from A to
Z, the higher the quality of the genotyping calls. Code A indicates
the highest quality and code D and I indicate no genotyping call
(reported as NA). The overall quality of the assay is summarized in
Table III. The total number of
genotyping calls are 5,658, which is a product of 138 (the number
of variations) and 41 (the number of samples). The percentage of
code A calls is 90. 84% and the sum of code A, code B and code C
calls is 98.09%. Overall, the assay achieved a good quality. The
assay quality was also investigated to see the distribution of no
call genotypes (quality code D and I) among the mutations
identified. Two variations (rs121912601, rs2717-192514) did not get
genotypes from >10% samples and genotyping of 4 variations
(rs121918230, rs121907911, rs28935487, rs267607183) failed in
>20% samples. As the Mass array genotyping assay is multiplexed,
these mutations/SNPs are likely to be susceptible to assay
condition variations. The performance of the assay may be improved
by redesigning the PCR primers and extension primers for these
variations.
| Table III.Analysis of overall quality of the
assay. |
Table III.
Analysis of overall quality of the
assay.
| Class | Count | Percentage, % |
|---|
| Conservative | 5,140 | 90.84 |
| Moderate | 363 | 6.42 |
| Aggressive | 47 | 0.83 |
| Low
possibility | 100 | 1.77 |
| Bad spectrum | 8 | 0.14 |
| Total | 5,658 | 100 |
The two affected sisters were selected for exome
sequencing. For each participant, 4,4017,835 bases were created and
covered on the target. The sequence data were generated with a ×177
average coverage for each subject. An average coverage of the
target region was 98.96 and 99.27% of the target region had at
least ×4 coverage. For each participant, 21,134 single-nucleotide
variants were identified, of which 9,986 were missense mutations
and 135 were nonsense (premature termination) mutations.
Clinical characteristics
A total of two female siblings presented with MH at
9 and 6 years-old, respectively. The oldest sibling was referred to
the Second Xiangya Hospital for persistent MH (8 months) with
macroscopic hematuria initially. The physical examination revealed
no abnormalities and the older sibling did not suffer from
hypertension, sensorineural deafness, or eye involvement.
Laboratory tests revealed only MH, which was demonstrated to be
glomerular hematuria by the urinary sediment test (erythrocytes
100,000/high power field; 70% of glomeruli; urinary protein 0
mg/dl), with normal renal function. Values obtained in the
hematological, biochemical and serological tests were: Serum
creatinine, 26.8 µmol/l; hemoglobin, 111 g/l; total protein, 61.5
g/l; uric acid, 83.6 µ/l; cholesterol, 3.5 µmol/l; complement
component, 31.16 g/l and blood urea nitrogen 4.58 µmol/l.
A renal biopsy was performed in hospital and this
demonstrated severe glomerular alterations consistent with FSGS
(Fig. 1). Due to continuous MH and
positive family history with renal disease, a renal biopsy was
performed at the Second Xiangya Hospital. On light microscopy,
characteristic lesions of focal glomerulosclerosis were present in
2 of 28 glomeruli (Fig. 1A).
Sclerotic glomeruli were present in 1 of 28 glomeruli and small
arteries exhibited loss of smooth muscle fibers. (Fig. 1B). There was mild mesangial matrix
proliferation. Vacuolation of the tubular epithelial cells, loss of
the brush border of lumen surface and inflammatory cell
infiltration was observed (Fig.
1C). Mitochondria in podocytes demonstrated normal morphology
(original magnification, ×10,000; data not shown). On
immunofluorescence, focal segmental coarse granular deposits of
immunoglobulin G(+) and proliferation of endothelial cells were
observed. Electron microscopy exhibited diffuse podocytic foot
process effacement (Fig. 1D). The
endothelium was swollen and hypertrophied, however the GBM
exhibited a normal structure and thickness. Paramesangial deposits
were noted. Massive tubules with swollen tubular epithelial cells,
edema in the interstitium and inflammatory cell infiltration were
noted (data not shown).
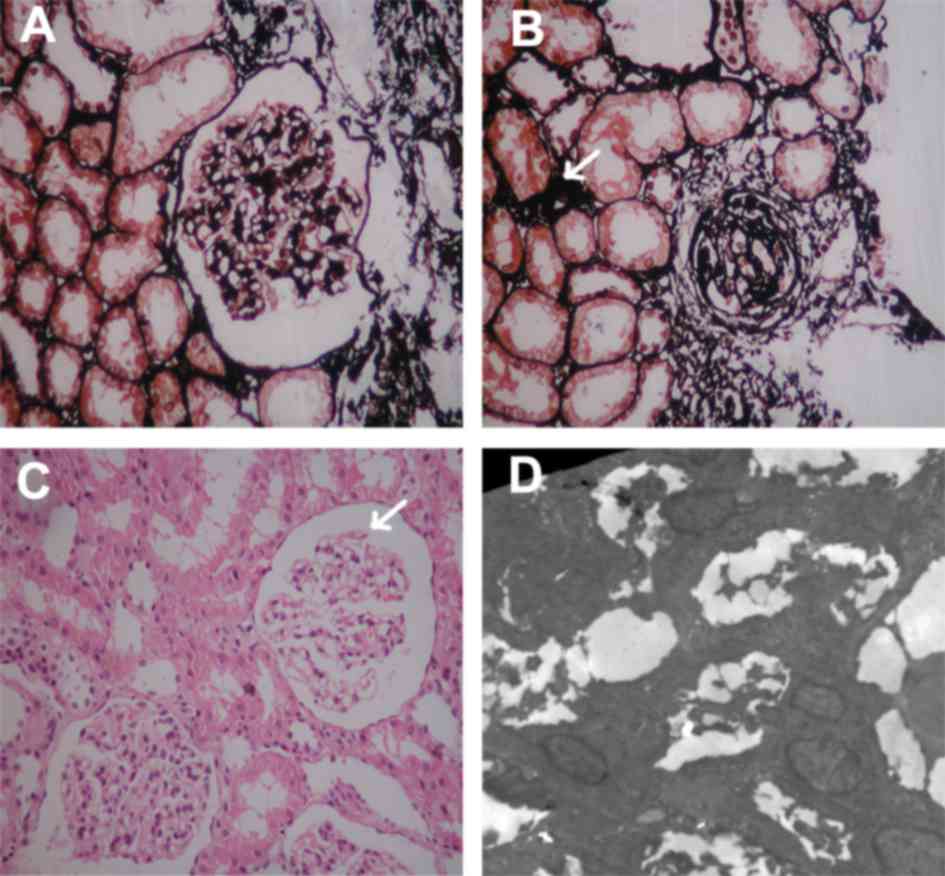 | Figure 1.Light and electron microscopic images
of the renal biopsy taken from the proband. (A) Light microscopy,
PASM staining. A glomerulus is exhibited with segmental sclerosis,
hyalinosis and adhesion to Bowman's capsule, consistent with an
FSGS lesion. (B) Light microscopy, PASM staining of the renal
tubules and glomeruli. Exhibited is globally sclerotic glomeruli
with small arteries demonstrating loss of smooth muscle fibres
(arrow). (C) Light microscopy, periodic acid-Schiff staining of the
renal biopsy taken from the proband. Exhibited is a glomerulus with
segmental sclerosis, hyalinosis (arrow), consistent with an FSGS
lesion, vacuolation of the tubular epithelial cells, effacement of
the brush border of lumen surface and inflammatory cell
infiltration. (D) Electron microscopy of kidney section, (original
magnification, ×5,000). Glomerular segment with extensive podocytes
foot process effacement. The endothelium was swollen and
hypertrophied. The glomerular basement membrane has normal
structure and thickness. Electron-dense deposits were observed in
the paramesangial regions. Massive tubules with swelling tubular
epithelial cells, edema in the interstitium and inflammatory cell
infiltration (not shown). FSGS, focal segmental glomerulosclerosis;
PASM, Periodic Schiff-Methenamine Silver. |
The patient was born following a full-term normal
pregnancy as the first child of unrelated Chinese parents. The
family history was remarkable in that multiple family members were
affected by isolated MH or other renal disease in her father's
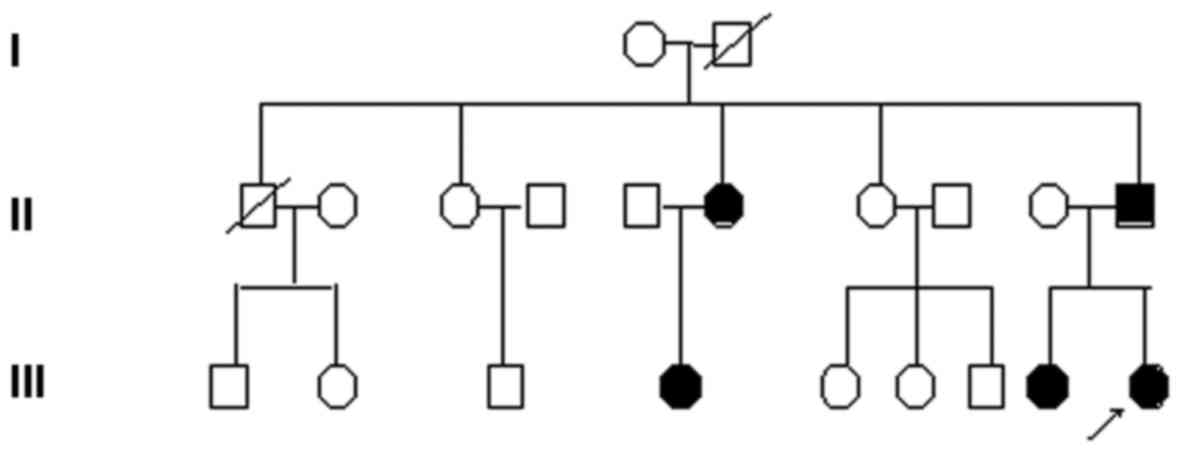
pedigree (Fig. 2). The family
history revealed that the parents were Chinese in origin and
non-consanguineous. Her sister was also identified as being
affected by isolated MH, histologically characterized as FSGS. Her
father was diagnosed with CKD by qualified doctors 8 years ago, in
another hospital. Her paternal aunts and paternal cousins have also
been identified as exhibiting hematuria. Urinalyses and blood
chemistries identified isolated MH. None of the affected
individuals had ESRD, sensorineural hearing loss, or eye
complications including lenticonus. Their mother was well and was
not known to have any kidney disease.
Variants of TRNL1 in the family
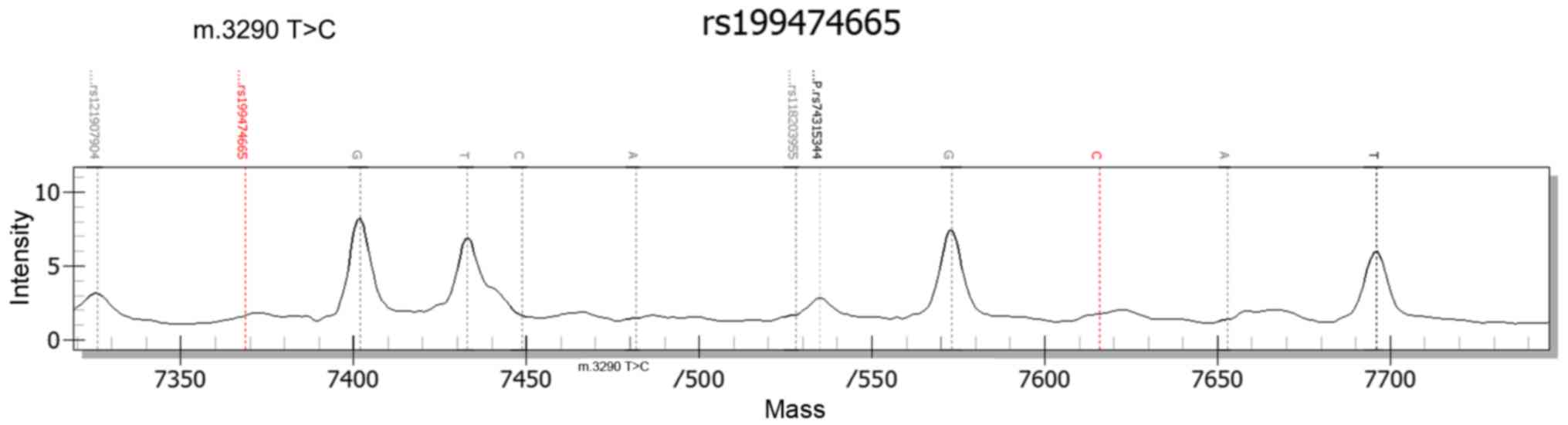
Using Massarray technology, the same mutation in
TRNL1 (m. 3290T>C) was identified in the two sisters, which was
not demonstrated by polyphen-2 and SIFT. However, it was predicted
to be a pathogenic alteration based on OMIM and Pubmed. This
mutation mtT3290C was first detected by Opdal et al
(38) in 1 of the sudden infant
death syndrome (SIDS) cases and it suggested that mutations in
mitochondrial DNA (mtDNA) may serve a role in certain cases of
SIDS. It was speculated that mtT3290C may segregate with FSGS
(38). The results are exhibited
in Fig. 3. No significant sequence
mutations were observed in the other 17 genes analyzed.
Variants of COL4A3 in two sisters with
familial FSGS
WES was performed on the proband and her sister, and
this identified a heterozygous candidate COL4A4 missense mutation
c. 4195A>T(p. M1399L) which was not identified by polyphen-2 and
SIFT. Examination of the mutation using 1,000 Genomes provided
evidence that the identified sequence variant is a rare
polymorphism with a MAF of 0.0022. In addition, the mutation that
was identified was in the NC1 (trimeric noncollagenous) domain. It
was previously demonstrated that mutations of COL4A3/COL4A4/COL4A5
in the NC1 domain disrupt heterotrimer formation in podocytes and
subsequently inhibit secretion into the GBM domain (39). These results indicate that the
substitution is pathogenic and may lead to FSGS.
Discussion
In the present study, a Chinese family presenting
with GMH, or CKD were investigated. Renal biopsies from the proband
and her sister demonstrated FSGS and normal GBM. Her father reached
CKD 8 years ago. A genetic analysis was performed on 15 genes
associated with FSGS in the proband and her father, and a
homozygous m. 3290T>C missense mutation in TRNL1 was identified
in the two siblings. Next generation sequencing of the siblings was
performed to reveal a second mutation, a heterozygous c. 4195A>T
missense variant in COL4A4. To the best of the authors knowledge,
this is the first report of the two aforementioned mutations that
may co-segregate with disease in familial hematuria, histologically
characterized by FSGS.
It has been demonstrated that podocyte damage is
sufficient to cause FSGS, which results from a number of
podocyte-specific gene mutations (7–24).
In the present study, Massarray sequencing of 18 podocyte-specific
genes in a family including two sisters and their father was
performed. In the family, no mutations were identified in the genes
most frequently reported including NPHS1, NPHS2, CD2AP, PLCE1,
ACTN4, TRPC6, INF2 and WT1.
mtDNA including COQ2 and PDSS2 (18–19),
has also been identified to be associated with focal
glomerulosclerosis. Human mtDNA which encode subunits of enzyme
complexes involved in oxidative energy metabolism may result in
various diseases and syndromes and the most severely affected
organs are the brain, heart, skeletal muscle, sensory organs and
the kidney, in mtDNA associated diseases (40). In recent years, the involvement of
the kidney has been concerned in mitochondrial cytopathies by
nephrologists. tRNALeu (UUR) gene also called mitochondrial
tRNAleucine 1, is a hotspot in mitochondrial disease and has a high
incidence of mutations (41). The
tRNALeu (UUR) mutation is associated with the mitochondrial
myopathy, encephalopathy, lactic acidosis and stroke-like episodes
(MELAS) syndrome. Renal tubular dysfunction and FSGS have been
associated with MELAS (42,43).
It has been reported that the mtDNA mutation may also cause
isolated renal disease in patients who were not diagnosed as MELAS
(44–46). It was reported by Lowik, et
al (17) that 3243A-G may be
identified in a steroid-resistant nephrotic syndrome with
histological signs of FSGS. They concluded that mtDNA abnormalities
lead to a steroid-resistant nephrotic syndrome with histological
signs of FSGS. In the present study, an mtT3290C mutation in TRNL1
in two siblings was identified. It was first reported by Opdal
et al (38) that the
mtT3290C mutation may serve a role in various patients with the
SIDS cases (40).
The T3290C mutation is located in the T ΨC loop of
the TRNL1 gene, disrupting the three-dimensional shape of this
tRNA. It has been proposed that a common pathogenic mechanism
associated with mutations in this particular mtDNA gene may be a
decreased steady-state level of tRNALeu (UUR) and a partial
impairment of mitochondrial protein synthesis (47). Opdal et al (40) proposed that any mtDNA mutation may
affect oxidative energy metabolism and thereby induce adenosine
triphosphate depletion (40), and
m. T3290C mutation may affect the complex structural podocyte
composition by affecting metabolic and energy requirements. Hence,
there are several reasons to suggest that m. 3290T>C in TRNL1
gene may be involved in FSGS. In addition, homozygous mutation was
identified in the two sisters and their father, whereas mtDNA
exhibited maternal inheritance. However, blood from the mother was
not available and it is unclear whether these mutations are somatic
or inherited. To the best of the authors knowledge, the present
study is the first to document the development of FSGS and isolated
hematuria with the mitochondrial T3290C transition.
However, the affected cases were in the father's
pedigree including paternal aunts and paternal cousins, and it were
not consistent with typical maternal inheritance.
This suggests that mutations in other genes may also
be involved in the development of FSGS. The list of genes
implicated in the development of FSGS is updated continuously. The
introduction of more comprehensive screening technologies including
WES allows simultaneous screens for mutations in other potentially
relevant genes and contributes to the detection of novel genes or
mutations (48,49), instead of testing for one gene at a
time, or screening for certain known mutations only.
As the sisters exhibited normal GBMs, the type IV
collagen-associated genes were not initially screened. However,
using WES technology a single heterozygous mutation c. A4195T (p.
M1399L) in the COL4A4 gene, which encodes the α4 chain of type IV
collagen, the most important structural component of the GBM, was
identified.
Though the variant was not demonstrated by SIFT and
polyphen-2, the COL4A4 mutation that was identified in the siblings
is most likely disease-causing. Firstly, certain studies suggest
that COL4A3 and COL4A4 mutations may cause a wide spectrum of
disease phenotypes from AS to FSGS (39,50,51).
Malone et al (51) was the
first to document COL4A3 and COL4A4 mutations associated with
primary FSGS. The authors identified seven variants in COL4A3 and
COL4A4 in a cohort of 70 families with a pathological diagnosis of
familial FSGS of unknown cause. Notably, each of these variants
were heterozygous and no mutations in known FSGS-associated genes
were identified (51). The authors
hypothesized that mutations in mature GBM collagen (IV) may have a
direct role in the pathogenesis of FSGS and that the phenotypes
induced by mutations in mature GBM collagen (IV) genes may
phenocopy primary FSGS. Secondly, the mutation that was identified
in these variants is exhibited at very low frequencies of 0.0022 by
1,000 Genomes. In addition, the mutation is located in the NC1
domain where the variant may disrupt heterotrimer formation in the
podocyte and subsequent secretion into the GBM domain (39). Molecular and bioinformatics
analyses suggested that the mutations in the conserved glycine-rich
regions or in the NC1 carboxy terminus of the involved proteins are
deleterious (52). It is now
recognized that the mature type IV collagen network, a3a4a5,
originates solely in the podocytes (53). Kruegel et al (54) proposed that podocyte receptors may
recognize the mutated COL4 leading to upregulation of podocyte
profibrotic factors, including transforming growth factor-β,
connective tissue growth factor and matrix metalloproteinases-2. −9
and −10. These data add support to the hypothesis that these
variants may cause disease.
The COL4A4 mutations follow an autosomal dominant or
recessive inheritance pattern. The patients with heterozygous
mutations in the COL4A3/COL4A4 are more common in the carrier state
of atherosclerotic renal artery stenosis and TBMN than autosomal
dominant AS, and familial hematuria and GBM morphology are typical
clinical features of these diseases (54,55).
The patients with COL4A4 mutations documented in the present study
had significant hematuria at diagnosis. Biopsies in the families in
the present study demonstrated the typical signs of FSGS on light
microscopy and foot process effacement on EM. However, in the
present study there were no consistent GBM ultrastructural
alterations in the siblings with COL4A4 variant and there was no
decrease in collagen (IV) staining in the GBM. In addition, these
phenotypes also lack extra-renal manifestations including deafness
or ocular symptoms, which are characteristic of AS. In the two
patients, there was not enough supportive evidence that was
consistent with AS or TBMN and the sisters were diagnosed as
familial hematuria rather than AS or TBMN.
Whether or not the reported heterozygous variant
(c4195A>T) alone in the present study is sufficient to cause
FSGS, or is only partially penetrant, the study by Malone et
al (51) demonstrated that the
variants in COL4A3/COL4A4 c may be associated with FSGS, however
the possibility of the presence of other modifier genes and/or
other acquired factors cannot be excluded (51). These genes or factors may determine
the phenotypic heterogeneity that leads to variability in disease
progression and results in an unpredictably benign course or
long-term progression of hematuria to proteinuria, and ESRD
(56). Podocyte foot process
effacement was a constant result in the present report, and it
suggests that the observed phenotype may be due to podocyte
abnormalities. So it is possible that the variable phenotypes
demonstrated in the present study may be due to variants in COL4A4
acting as disease modifiers for FSGS and this is consistent with
the view of Bullich et al (57).
FSGS-associated genes frequently follow an autosomal
dominant or recessive inheritance pattern, therefore a mutation in
mtDNA may have been overlooked. In addition, the WES cannot be
performed to analyze mtDNA mutations. The results of the present
study demonstrated that Massarray technology and WES technology
were complementary, each with its own advantage. The combination of
Massarray technology and WES may improve the detection rate of
genetic mutation with an increased level of accuracy.
At present, monogenic FSGS subtypes have been
reported by genetic studies primarily focusing on familial FSGS.
However, a rare study on the potential role of combinations of
mutations in different genes was reported in FSGS (12,13).
The present study, to the best of the authors knowledge, is the
first report to document two relevant genes co-segregated with
FSGS.
It has been proposed that hematuria is the forgotten
CKD factor (32). In a number of
families carrying these mutations, certain members continue to
exhibit chronic and isolated MH for the rest of their lives,
whereas others develop proteinuria later on in life, usually with
hypertension and a variable gradual progression to CRF leading to
ESRD (58,59).
Therefore, the term familial hematuria (FM) would be
appropriate to use instead of misnomer benign familiar hematuria
and the pediatric nephrologist must give a correct prediction of
prognosis to the children with hematuria and to avoid misdiagnosis.
Genetic testing benefits include early diagnosis, highly-targeted
therapy and an ESRD onset delay. Genetic investigations may be more
definitive and diagnostic than renal biopsies.
For the initial treatment of FSGS, the Kidney
Disease Improving Global Outcomes 2012 guideline (60) recommends that corticosteroid and
immunosuppressive therapy be considered only in idiopathic FSGS
associated with clinical features of nephrotic syndrome (17). There is no evidence to suggest
corticosteroids or immunosuppressive therapy in the treatment of
the mutation induced FSGS.
The treatment for the two sisters consisted of
Chinese traditional medicines including huaiqihuang and
shenyansiwei capsules. Regular follow-up surveys were carried out
in the clinic. The older sister has had enalapril administered up
to this point as proteinuria was detected five months following
diagnosis with FH. Currently, the proteinuria is in remission and
hematuria is reducing gradually. Blood pressure was relatively well
regulated and renal function was normal therefore steroid, and
immunosuppressive therapy was not instituted.
The results of the present study demonstrate that it
may not be possible to take a detailed three generational family
history in every pediatric out-patient clinic, however it is always
worth asking if there is a family history of kidney problems,
especially if these have occurred in relatively young people.
Screening for COL4A mutations in FSGS, particularly when presenting
with FH, is recommended.
Whether the variants in COL4A4 were inherited from
their father and TRNL1 were inherited from the healthy mother, has
not been resolved. Next, the blood of the proband's parent and
other affected family members should be obtained to screen for
COL4A4 and TRNL1 genes. Then, the pathogenic mechanism of two
variants should be verified by animal or in vitro
experiments.
In the present study, the sisters with mtDNA
mutation did not manifest features including hearing loss, diabetes
mellitus, neuromuscular symptoms or cardiomyopathy. The family
members should be followed closely to identify the development of
associated conditions including diabetes mellitus and
cardiomyopathy.
Heterozygous carriers of COL4A3 or COL4A4 mutations,
irrespective of gender, may be asymptomatic, may have hematuria
(carriers of recessive disease) or may progress to ESRD (58,59).
Therefore, the family members require long-term follow-up.
In the present study, a missense mutation in the
COL4A4 and TRNL1 genes were identified, which may be responsible
for MH with FSGS in this family. Screening for COL4A mutations in
familial FSGS patients is recommended. Genetic investigations of
families with similar clinical phenotypes should be a priority for
nephrologists. The combination of Massarray technology and WES may
improve the detection rate of genetic mutation with a high level of
accuracy.
Acknowledgements
The present study was supported partially by the
National ‘Twelfth Five-Year’ Science and Technology Support Project
(2012BAI03B02), Ministry of Science and Technology of China, and by
Xiangya Excellent Physician Award (ZWY, 2013), Central South
University, China.
References
|
1
|
Korbet SM: Treatment of primary FSGS in
adults. J Am Soc Nephrol. 23:1769–1776. 2012. View Article : Google Scholar
|
|
2
|
Ponticelli C and Graziani G: Current and
emerging treatments for idiopathic focal and segmental
glomerulosclerosis in adults. Expert Rev Clin Immunol. 9:251–261.
2013. View Article : Google Scholar
|
|
3
|
Chen YM and Liapis H: Focal segmental
glomerulosclerosis: Molecular genetics and targeted therapies. BMC
Nephrol. 16:1012015. View Article : Google Scholar :
|
|
4
|
Taylor J and Flinter F: Familial
haematuria: When to consider genetic testing. Arch Dis Child.
99:857–861. 2014. View Article : Google Scholar
|
|
5
|
Kim YH, Goyal M, Kurnit D, Wharram B,
Wiggins J, Holzman L, Kershaw D and Wiggins R: Podocyte depletion
and glomerulosclerosis have a direct relationship in the
PAN-treated rat. Kidney Int. 60:957–968. 2001. View Article : Google Scholar
|
|
6
|
Wharram BL, Goyal M, Wiggins JE, Sanden
SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman
LB and Wiggins RC: Podocyte depletion causes glomerulosclerosis:
Diphtheria toxin-induced podocyte depletion in rats expressing
human diphtheria toxin receptor transgene. J Am Soc Nephrol.
16:2941–2952. 2005. View Article : Google Scholar
|
|
7
|
Santín S, García-Maset R, Ruíz P, Giménez
I, Zamora I, Peña A, Madrid A, Camacho JA, Fraga G, Sánchez-Moreno
A, et al: Nephrin mutations cause childhood- and adult-onset focal
segmental glomerulosclerosis. Kidney Int. 76:1268–1276. 2009.
View Article : Google Scholar
|
|
8
|
Tonna SJ, Needham A, Polu K, Uscinski A,
Appel GB, Falk RJ, Katz A, Al-Waheeb S, Kaplan BS, Jerums G, et al:
NPHS2 variation in focal and segmental glomerulosclerosis. BMC
Nephrol. 9:132008. View Article : Google Scholar :
|
|
9
|
Gigante M, Pontrelli P, Montemurno E, Roca
L, Aucella F, Penza R, Caridi G, Ranieri E, Ghiggeri GM and
Gesualdo L: CD2AP mutations are associated with sporadic nephritic
syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol
Dial Transplant. 24:1858–1864. 2009. View Article : Google Scholar
|
|
10
|
Hinkes B, Wiggins RC, Gbadegesin R,
Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins
BE, et al: Positional cloning uncovers mutations in PLCE1
responsible for a nephrotic syndrome variant that may be
reversible. Nat Genet. 38:1397–1405. 2006. View Article : Google Scholar
|
|
11
|
Santín S, Ars E, Rossetti S, Salido E,
Silva I, García-Maset R, Giménez I, Ruíz P, Mendizábal S, Nieto
Luciano J, et al: TRPC6 mutational analysis in a large cohort of
patients with focal segmental glomerulosclerosis. Nephrol Dial
Transplant. 24:3089–3096. 2009. View Article : Google Scholar
|
|
12
|
Hall G, Gbadegesin RA, Lavin P, Wu G, Liu
Y, Oh EC, Wang L, Spurney RF, Eckel J, Lindsey T, et al: A novel
missense mutation of Wilms'tumor1 causes autosomal dominant FSGS. J
Am Soc Nephrol. 26:831–843. 2015. View Article : Google Scholar
|
|
13
|
Boyer O, Woerner S, Yang F, Oakeley EJ,
Linghu B, Gribouval O, Tête MJ, Duca JS, Klickstein L, Damask AJ,
et al: LMX1B mutations cause hereditary FSGS without extrarenal
involvement. J Am Soc Nephrol. 24:1216–1222. 2013. View Article : Google Scholar :
|
|
14
|
Boerkoel CF, Takashima H, John J, Yan J,
Stankiewicz P, Rosenbarker L, André JL, Bogdanovic R, Burguet A,
Cockfield S, et al: Mutant chromatin remodeling protein SMARCAL1
causes Schimke immuno-osseous dysplasia. Nat Genet. 30:215–220.
2002. View Article : Google Scholar
|
|
15
|
Choi HJ, Lee BH, Cho HY, Moon KC, Ha IS,
Nagata M, Choi Y and Cheong HI: Familial focal segmental
glomerulosclerosis associated with an ACTN4 mutation and paternal
germline mosaicism. Am J Kidney Dis. 51:834–838. 2008. View Article : Google Scholar
|
|
16
|
Gbadegesin RA, Lavin PJ, Hall G,
Bartkowiak B, Homstad A, Jiang R, Wu G, Byrd A, Lynn K, Wolfish N,
et al: Inverted formin 2 mutations with variable expression in
patients with sporadic and hereditary focal and segmental
glomerulosclerosis. Kidney Int. 81:94–99. 2012. View Article : Google Scholar
|
|
17
|
Lowik MM, Hol FA, Steenbergen EJ, Wetzels
JF and van den Heuvel LP: Mitochondrial tRNALeu (UUR) mutation in a
patient with steroid-resistant nephrotic syndrome and focal
segmental glomerulosclerosis. Nephrol Dial Transplant. 20:336–341.
2005. View Article : Google Scholar
|
|
18
|
Diomedi-Camassei F, Di Giandomenico S,
Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer
L, Barisoni L, Pastore A, et al: COQ2 nephropathy: A newly
described inherited mitochondriopathy with primary renal
involvement. J Am Soc Nephrol. 18:2773–2780. 2007. View Article : Google Scholar
|
|
19
|
López LC, Schuelke M, Quinzii CM, Kanki T,
Rodenburg RJ, Naini A, Dimauro S and Hirano M: Leigh syndromewith
nephropathy and CoQ10 deficiency due to decaprenyl diphosphate
synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 79:1125–1129.
2006. View
Article : Google Scholar :
|
|
20
|
Matejas V, Hinkes B, Alkandari F,
Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Bláhová K,
Bockenhauer D, Cheong HI, et al: Mutations in the human laminin
beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum
Mutat. 31:992–1002. 2010. View Article : Google Scholar :
|
|
21
|
Hata D, Miyazaki M, Seto S, Kadota E, Muso
E, Takasu K, Nakano A, Tamai K, Uitto J, Nagata M, et al: Nephrotic
syndrome and aberrant expression of laminin isoforms in glomerular
basement membranes for an infant with Herlitz junctional
epidermolysis bullosa. Pediatrics. 116:e601–e607. 2005. View Article : Google Scholar
|
|
22
|
Kambham N, Tanji N, Seigle RL, Markowitz
GS, Pulkkinen L, Uitto J and D'Agati VD: Congenital focal segmental
glomerulosclerosis associated with beta4 integrin mutation and
epidermolysis bullosa. Am J Kidney Dis. 36:190–196. 2000.
View Article : Google Scholar
|
|
23
|
Berkovic SF, Dibbens LM, Oshlack A, Silver
JD, Katerelos M, Vears DF, Lüllmann-Rauch R, Blanz J, Zhang KW,
Stankovich J, et al: Array-based gene discovery with three
unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus
epilepsy and glomerulosclerosis. Am J Hum Genet. 82:673–684. 2008.
View Article : Google Scholar :
|
|
24
|
Serebrinsky G, Calvo M, Fernandez S, Saito
S, Ohno K, Wallace E, Warnock D, Sakuraba H and Politei J: Late
onset variants in Fabry disease: Results in high risk population
screenings in Argentina. Mol Genet Metab Rep. 4:19–24. 2015.
View Article : Google Scholar :
|
|
25
|
Rood IM, Deegens JK and Wetzels JF:
Genetic causes of focal segmental glomerulosclerosis: Implications
for clinical practice. Nephrol Dial Transplant. 27:882–890. 2012.
View Article : Google Scholar
|
|
26
|
Bierzynska A, Soderquest K and Koziell A:
Genes and podocytes-new insights into mechanisms of podocytopathy.
Front Endocrinol (Lausanne). 5:2262015.
|
|
27
|
Kashtan CE: Familial haematuria. Pediatr
Nephrol. 24:1951–1958. 2009. View Article : Google Scholar
|
|
28
|
Piqueras AI, White RH, Raafat F, Moghal N
and Milford DV: Renal biopsy diagnosis in children presenting with
haematuria. Pediatr Nephrol. 12:386–391. 1998. View Article : Google Scholar
|
|
29
|
Vivante A, Afek A, Frenkel-Nir Y, Tzur D,
Farfel A, Golan E, Chaiter Y, Shohat T, Skorecki K and
Calderon-Margalit R: Persistent asymptomatic isolated microscopic
hematuria in Israeli adolescents and young adults and risk for
end-stage renal disease. JAMA. 306:729–736. 2011. View Article : Google Scholar
|
|
30
|
Collar JE, Ladva S, Cairns TD and Cattell
V: Red cell traverse through thin glomerular basement membranes.
Kidney Int. 59:2069–2072. 2001. View Article : Google Scholar
|
|
31
|
Chow KM, Kwan BC, Li PK and Szeto CC:
Asymptomatic isolated microscopic haematuria: Long-term follow-up.
QJM. 97:739–745. 2004. View Article : Google Scholar
|
|
32
|
Moreno JA, Martín-Cleary C, Gutiérrez E,
Rubio-Navarro A, Ortiz A, Praga M and Egido J: Haematuria: The
forgotten CKD factor? Nephrol Dial Transplant. 27:28–34. 2012.
View Article : Google Scholar
|
|
33
|
Gale DP: How benign is hematuria? Using
genetics to predict prognosis. Pediatr Nephrol. 28:1183–1193. 2013.
View Article : Google Scholar
|
|
34
|
Deltas C, Pierides A and Voskarides K:
Molecular genetics of familial hematuric diseases. Nephrol Dial
Transplant. 28:2946–2960. 2013. View Article : Google Scholar
|
|
35
|
Pei Y and Watnick T: Diagnosis and
screening of autosomal dominant polycystic kidney disease. Adv
Chronic Kidney Dis. 17:140–152. 2010. View Article : Google Scholar :
|
|
36
|
Renkema KY, Stokman MF, Giles RH and
Knoers NV: Next-generation sequencing for research and diagnostics
in kidney disease. Nat Rev Nephrol. 10:433–444. 2014. View Article : Google Scholar
|
|
37
|
Buetow KH, Edmonson M, MacDonald R,
Clifford R, Yip P, Kelley J, Little DP, Strausberg R, Koester H,
Cantor CR and Braun A: High-throughput development and
characterization of a genomewide collection of gene-based single
nucleotide polymorphism markers by chip-based matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry. Proc Natl
Acad Sci USA. 98:581–584. 2001. View Article : Google Scholar :
|
|
38
|
Opdal SH, Rognum TO, Torgersen H and Vege
A: Mitochondrial DNA point mutations detected in four cases of
sudden infant death syndrome. Acta Paediat. 88:957–960. 1999.
View Article : Google Scholar
|
|
39
|
Xie J, Wu X, Ren H, Wang W, Wang Z, Pan X,
Hao X, Tong J, Ma J, Ye Z, et al: COL4A3 mutations cause focal
segmental glomerulosclerosis. J Mol Cell Biol. 7:1842015.
View Article : Google Scholar
|
|
40
|
Opdal SH, Vege A, Egeland T, Musse MA and
Rognum TO: Possible role of mtDNA mutations in sudden infant death.
Pediatr Neurol. 27:23–29. 2002. View Article : Google Scholar
|
|
41
|
Moares CT, Ciacci F, Bonilla E, Jansen C,
Hirano M, Rao N, Lovelace RE, Rowland LP, Schon EA and DiMauro S:
Two novel pathogenic mitochondrial DNA mutations affecting
organelle number and protein synthesis. Is the tRNA(Leu(UUR)) gene
an etiologic hot spot? J Clin Invest. 92:2906–2915. 1993.
View Article : Google Scholar :
|
|
42
|
Mochizuki H, Joh K, Kawame H, Imadachi A,
Nozaki H, Ohashi T, Usui N, Eto Y, Kanetsuna Y and Aizawa S:
Mitochondrial encephalomyopathies preceded by de-Toni-Debré-Fanconi
syndrome or focal segmental glomerulosclerosis. Clin Nephrol.
46:347–352. 1996.
|
|
43
|
Kurogouchi F, Oguchi T, Mawatari E,
Yamaura S, Hora K, Takei M, Sekijima Y, Ikeda SI and Kiyosawa K: A
case of mitochondrial cytopathy with a typical point mutation for
MELAS, presenting with severe focal-segmental glomerulosclerosis as
main clinical manifestation. Am J Nephrol. 18:551–556. 1998.
View Article : Google Scholar
|
|
44
|
Hotta O, Inoue CN, Miyabayashi S, Furuta
T, Takeuchi A and Taguma Y: Clinical and pathologic features of
focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR)
gene mutation. Kidney Int. 59:1236–1243. 2001. View Article : Google Scholar
|
|
45
|
Yamagata K, Muro K, Usui J, Hagiwara M,
Kai H, Arakawa Y, Shimizu Y, Tomida C, Hirayama K, Kobayashi M and
Koyama A: Mitochondrial DNA mutations in focal segmental
glomerulosclerosis lesions. J Am Soc Nephrol. 13:1816–1823. 2002.
View Article : Google Scholar
|
|
46
|
Guéry B, Choukroun G, Noël LH, Clavel P,
Rötig A, Lebon S, Rustin P, Bellané-Chantelot C, Mougenot B,
Grünfeld JP and Chauveau D: The spectrum of systemic involvement in
adults presenting with renal lesion and mitochondrial tRNA(Leu)
gene mutation. J Am Soc Nephrol. 14:2099–2108. 2003. View Article : Google Scholar
|
|
47
|
Hao H and Moares CT: Functional and
molecular mitochondrial abnormalities associated with a C->T
transition at position 3256 of the human mitochondrial genome. The
effects of a pathogenic mitochondrial tRNA point mutation in
organelle translation and RNA processing. J Biol Chem.
271:2347–2352. 1996. View Article : Google Scholar
|
|
48
|
Sadowski CE, Lovric S, Ashraf S, Pabst WL,
Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J,
et al: A single-gene cause in 29.5% of cases of steroid-resistant
nephrotic syndrome. J Am Soc Nephrol. 26:1279–1289. 2015.
View Article : Google Scholar
|
|
49
|
Halbritter J, Baum M, Hynes AM, Rice SJ,
Thwaites DT, Gucev ZS, Fisher B, Spaneas L, Porath JD, Braun DA, et
al: Fourteen monogenic genes account for 15% of
nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol. 26:543–551.
2015. View Article : Google Scholar
|
|
50
|
Deltas C, Pierides A and Voskarides K: The
role of molecular genetics in diagnosing familial hematuria(s).
Pediatr Nephrol. 27:1221–1231. 2012. View Article : Google Scholar
|
|
51
|
Malone AF, Phelan PJ, Hall G, Cetincelik
U, Homstad A, Alonso AS, Jiang R, Lindsey TB, Wu G, Sparks MA, et
al: Rare hereditary COL4A3/COL4A4 variants maybe mistaken for
familial focal segmental glomerulosclerosis. Kidney Int.
86:1253–1259. 2014. View Article : Google Scholar :
|
|
52
|
Liapis H and Jain S: The interface of
genetics with pathology in alport nephritis. J Am Soc Nephrol.
24:1925–1927. 2013. View Article : Google Scholar :
|
|
53
|
Abrahamson DR, Hudson BG, Stroganova L,
Borza DB and St John PL: Cellular origins of type IV collagen
networks in developing glomeruli. J Am Soc Nephrol. 20:1471–1479.
2009. View Article : Google Scholar :
|
|
54
|
Kruegel J, Rubel D and Gross O: Alport
syndrome-insights from basic and clinical research. Nat Rev
Nephrol. 9:170–178. 2013. View Article : Google Scholar
|
|
55
|
Pierides A, Voskarides K, Athanasiou Y,
Ioannou K, Damianou L, Arsali M, Zavros M, Pierides M, Vargemezis
V, Patsias C, et al: Clinico-pathological correlations in 127
patients in 11 large pedigrees, segregating one of three
heterozygous mutations in the COL4A3/COL4A4 genes associated with
familial haematuria and significant late progression to proteinuria
and chronic kidney disease from focal segmental glomerulosclerosis.
Nephrol Dial Transplant. 24:2721–2729. 2009. View Article : Google Scholar
|
|
56
|
Ramzan K, Imtiaz F, Taibah K, Alnufiee S,
Akhtar M, Al-Hazzaa SA and Al-Owain M: COL4A4-related nephropathy
caused by a novel mutation in a large consanguineous Saudi family.
Int J Pediatr Otorhinolaryngol. 78:427–432. 2014. View Article : Google Scholar
|
|
57
|
Bullich G, Trujillano D, Santín S,
Ossowski S, Mendizábal S, Fraga G, Madrid Á, Ariceta G, Ballarín J,
Torra R, et al: Targeted next-generation sequencing in
steroid-resistant nephrotic syndrome: Mutations in multiple
glomerular genes may influence disease severity. Eur J Hum Genet.
23:1192–1199. 2015. View Article : Google Scholar
|
|
58
|
Artuso R, Fallerini C, Dosa L, Scionti F,
Clementi M, Garosi G, Massella L, Epistolato MC, Mancini R, Mari F,
et al: Advances in Alport syndrome diagnosis using next-generation
sequencing. Eur J Hum Genet. 20:50–57. 2012. View Article : Google Scholar
|
|
59
|
Savige J, Gregory M, Gross O, Kashtan C,
Ding J and Flinter F: Expert guidelines for the management of
Alport syndrome and thin basement membrane nephropathy. J Am Soc
Nephrol. 24:364–375. 2013. View Article : Google Scholar
|
|
60
|
Chapter 6: Idiopathic focal segmental
glomerulosclerosis in adults. Kidney Int Suppl (2011). 2:181–185.
2012. View Article : Google Scholar :
|