Introduction
Cell sheet technology, as the main research
direction of modern regenerative medicine, refers to the continuous
culture of high-density inoculated cells in vitro for
multi-layer growth to form a membranous structure that is rich in
cellular and extracellular matrices, and to harvest it in a
non-enzymatic way. Thus, the integrity of cell surface proteins,
ion channels and intercellular connexins is preserved (1). This harvest method markedly improves
the utilization of cells and increases the plasticity of the cell
sheet. Thus, cell sheets may be implanted directly into the defect
site for tissue repair, or multiple homogeneous or heterogeneous
cell sheets are constructed by three-dimensional superposition
in vitro (2) to repair the
defect site. Based on the above-mentioned advantages, the
technology has been widely used in tissue and organ repair and
regeneration, including myocardial repair, corneal epithelial
repair, esophageal endothelial repair and periodontal repair,
amongst others (3–5), with certain methods already being
applied in clinical practice. In addition, as cell sheet repair
does not require scaffold materials, various problems, such as
inflammatory response caused by acid production for material
degradation, are avoidable.
Gene modification of stem cells refers to the
introduction of DNA or RNA into target cells to change their
direction of differentiation or secretion of a certain protein, so
as to promote the effects of stem cell therapy and tissue
regeneration (6). Various
micromolecular nucleic acids, including DNA, antisense
oligonucleotides, siRNA and miRNA, may all be specifically
transfected into cells to exert therapeutic effects on the
molecular level (7). Although
viral vectors efficiently transfect multiple types of cell, with
stable gene expression for a long time, the safety issues
concerning clinical immunogenicity and carcinogenicity hinder
further development. By contrast, non-viral vectors have many
advantages, such as low immune response, easy in vitro
synthesis, and modification and safety, so they are preferred to
viral vectors in gene therapy. At present, although gene
transfection has been widely applied in the gene modification of
stem cells, it has seldom been used to modify cell sheets.
Meanwhile, only certain complicated methods, such as sheet
transfection by magnetic force with magnetic bead-modified virus as
a vector, have been demonstrated to be effective. Therefore, it is
necessary to identify a simple and practical method for
transfection if cell sheets are modifiable using genetic
engineering technology to facilitate their specific differentiation
into a regenerative target.
miRNAs, as a class of non-coding RNAs, inhibit
translation through incomplete matching and binding with target
mRNA, enabling individual miRNAs to act simultaneously on multiple
genes and inhibit their expression (8). Furthermore, each gene is regulated by
multiple miRNAs. Accordingly, miRNA is involved in many cell
functions, including a variety of metabolic processes, such as
proliferation, differentiation and apoptosis (9–11).
Different miRNAs promote bone marrow mesenchymal stem cells
(BMMSCs) to differentiate in a variety of ways, including
osteogenic differentiation. miR-122 is a negative regulatory factor
for the osteogenic differentiation of BMMSCs (12), and its inhibitor significantly
reduces the level of endogenous miR-122 and improves the osteogenic
differentiation capability of stem cells (13).
The aim of the present study was to complex an
miR-122-modified BMMSC sheet with a micro-arc titanium oxide
implant to construct a gene-modified tissue-engineered implant. By
using an unmodified sheet-implant complex as a control, the
osteogenic activity and induction ability of the implanted bone
were evaluated in vitro and in vivo. The aim of the
study was to provide novel ideas and methods for solving clinical
problems, including long, poor healing of implanted bones and high
failure rate in patients with bone metabolic diseases.
Materials and methods
Experimental animals and grouping
A total of 18 one-week-old Sprague-Dawley rats
(weight, 20–25 g) were obtained from the Experimental Animal Center
of First People's Hospital of Qingdao Economic and Technological
Development Zone (Qingdao, China), and divided into a control
group, an miR-122 control group and an miR-122 group (n=6). The
present study was approved by the Ethics Committees on Animal
Experimentation at the First People's Hospital of Qingdao Economic
and Technological Development Zone and efforts were made to
minimize suffering. All rats were fed under specific pathogen-free
conditions at 22–25°C, and given free access to water and food.
Isolation and culture of BMMSCs
The rats were euthanized by cervical dislocation and
immersed in 75% ethanol for 10 min. The femur and tibia were
isolated in a culture dish. The contents of the bone marrow cavity
were washed out using a 5-ml syringe and α-minimum essential medium
(α-MEM) with 15% fetal bovine serum (FBS) and 1%
penicillin-streptomycin, which were all purchased from Gibco;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA), and seeded in a
75-ml culture flask. Subsequently, α-MEM was added to 10 ml, and
routinely cultured in 5% CO2 and saturated humidity at
37°C. The medium was refreshed for the first time 72 h after
inoculation to remove non-adherent cells and tissue residues, and
the adherent cells were continuously cultured at 37°C. The medium
was refreshed once every two days, and the cells were digested and
passaged with 0.25% trypsin until 70–85% confluence.
Flow cytometry
First-generation BMMSCs were digested with trypsin,
centrifuged at 12,000 × g and 4°C for 5 min to discard the
supernatant, washed twice with phosphate-buffered saline (PBS)
containing 3% FBS, then resuspended with this solution and counted
under a light microscope. The cells were subpackaged into eight EP
tubes at a density of 1×105 cells/200 µl, and 2 µl
(1:500) of cluster of differentiation 29 (CD29) (cat. no. 555005),
CD90 (cat. no. 554897), CD105 (cat. no. 550546), CD34 (cat. no.
560238), CD45 (cat. no. 553091) and CD31 (cat. no. 555027)
antibodies (BD Pharmingen, San Diego, CA, USA) was added into
individual tubes. The cells were incubated in the dark at 4°C for 1
h, centrifuged at 12,000 × g and 4°C for 5 min to rinse off excess
antibodies, resuspended with PBS containing 3% FBS to 200 µl and
detected by flow cytometer (BD Biosciences, San Jose, CA, USA).
Adipogenic differentiation
Second-generation BMMSCs were seeded in a six-well
culture plate at a density of 3×105 cells/well to
observe their adherent growth. Upon >85% confluence, the medium
was replaced with adipogenesis-inducing solution [α-MEM containing
5% FBS and 100 nM dexamethasone (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), 0.5 mM 3-isobutyl-1-methylxanthine, 50 mM
indomethacin, 0.01 mg/ml insulin (Sigma-Aldrich; Merck KGaA), 50
µg/ml ascorbic acid (Sigma-Aldrich; Merck KGaA)] and induced for
~10 days. The culture medium was refreshed once every 3 days. After
lipid droplets formed, the cells were washed three times with PBS,
fixed with 4% paraformaldehyde, stained with Oil Red O for 1 h at
room temperature, observed under an inverted microscope (Olympus,
Tokyo, Japan) and photographed.
Osteogenic differentiation
Second-generation BMMSCs were seeded in two six-well
culture plates at a density of 3×105 cells/well for
their adherent growth. Upon confluence of >85%, the medium was
replaced with osteogenesis-inducing solution and refreshed once
every 2 days. Alkaline phosphatase (ALP) formation was observed
under an inverted microscope by staining using an ALP kit (cat. no.
86C-1KT, Sigma-Aldrich; Merck KGaA). Cells in the other plate were
continuously induced until day 28 and stained with 1% alizarin red
for 5 min at room temperature. Formation of mineralized nodules was
observed under an inverted microscope and photographed.
Detection of collagen secretion
The sheet was fixed at each time-point of osteogenic
induction, stained with 1% Sirius Red/picric acid for 1 h at room
temperature (R&D Systems GmbH, Wiesbaden, Germany) for 18 h,
after the excess staining solution was washed away using 0.1 M
acetic acid, the sheet was observed under an inverted microscope
and photographed. Semi-quantitative analysis of collagen staining
was performed as follows: Destaining solution (1 ml) was added into
each well (Miltenyi Biotec GmbH, Gladbach, Germany) while shaking
for 15 min, and the optical density (OD) was measured at 540 nm
(Thermo Fisher Scientific, Inc.).
Detection of extracellular matrix
mineralization
The sheet was fixed with citrate concentrated
solution and acetone at each time-point of osteogenic induction,
and 1% alizarin red (Beyotime Institute of Biotechnology, Haimen,
China) was added and stained for 3 min at room temperature.
Subsequent to washing with PBS to remove the excess staining
solution, the sheet was observed under an inverted microscope and
photographed. Semi-quantitative analysis of mineralized staining
was performed as follows: Destaining solution (1 ml; pH 7.0) was
added to each well, while shaking for 15 min, and the optical
density was measured at a wavelength of 620 nm (Thermo Fisher
Scientific, Inc.).
Construction of the miR-122-modified
BMMSC sheet
First-generation BMMSCs were seeded in six-well
culture plates at a density of 3×105 cells/well. When
the confluence reached 100%, basic culture medium in the plate was
replaced with a sheet-inducing medium, and continuously cultured at
37°C for 6 days. The culture medium was refreshed once every two
days. Liquid in the plate well was replaced with sheet-inducing
medium without antibiotics (Sigma-Aldrich; Merck KGaA) on day 7,
and a mixture of four types of transfection vectors was added on
day 8 to complete the transfection (5% CO2 and saturated
humidity for 24 h at <37°C). The medium was replaced with
sheet-inducing medium and continuously incubated at 37°C for 24 h
to complete sheet construction.
Scanning electron microscopy
(SEM)
The obtained sheet was washed with PBS three times,
and 3% glutaraldehyde (pH 7.4) was added and maintained overnight
at 4°C. Gradient dehydration was conducted using 50, 70, 80, 90 and
100% ethanol solutions. After critical-point drying and gold
spraying, the morphology of the sheet was observed under a field
emission scanning electron microscope (Hitachi, Ltd., Tokyo,
Japan).
Hematoxylin and eosin staining H&E
staining
The sheet harvested from each group was fixed with
4% paraformaldehyde, conventionally dehydrated, embedded in
paraffin, deparaffinized with xylene, soaked in gradient ethanol
solutions (3 times, 3 min), deparaffinized by series concentrations
of ethanol solutions, stained with hematoxylin at room temperature
for 10 min, washed with distilled water, differentiated with 0.5%
hydrochloric acid-ethanol solution for 30 sec, colored blue with
concentrated aqueous ammonia, washed with distilled water, stained
with 0.5% eosin at room temperature for 3 min and washed with
distilled water. Subsequently, the sheet was routinely dehydrated,
transparentized using xylene, sealed with neutral gum, and observed
under an optical microscope (Olympus, Tokyo, Japan).
Quantitative polymerase chain reaction
(qPCR) detection
All reagents were purchased from Takara Bio, Inc.
(Otsu, Japan) and the cell sheets were washed three times with PBS.
Cells (106) were directly added to 1 ml TRIzol, mixed in
a vortex oscillator, left still at room temperature for 5 min,
mixed with 1/5 volume of chloroform for 1 min, left still at room
temperature for 5 min, centrifuged at 4°C and 12,000 × g for 15
min, mixed with an equal volume of isopropanol, left still at room
temperature for 10 min and centrifuged again at 4°C and 12,000 × g
for 10 min. After the supernatant was discarded, 1 ml of 75%
ethanol was added, and an appropriate quantity of
diethylpyrocarbonate-water was added to completely dissolve the
precipitate. The 25-µl reaction system contained the following:
Fluorescent reverse transcription (RT)-PCR reaction solution (20
µl), 1 µl DNA polymerase, 0.35 µl reverse transcriptase and 5 µl
template RNA. The mixture was centrifuged at 6,000 × g for 10 sec.
The reaction conditions for miRNA RT were as follows: One cycle of
pre-denaturation at 95°C for 30 sec. The PCR reaction was as
follows: Forty cycles at 95°C for 5 sec, at 55°C for 30 sec and at
72°C for 30 sec, and melting curve analysis was conducted at 95°C
for 5 sec. The procedure for quantitative fluorescent RT-PCR was as
follows: RT at 50°C for 30 min, pre-denaturation at 95°C for 3 min,
denaturation at 95°C for 15 sec, annealing at 50°C for 30 sec and
extension at 72°C for 30 min (5 cycles in total); denaturation at
95°C for 10 sec and annealing at 55°C for 40 sec (40 cycles in
total). The relative expression level was calculated according to
X=2−∆∆Cq with U6 serving as the internal reference
(14). Primer sequences for the
PCR system (Roche Innovatis, Bielefeld, Germany) were as follows.
Upstream, 5′-CCACTACGGTCTTCACAGGACCT-3′ and downstream,
5′-TCTTCAGTTGCCTTCTTGGTTC-3′ for runt-related transcription factor
2 (RUNX2); upstream, 5′-AAGGCAGTTGGCAGTAGTGG-3′ and downstream,
5′-TGAATGGGCTTCTTCCTCAG-3′ for osterix (OSX); upstream,
5′-AACGTGGCCAAGAACATCATCA-3′ and downstream,
5′-TGTCCATCTCCAACCTGCAC-3′ for ALP; upstream,
5′-GCCTCCCAGAACATCACCTA-3′ and downstream,
5′-AGTCGTGTCGATCCGTAGC-3′ for collagen I (COL-I); upstream,
5′-CATGGTGCTAGCTAGAGCTTCC-3′ and downstream,
5′-TTCCTGACGTGATCGAAGTTCC-3′ for bone morphogenetic protein 2
(BMP-2); upstream, 5′-GGTGCAGACCTAGCAGACACCA-3′ and downstream,
5′-AGGTAGCGCCGGAGTCTATTCA-3′ for osteocalcin (OCN); upstream,
5′-GGCACAGTCAAGGCTGAGAATG-3′ and downstream,
5′-ATGGTCATGCAAGACGCCAGTA-3′ for GAPDH:
Western blot analysis
Sheet protein (1 mg, quantified by the bicinchoninic
acid method) from each group was subjected to 12% SDS-PAGE. Then
two pieces of gel were carefully peeled off, and sequentially paved
sponge pad, filter paper, gel, sheet, filter paper and sponge pad
to prepare a gel transfer interlayer from which bubbles were
removed using a glass rod. Subsequently, it was placed into an
electrical slot and transferred for 90 min at 90 V (330 mA). The
membrane was transferred to a plate containing 25 ml blocking
solution using forceps, shaken slowly for 1 h at room temperature,
and ALP (cat. no. ab67228), COL-1 (cat. no. ab34710), RUNX2 (cat.
no. ab76956), OSX (cat. no. ab22552), OCN (cat. no. ab13420),
extracellular signal-regulated kinases (ERKs) 1/2 (cat. no.
ab17942) and phosphorylated (p)-ERK 1/2 (cat. no. ab201015)
antibodies (all 1:1,000 diluted; Abcam, Cambridge, UK) were added
and incubated overnight at 4°C. The membrane was washed with
Tris-buffered saline and Tween-20 (TBST) three times (5 min each
time) at room temperature in a shaker and the next day horseradish
peroxidase-conjugated secondary antibody (1:1,000 diluted, cat. no.
BA1058; Wuhan Boster Biological Technology, Ltd., Wuhan, China) was
added, incubated in a shaker at 37°C for 1 h, washed with TBST
three times (5 min each time) and color-developed by
diaminobenzidine solution (Thermo Fisher Scientific, Inc.).
Immunohistochemical staining
Seven days after osteogenic induction, cell sheets
of the three groups were washed with PBS, fixed with 4%
paraformaldehyde for 24 h, dehydrated using a dehydrator, embedded
in paraffin, serially sectioned (5 µm) using a slicing machine, and
deparaffinized with gradient xylene and ethanol solutions to water.
After a paraffin circle was made, the sheets were repaired with 1%
trypsin at 37°C for 30 min, washed with PBS three times (5 min each
time), treated with 3% hydrogen peroxide for 15 min, washed with
PBS three times (5 min each time) and blocked with goat serum
(Sigma-Aldrich; Merck KGaA) at 37°C for 1 h. Suitable
concentrations of ALP, COL-1, OCN and RUNX2 primary antibodies
(1:150; Cell Signaling Technology, Inc., Danvers, MA, USA) were
added and incubated at 4°C overnight, rinsed with PBS three times
(5 min each time), and universal secondary antibody (1:50)
(Biosynthesis, Beijing, China) was added and incubated at 37°C for
2 h, washed with PBS three times (5 min each time), stained with
3,3-diaminobenzidine color-developing solution for 15 min at room
temperature, observed under a microscope and photographed.
Preparation of micro-arc titanium
oxide implant
Pure titanium samples were polished with silicon
carbide abrasive papers (Agilent Technologies, Inc., Santa Clara,
CA, USA) from 400 to 1,000 meshes sequentially, ultrasonically
cleaned in deioni zed water (Thermo Fisher Scientific, Inc.) and
dried in air at room temperature. A micro-arc oxidation electrolyte
solution (GE Healthcare Life Sciences, Little Chalfont, UK) was
formulated with 0.02 M β-glycerol phosphate and 0.2 M citric acid.
The samples were subjected to micro-arc oxidation treatment for 5
min at 400 V, 100 Hz and 20% duty ratio. Subsequently, ultrasonic
cleaning was conducted successively in acetone, absolute ethanol
and deionized water for 10 min each, and dried in air at room
temperature. Finally, the samples were sealed and packaged for Co60
(Thermo Fisher Scientific, Inc.) irradiation sterilization.
Statistical analysis
All data were expressed as means ± standard
deviation and analyzed using SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA). Groups were compared by one-way analysis of variance, and
pairwise comparison was performed using Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Isolation, culture and identification
of BMMSCs
Primary BMMSCs were cultured using the whole bone
marrow adherence method, which almost reached complete confluence 7
days after inoculation. The cells were mostly spindle- or
triangle-shaped, with large nuclei and high refractive indices.
Flow cytometry demonstrated that surface markers, CD29 (99.6%),
CD90 (99.2%) and CD105 (95%) of BMMSCs were positively expressed,
whereas blood cell markers, CD34 (1.7%), CD45 (2.9%) and CD31
(1.0%) were negatively expressed. The results conformed to the
characteristics of mesenchyme-derived stem cells. After 10 days of
adipogenic induction, Oil Red O staining exhibited the scattered
distribution of red lipid droplets in and between the cells. ALP
staining had positive results after 7 days of osteogenic induction,
showing bluish purple intracellular crystals. Calcified nodules
formed on the cell surface after three weeks of induction, as
indicated by alizarin red staining (Fig. 1).
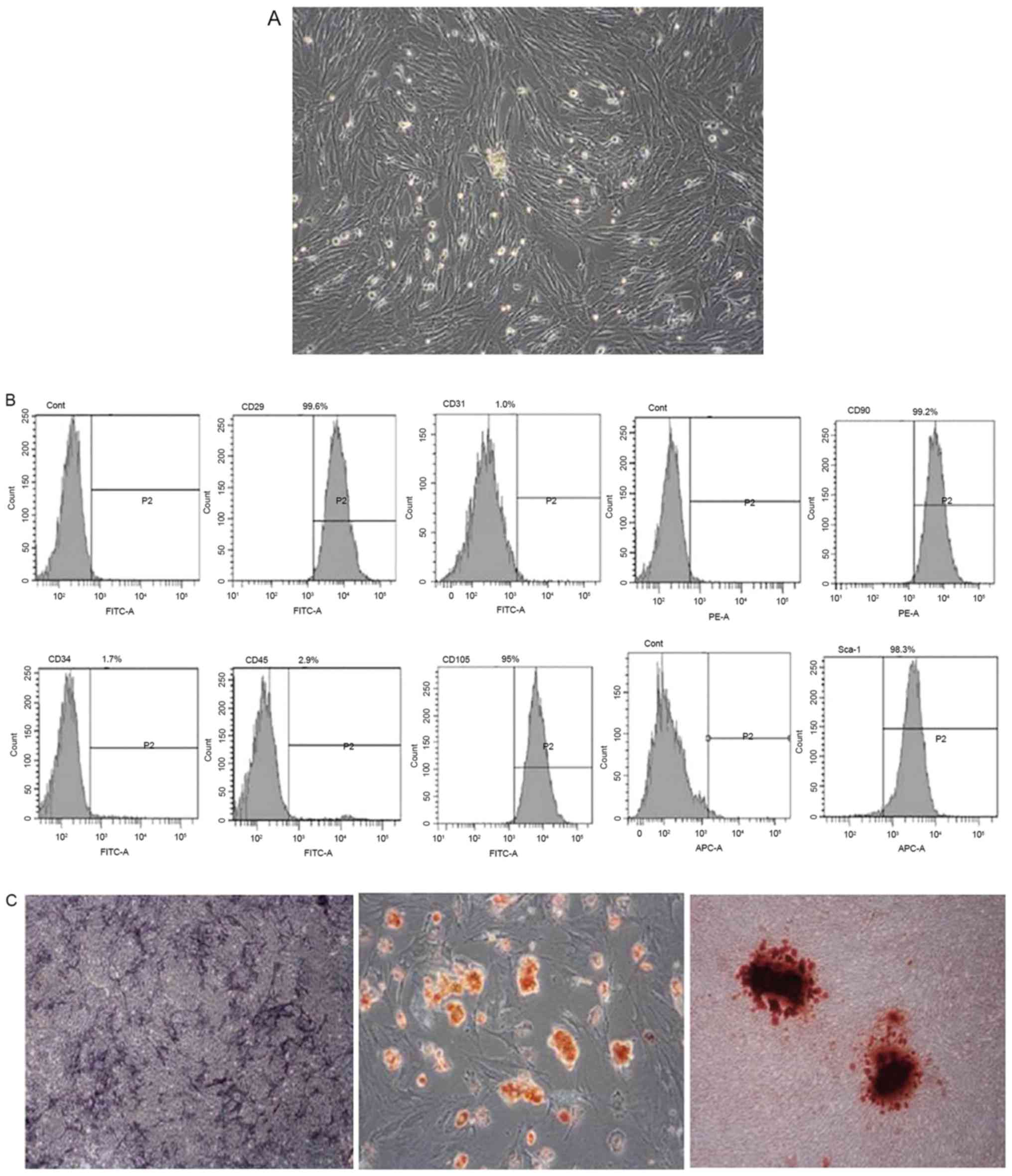 | Figure 1.Isolation, culture and identification
of BMMSCs. (A) Rat BMMSCs (magnification, ×400). (B) Flow cytometry
results of cell surface markers (C) left, alkaline phosphatase
staining results after 7 days of osteogenic induction; middle, Oil
Red O staining results after 10 days of adipogenic induction;
right, alizarin red staining results after three weeks of
osteogenic induction (magnification, ×400). BMMSCs, bone marrow
mesenchymal stem cells; CD, cluster of differentiation; Cont,
control; FITC, fluorescein isothiocyanate; PE, phycoerythrin; APC,
allophycocyanin. |
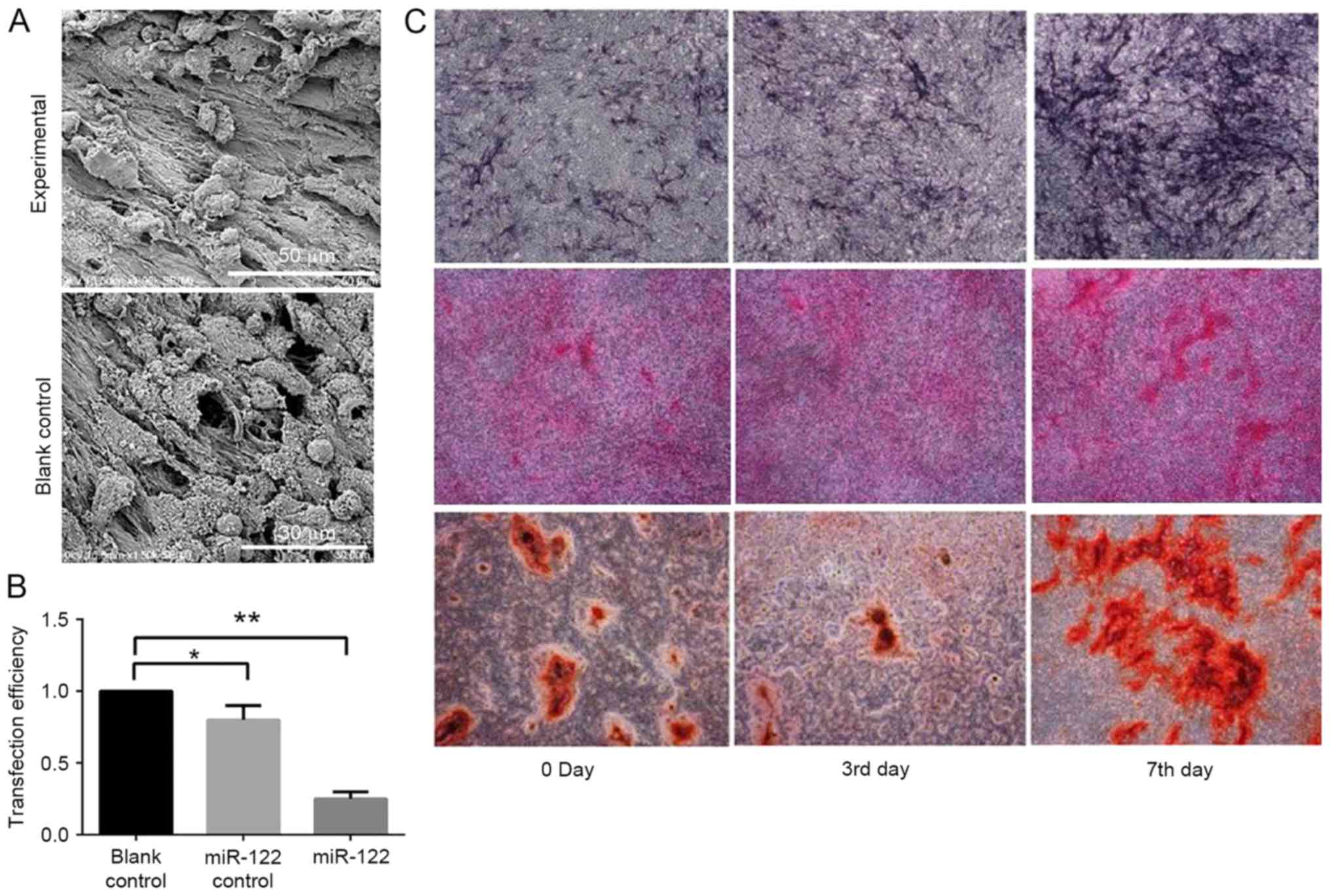
Morphology of miR-122-modified
sheet
SEM images demonstrated that miR-122-modified and
unmodified sheets were rich in intracellular and extracellular
matrices. Flow cytometry identified that miR-122 transfection was
dose-dependent. In addition, ALP staining was conducted after 3 and
7 days of osteogenic induction. ALP expression levels in the
miR-122-modified sheet exceeded those in the blank control and
negative control groups. Furthermore, Sirius Red staining and
semi-quantitative analysis revealed that the experimental group
secreted significantly more collagen than the control groups
(Fig. 2).
Expression levels of
osteogenesis-associated functional genes and proteins
RUNX2 and OSX are osteogenesis-associated genes. ALP
and COL-1 are early osteogenic markers, while OCN and BMP-2 are
late ones. On day 3 of osteogenic induction, the RUNX2, OSX, OCN,
COL-1, ALP and BMP-2 expression levels of the experimental group
were 2.0, 3.1, 4.6, 3.2, 10.5 and 4.5 times those of the blank
control group, respectively. Western blotting and
immunohistochemical assay demonstrated that significantly more
RUNX2, OSX, ALP and OCN, as well as significantly less COL-1, were
expressed in the experimental group when compared with those of the
two control groups (P<0.05). As evidenced by western blotting,
the p-ERK 1/2 expression level was only upregulated in the
experimental group following miR-122 transfection (Fig. 3).
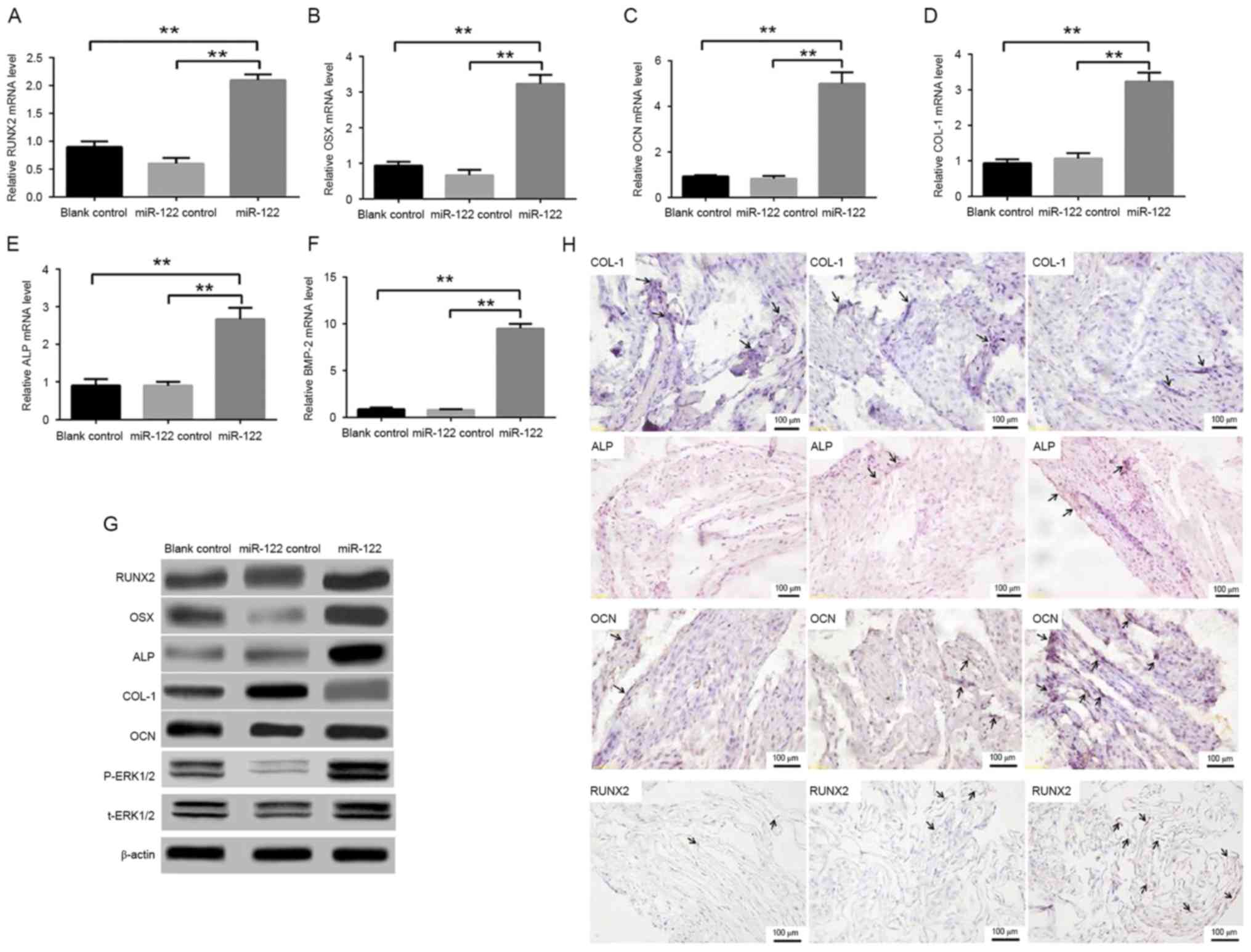 | Figure 3.Expression levels of
osteogenesis-associated functional genes and proteins. (A-F)
Quantitative polymerase chain reaction results for RUNX2, OSX, ALP,
COL-1, OCN and BMP-2 after three days of osteogenic induction. (G)
Expression levels of osteogenesis-associated proteins (RUNX2, OSX,
ALP and OCN) and t-ERK 1/2, p-ERK 1/2 and ERK 1/2 signaling
pathways after three days of osteogenic induction. (H)
Immunohistochemical assay results of COL-1, ALP, OCN and RUNX2
proteins after three days of osteogenic induction. **P<0.01 vs.
miR-122 group. RUNX2, runt-related transcription factor 2; OSX,
osterix; ALP, alkaline phosphatase; COL-1, collagen I; OCN,
osteocalcin; BMP-2, bone morphogenetic protein 2; OSX, osterix;
ERK, extracellular signal-regulated kinases; t, total; p,
phosphorylated. |
Underlying mechanism for osteogenic
differentiation of BMMSC sheet promoted by miR-122
SEM was performed 48 h after complexation between
the miR-122-transfected cell sheet and implant. Numerous
collagenous fibers and cells containing abundant extracellular
matrices were interlinked into a layer of network structure that
closely adhered to the porous surface of the implant. Under high
magnification, the cells were extended, with a large quantity of
pseudopodia protruding toward and fitting the extracellular
matrices. On day 3 of osteogenic induction after complexation of
the sheet and implant, the BMP-2, RUNX2, ALP, OSX and COL-1
expression levels of the experimental group were 1.2, 1.6, 2.2,
2.1, 1.3 and 3.3 times those of the blank control group,
respectively. In the 4th week, large-area, paralleling bone-like
mineralized tissues formed near the implant of the experimental
group, in which there were considerable bone lacunae and osteocytes
containing new blood vessels. By contrast, the implants of the two
control groups were primarily subjected to fibrous repair, only
accompanied by formation of a small quantity of bones. In the 8th
week, there were widely distributed, dense and well mineralized
mature bone tissues on the implant surface of the experimental
group, which formed good osseointegration with the implant. The new
bones had typical lacunae, osteocytes and bone lining cells, which
were accompanied by angiogenesis. In addition, the bones
predominantly formed through endochondral ossification. As to the
implants of the two control groups, significantly fewer bones
formed and the degree of mineralization was low (Fig. 4).
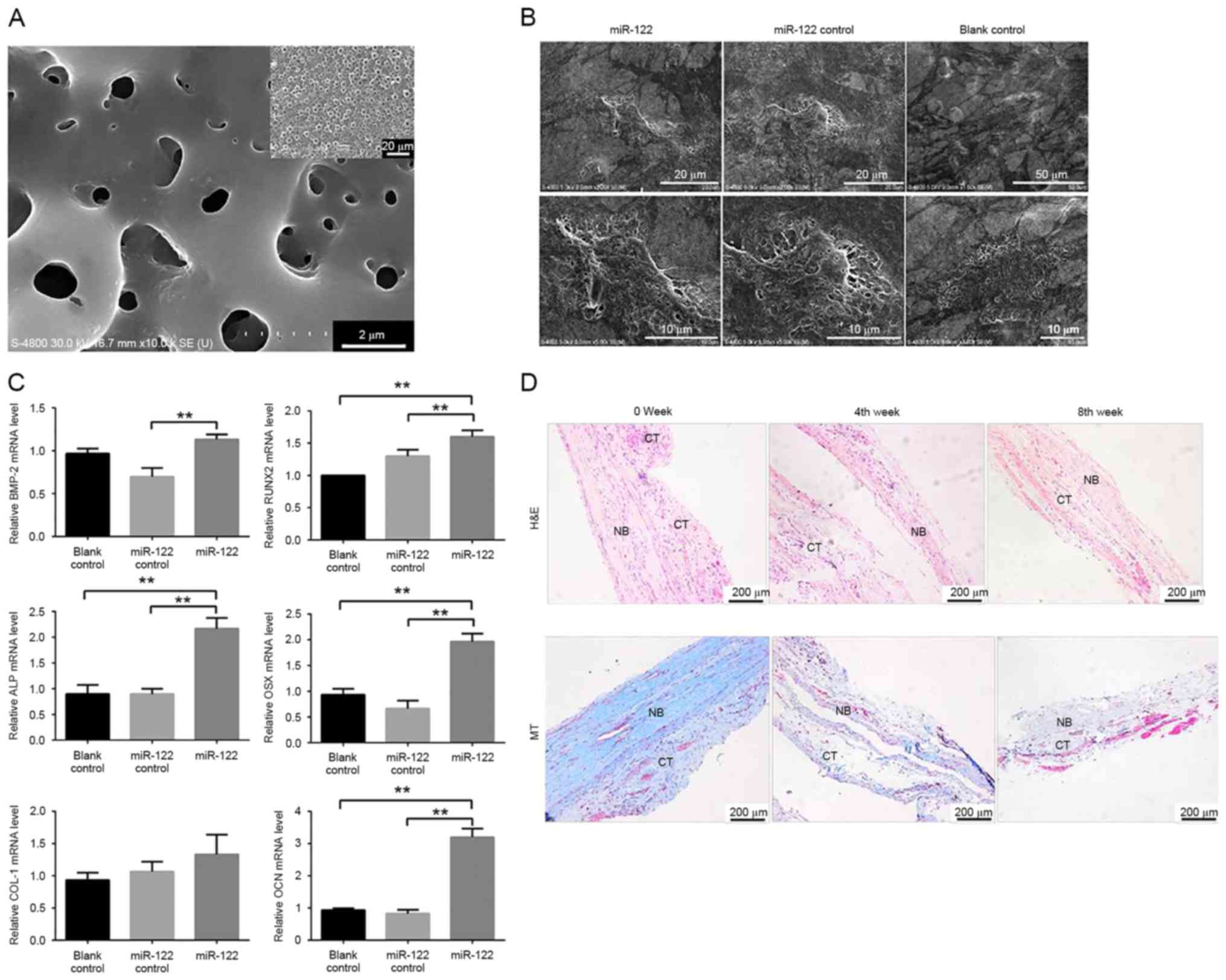 | Figure 4.Underlying mechanism for osteogenic
differentiation of the bone marrow mesenchymal stem cells sheet
promoted by miR-122. (A and B) Scanning electron microscopy was
performed 48 h after complexation between the miR-122-transfected
cell sheet and implant. (C) Quantitative polymerase chain reaction
results of BMP-2, RUNX2, ALP, OSX and COL-1 expression levels after
3 days of osteogenic induction. (D) Histological observation of
implants after 4 and 8 weeks of induction using hematoxylin and
eosin and Masson's trichrome **P<0.01 vs. miR-122 group. BMP-2,
bone morphogenetic protein 2; RUNX2, runt-related transcription
factor 2; ALP, alkaline phosphatase; OSX, osterix; COL-1, collagen
I; OCN, osteocalcin; OSX, osterix. |
Discussion
In the present study, BMMSCs were isolated and
cultured using the whole bone marrow adherence method, as
fibroblast-like cells with clonal or whirlpool growth patterns.
Flow cytometry indicated that surface markers, CD29, CD90 and CD105
were positively expressed on the surface of BMMSCs, although blood
cell markers, CD34, CD45 and CD31 were negatively expressed.
Therefore, the obtained BMMSCs were mesenchyme-derived cells.
Multi-lineage differentiation potential in
vitro is one of the important features of stem cells (15), which was detected in the present
study by in vitro adipogenic and osteogenic induction tests.
As ALP staining was strongly positive after 7 days of osteogenic
induction, BMMSCS began to transform into osteoblasts and
thereafter became typical ones, as indicated by the strongly
positive staining of calcified nodules 14 days later. Thus, the
obtained BMMSCs were able to differentiate into osteoblasts. In the
meantime, after 10 days of adipogenic induction, the cells
transformed from spindle- to polygonal-shaped, and lipid droplets
(similar in appearance to a string of beads) were observed in the
cytoplasm and between the cells. Given that Oil Red O staining
showed positive results, BMMSCs could differentiate into
adipocytes.
RNA interference refers to specific binding of
exogenous or endogenous siRNA or miRNA to corresponding target gene
mRNA after entering cells, thereby inhibiting protein translation
(16,17) and expressions of specific genes.
Typically, siRNA leads to complete silencing of specific target
genes, while miRNA acts on multiple genes and inhibits their
expression levels (18).
Additionally, miRNA regulates numerous metabolic processes, such as
cell proliferation, differentiation and apoptosis, managing to
promote different directions of differentiation of MSCs (including
osteogenic differentiation of stem cells) (19–22).
In the present study, the osteogenic capability of
miR-122-transfected BMMSC sheet was comprehensively evaluated by
ALP staining, analyses of collagen secretion and extracellular
matrix mineralization, as well as determination of
osteogenesis-associated gene and protein expression levels. Based
on the osteogenic parameters after 3 and 7 days of induction,
miR-122 transfection significantly promoted the osteogenic
differentiation of BMMSC sheets.
It is well-documented that miRNA exerts key
regulatory effects on the proliferation and differentiation of stem
cells (23), although the detailed
molecular mechanisms remain elusive. Furthermore, in the present
study, endogenous miR-122 expression levels were detected in the
sheets of different groups at various stages of osteogenic
induction using PCR. miR-122 modification continuously inhibited
such expression throughout the process. ERK 1/2 is a regulatory
kinase for the mitogen-activated protein kinases (MAPK) signaling
pathway. Notably, p-ERK 1/2 is involved in various biological
processes of osteoblasts, such as proliferation, differentiation
and apoptosis (24).
In the present study, the expression levels of p-ERK
1/2, RUNX2 and osteogenesis-associated genes in the cell sheet of
an experimental group were detected. As a key transcription factor
for the osteogenic differentiation of BMMSCs, RUNX2 directly
regulates the transcription of OCN, COL-I and osteopontin, with its
activity regulated by an MAPK-dependent phosphorylation cascade and
activated by ERK 1/2. Furthermore, miR-122 suppresses the
osteogenic differentiation of human BMMSCs by inhibiting the ERK
1/2 signaling pathway (25).
In conclusion, it was verified that genetically
modifying BMMSC sheets with a non-viral vector is feasible. The
resulting tissue-engineered implant provides novel strategies for
circumventing long, poor healing implanted bones and their high
failure rate. Further studies using more animals and human subjects
should be performed to validate the results of the present
study.
References
|
1
|
Masuda S and Shimizu T: Three-dimensional
cardiac tissue fabrication based on cell sheet technology. Adv Drug
Deliv Rev. 96:103–109. 2016. View Article : Google Scholar
|
|
2
|
Clark AG and Vignjevic DM: Modes of cancer
cell invasion and the role of the microenvironment. Curr Opin Cell
Biol. 36:13–22. 2015. View Article : Google Scholar
|
|
3
|
Chian KS, Leong MF and Kono K:
Regenerative medicine for oesophageal reconstruction after cancer
treatment. Lancet Oncol. 16:e84–e92. 2015. View Article : Google Scholar
|
|
4
|
Westrate LM, Lee JE, Prinz WA and Voeltz
GK: Form follows function: The importance of endoplasmic reticulum
shape. Annu Rev Biochem. 84:791–811. 2015. View Article : Google Scholar
|
|
5
|
Pocha SM and Montell DJ: Cellular and
molecular mechanisms of single and collective cell migrations in
Drosophila: Themes and variations. Annu Rev Genet.
48:295–318. 2014. View Article : Google Scholar
|
|
6
|
Sugimura R: Bioengineering hematopoietic
stem cell niche toward regenerative medicine. Adv Drug Deliv Rev.
99:212–220. 2016. View Article : Google Scholar
|
|
7
|
Flor TB and Blom B: Pathogens use and
abuse microRNAs to deceive the immune system. Int J Mol Sci.
17:5382016. View Article : Google Scholar :
|
|
8
|
Hayes CN and Chayama K: MicroRNAs as
biomarkers for liver disease and hepatocellular carcinoma. Int J
Mol Sci. 17:2802016. View Article : Google Scholar :
|
|
9
|
Huang JT, Liu SM, Ma H, Yang Y, Zhang X,
Sun H, Zhang X, Xu J and Wang J: Systematic review and
meta-analysis: Circulating miRNAs for diagnosis of hepatocellular
carcinoma. J Cell Physiol. 231:328–335. 2016. View Article : Google Scholar
|
|
10
|
Farooqi AA, Fayyaz S, Shatynska-Mytsyk I,
Javed Z, Jabeen S, Yaylim I, Gasparri ML and Panici PB: Is miR-34a
a well-equipped swordsman to conquer temple of molecular oncology?
Chem Biol Drug Des. 87:321–334. 2016. View Article : Google Scholar
|
|
11
|
Chmielewska AM, Rychłowska M, Król E,
Solarz K and Bieńkowska-Szewczyk K: Novel methods of hepatitis C
treatment and prevention. Postepy Hig Med Dosw (Online).
69:946–963. 2015. View Article : Google Scholar
|
|
12
|
Stachowiak G, Zajac A, Nowak M,
Stetkiewicz T and Wilczyński JR: Hemostatic disorders of the
menopausal period: The role of microRNA. Prz Menopauzalny.
14:144–148. 2015.
|
|
13
|
Casas-Agustench P, Iglesias-Gutiérrez E
and Dávalos A: Mother's nutritional miRNA legacy: Nutrition during
pregnancy and its possible implications to develop cardiometabolic
disease in later life. Pharmacol Res. 100:322–334. 2015. View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Ivanovska IL, Shin JW, Swift J and Discher
DE: Stem cell mechanobiology: Diverse lessons from bone marrow.
Trends Cell Biol. 25:523–532. 2015. View Article : Google Scholar :
|
|
16
|
Willeit P, Skroblin P, Kiechl S,
Fernández-Hernando C and Mayr M: Liver microRNAs: Potential
mediators and biomarkers for metabolic and cardiovascular disease?
Eur Heart J. 37:3260–3266. 2016. View Article : Google Scholar :
|
|
17
|
McGill MR and Jaeschke H: MicroRNAs as
signaling mediators and biomarkers of drug- and chemical-induced
liver injury. J Clin Med. 4:1063–1078. 2015. View Article : Google Scholar :
|
|
18
|
Sedano CD and Sarnow P: Interaction of
host cell microRNAs with the HCV RNA genome during infection of
liver cells. Semin Liver Dis. 35:75–80. 2015. View Article : Google Scholar :
|
|
19
|
Motavaf M, Safari S and Alavian SM:
Targeting microRNA-122: Walking on cutting edge of hepatitis C
virus infection therapy. Acta Virol. 58:301–308. 2014. View Article : Google Scholar
|
|
20
|
He L, Tian DA, Li PY and He XX: Mouse
models of liver cancer: Progress and recommendations. Oncotarget.
6:23306–23322. 2015. View Article : Google Scholar :
|
|
21
|
Gibson NW: Engineered microRNA
therapeutics. J R Coll Physicians Edinb. 44:196–200. 2014.
View Article : Google Scholar
|
|
22
|
Wilson JA and Sagan SM: Hepatitis C virus
and human miR-122: Insights from the bench to the clinic. Curr Opin
Virol. 7:11–18. 2014. View Article : Google Scholar
|
|
23
|
Gupta P, Cairns MJ and Saksena NK:
Regulation of gene expression by microRNA in HCV infection and
HCV-mediated hepatocellular carcinoma. Virol J. 11:642014.
View Article : Google Scholar :
|
|
24
|
Baek J, Kang S and Min H:
MicroRNA-targeting therapeutics for hepatitis C. Arch Pharm Res.
37:299–305. 2014. View Article : Google Scholar
|
|
25
|
Qiu Z and Dai Y: Roadmap of
miR-122-related clinical application from bench to bedside. Expert
Opin Investig Drugs. 23:347–355. 2014. View Article : Google Scholar
|