Introduction
Chimeric antigen receptor (CAR) T cells, an example
of adoptive cellular immunotherapy, and is a potentially curative
therapy for a multitude of cancer types (1). CARs are engineered fusion proteins
that generally consist of an extracellular single-chain variable
fragment (scFv) of an antibody for target recognition, the
transmembrane domain that is fused with co-stimulation signaling
domains, such as cluster of differentiation (CD) 28 or 4-1BB, and a
CD3ζ signaling domain to provide T-cell activation signals
(2–4). Additionally, antigen recognition by
CARs occurs in a major histocompatibility complex (MHC)-independent
manner, in order to overcome the tumor's immune escape by
downregulation of MHC molecules on the cell surface (5,6).
Targeted immunotherapy using chimeric antigen
receptor (CAR) molecules to redirect the specificity of cytotoxic
T-cells has emerged as a promising strategy for the treatment of a
broad range of malignancies (7,8).
However, despite encouraging outcomes, accumulating evidence has
demonstrated that the immunosuppressive microenvironment induced by
tumors and host regulatory cells may limit the full potential of
adoptive T-cell immunotherapy (9).
Tumors may evade immune surveillance by stimulating immune
inhibitory receptors, including hepatitis A virus cellular receptor
2 (TIM-3), cytotoxic T-lymphocyte protein-4 (CTLA-4) and programmed
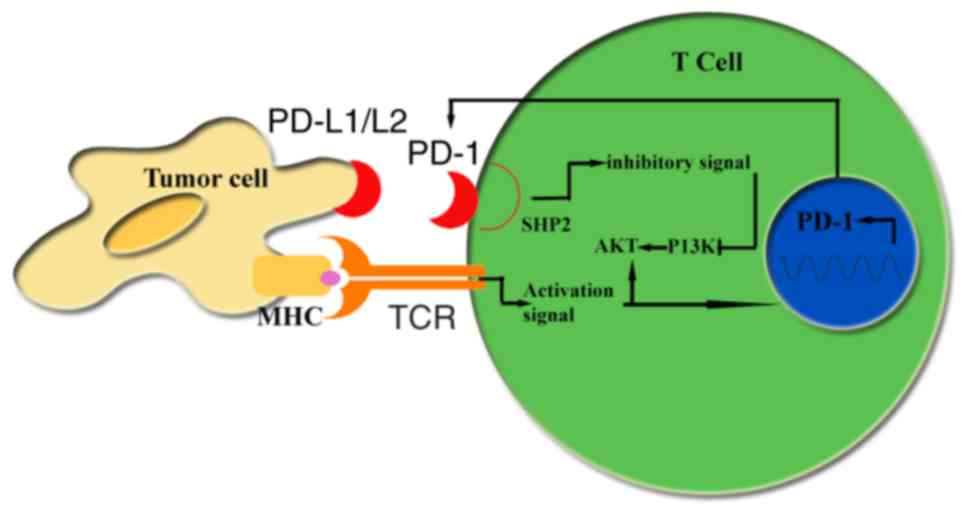
cell death protein-1 (PD-1), on T-cells (10). An example regulatory pathway
includes PD-1/programmed cell death protein ligand 1 (PD-L1), which
acts as a negative feedback loop to switch off adaptive immunity
following the initial immune response (11). The CAR-T and PD-1 blockade
techniques have achieved notable results in the therapy numerous
types of cancers (12,13). However, a number of clinical trials
have demonstrated that the efficacy of CAR-T and PD-1 blockade
therapy remains limited (14–16).
Due to these reported issues, the present review aimed to discuss
the status of combination therapies using a combination of CAR-T
and PD-1 blockade.
Current monotherapy status
CAR-T technology, a promising immunotherapy tool,
utilizes artificial T-cell surface receptors that stimulate the
physiological functions of the native T-cell receptor (TCR)
(16). The CAR is composed of an
extracellular antigen recognition domain, a spacer, a transmembrane
domain and an intracellular T-cell activation domain (5,17).
Through the use of genetic modification techniques, effector
T-cells may be induced to exhibit improved properties with regard
to targeting, killing activity and durability, compared with
conventional immunotherapies. CARs combine the effector functions
of T lymphocytes with the ability of antibodies to recognize
predefined surface antigens with high specificity and avidity,
independent of major histocompatibility complex restriction
(18,19). Additionally, compared with other
T-cell immunotherapy strategies, CAR-T-cells may overcome the local
immunosuppressive tumor microenvironment and break down host immune
tolerance to tumor cells. CAR-T therapies have generated
encouraging results for treating malignant tumors in clinical
trials, including cluster of differentiation (CD) 20 for the
treatment of non-Hodgkin's lymphoma (20), GD2 for neuroblastoma (21), and CD19 for chronic lymphocytic
leukemia (22). However, the
application of such therapies to solid tumors has been less
encouraging due to a number of factors, including the difficulty in
identifying unique tumor-associated antigens, inefficient homing of
CAR-T-cells to tumor locations, low persistence of CAR-T-cells
following infusion and their functional impairment in the
immunosuppressive microenvironment of solid tumors (23,24).
Meanwhile, numerous additional potential risks and challenges must
be addressed, including the potential for off-target effects,
insertion mutations, immune evasion, tumor lysis syndromes and
B-cell aplasia.
Tumors are associated with the immune system, and
may evade immune surveillance by stimulating immune inhibitory
receptors (25). TIM-3, CTLA-4 and
PD-1 are all inhibitory receptors with sustained expression in
T-cells which may be involved in tumor immune evasion (26). The PD-1/PD-L1 axis, a potential
barrier to adoptive T-cell immunotherapeutic strategies, is rapidly
emerging as a clinically important immune inhibitory pathway
(27). PD-1 is an inhibitory
receptor expressed by activated T-cells, activated B cells, natural
killer cells and myeloid cells (28). PD-1 inhibits T-cell activation when
engaged by its ligands PD-L1 or PD-L2, which are expressed on tumor
cells and stromal cells (29). The
interaction of PD-L1 with PD-1 may provide an inhibitory signal to
induce apoptosis and to suppress the activation or proliferation of
T-cells, meaning that immune-checkpoint inhibitors may block the
inhibitory signal of T-cells to prevent T-cell anergy (30–32).
Previously, checkpoint inhibitor therapies, including PD-1 blockade
(Fig. 1), which promote T-cell
responses by preventing T-cell exhaustion and anergy, have been
reported to exert marked antitumor responses in patients with renal
clear cell carcinoma (ccRCC) (33), non-small-cell lung cancer (34), advanced melanoma (35), urothelial carcinoma (36) and other solid tumors (37,38),
in addition to lymphoid malignancies (39). Similar to other cancer therapies,
toxicity remains a concern. Toxicity associated with PD-1 blockade
is typically immune-associated, and may include pneumonitis,
colitis, hepatitis, hypophysitis and thyroiditis (34,40).
Rationale for the combination of CAR-T and
PD-1 blockade
A prominent example of a clinically successful CAR-T
therapy is the treatment of hematological malignancies using a
second-generation CD19-specific CAR, which has demonstrated
antitumor activity in clinical trials (41,42).
However, the application of CAR-T-cells in the treatment of solid
tumors is associated with a number of challenges; one important
obstacle is the immunosuppressive effects of tumors (43). The early success of checkpoint
inhibitors in enhancing T-cell immunity presented the possibility
that these reagents may be used to enhance the antitumor activity
of genetically-modified T-cells (44). The most successful cases reported
for CAR-T have involved hematological lymphoid malignancies
(45), whereas blockade of the
PD-1/PD-L1 pathway has demonstrated signs of efficacy against solid
tumors (46,47). It was hypothesized that CAR-T in
combination with PD-1 blockade may be a promising immunotherapeutic
strategy for tumors, which may enhance the antitumor efficacy and
extend the scope of treatment.
An improved understanding of the mechanisms of
action of CAR-T may aid the design of novel CAR-T-based combination
therapies. CAR is an artificial T-cell surface receptor which
stimulates the physiological functions of the native TCR (17). Common elements of all CARs include
a single-chain antibody for antigen recognition on the surface of
tumor cells, and a membrane domain and intracellular signaling
domains borrowed from the CD3ζ chain and costimulatory receptors,
including CD28, CD137 and CD27, to supply a costimulatory signal,
which appears to be important for expansion and persistence in
vivo (48,49).
Additionally, the mechanisms of action for PD-1
blockade merit further investigation. PD-1 is an inhibitory
receptor expressed by activated T-cells, activated B cells, natural
killer cells and myeloid cells (29). Engagement of the PD-1/PD-L1 pathway
results in the phosphorylation of tyrosine-based motifs in the
cytoplasmic tail of the PD-1 inhibitory receptor, which promotes
the recruitment of tyrosine-protein phosphatase non-receptor type
11 (SHP-1), leading to dephosphorylation of phosphatidylinositol
3-kinase (PI3K). The resulting inhibition of PI3K generates
downstream activation of RAC-α serine/threonine protein kinase,
decreasing T-cell activation, proliferation and survival (50) (Fig.
2).
In order to further examine the combination
strategy, it is necessary to understand in detail the mechanism of
action of the combination approach, which is not completely clear
at present. However, certain insights may be obtained from previous
studies. For instance, John et al (51) observed a significant decrease in
the percentage of Gr1+CD11b+ myeloid-derived
suppressor cells (MDSCs) in the tumor microenvironment of mice
treated with the combination therapy. L-MDSC may circumvent the
effects of PD-L1 blockade by exploiting alternative suppressive
pathways, including indolamine 2,3-dioxygenase (52), arginase or inducible NO synthase
(53). In addition, CAR-T
proliferation in the presence of L-MDSC was rescued by SHP-1 and
SHP-2 inhibition, which prevented PD-1 signaling within CAR-T
(54). The results of ongoing and
future studies may facilitate the understanding of the mechanism of
action for this combination modality in order to improve patient
prognosis.
Current status of the combination of CAR-T
and PD-1 blockade
Immunotherapy frequently utilizes combination
approaches to increase efficacy. There are two individual
approaches which are currently leading this field: CAR-T and PD-1
blockade (46). Given the
promising results from CAR-T and PD-1 blockade monotherapies, it is
of importance to investigate whether a combined immunotherapeutic
approach involving blockade of the PD-1 pathway may enhance the
function of genetically-modified T-cells expressing a CAR, leading
to enhanced tumor eradication. Studies into the combination
strategy have primarily focused on PD-1 blockade using monoclonal
antibody (mAb) and genetic approaches.
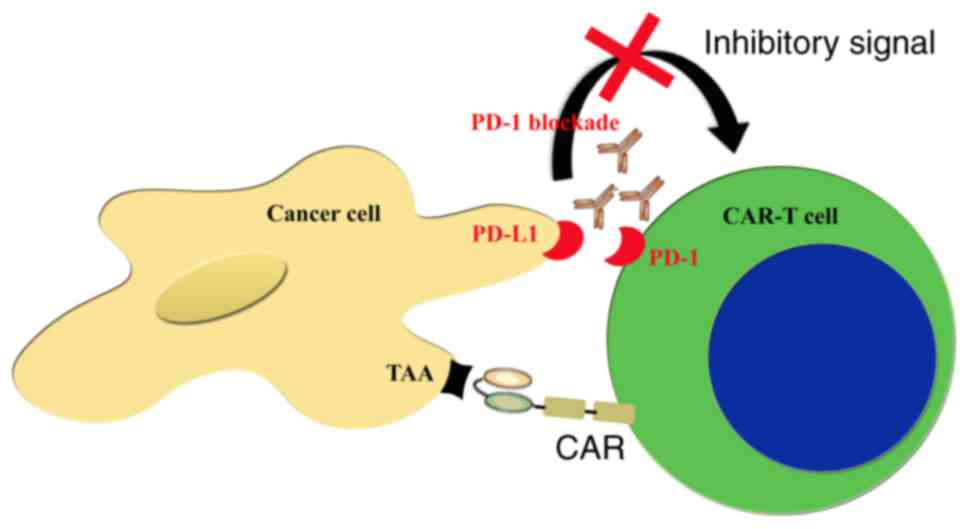
An area of interest is CAR-T therapy in combination
with PD-1 blockade using mAbs. A study from an Australian group
provided promising results; their work demonstrated for the first
time that PD-1 blockade was able to enhance the efficacy of
CAR-T-cell therapy against established solid tumors in vitro
and in vivo (51). The
researchers generated primary mouse T-cells expressing an
anti-receptor tyrosine-protein kinase erbB-2 (Her-2) CAR containing
an extracellular scFv-anti-Her-2 human mAb region, fused to a
transmembrane, intracellular costimulatory CD28 domain and
intracellular TCR-ζ domain. The study examined whether
administration of anti-PD-1 monoclonal antibodies was able increase
the therapeutic activity of CAR-T-cells against two different
Her-2+PD-L1+ tumors. Preclinical evidence for
the synergistic combination of adoptive T-cell therapy with T-cells
expressing CARs and anti-PD-1 mAbs was reported (51). A similar result obtained in a study
from Moon et al (55)
indicated that the addition of a blocking PD-L1 antibody to an
ex vivo CAR tumor infiltrating lymphocyte killing assay was
able to restore the defect in tumor cell killing, suggesting that
the PD-1 pathway serves a role in maintaining the dysfunction of
exhausted CAR-T-cells. An additional similar result reported by
Burga et al (56) on the
combination of CAR-T and anti-PD-L1 antibodies supported the
potential clinical merit of neutralizing L-MDSC in order to allow
for optimal antitumor efficacy. The researchers demonstrated that
CAR-T therapy in combination with PD-1 blockade through mAbs may be
highly synergistic.
A second area of interest is CAR-T therapy in
combination with PD-1 blockade through genetic approaches. The
results of a previous preclinical trial indicated that
anti-carbonic anhydrase IX (CAIX) CAR-T-cells secreting anti-PD-L1
antibodies were able to diminish T-cell exhaustion in vitro
and further decrease tumor growth in an orthotopic mouse model of
human renal cell carcinoma (RCC). Suarez et al (57) developed a novel CAR therapy for
CAIX+RCC that was able to block T-cell exhaustion. The group
engineered a bicistronic lentiviral vector to express the anti-CAIX
scFv bound to CD28 and CD3ζ signaling domains in one cassette, and
anti-PD-L1 immunoglobulin G1 (IgG1) or IgG4 in a second expression
cassette subsequent to an internal ribosome entry site site, thus
engineering human anti-CAIX-targeted CAR-T-cells that secreted
human anti-PD-L1 antibodies at the tumor site. Compared with the
anti-CAIX CAR-T-cells alone in a humanized mouse model of ccRCC,
tumor growth was decreased 5-fold and tumor weight was decreased by
50–80% (57). The results of a
preclinical trial performed by Liu et al (43) demonstrated that, while PD-1
blockade augmented the antitumor efficacy of CAR-T-cells, the use
of CAR-T-cells expressing PD1CD28 was superior in controlling tumor
burden. In order to address this possibility, the researchers used
anti-PD1 antibodies in combination with CAR-T-cells, followed by a
genetic approach described by others, in which T-cells were
transduced with a CAR and a chimeric switch-receptor containing the
extracellular domain of PD1 fused to the transmembrane and
cytoplasmic domain of the costimulatory molecule CD28. When the PD1
portion of this switch-receptor engages its ligand, PD-L1, it
transmits an activating signal via the CD28 cytoplasmic domain
instead of the inhibitory signal generally transduced by the PD1
cytoplasmic domain. The aforementioned previous study tested the
effect of this PD1CD28 supplement on human CAR-T-cells targeting
aggressive models of human solid tumors expressing relevant tumor
antigens. Treatment of mice bearing large, established solid tumors
with PD1CD28 CAR-T-cells led to a significant regression in tumor
volume due to enhanced CAR-T-cell infiltration, decreased
susceptibility to tumor-induced hypofunction and attenuation of
insulin receptor expression, compared with treatment with
CAR-T-cells alone or PD-1 antibodies (43). The group demonstrated that CAR-T
therapy in combination with PD-1 blockade through genetic
approaches may be synergistic.
Combination therapy with CAR-T and PD-1 blockade has
been further evaluated in clinical trials. Gargett et al
(58) demonstrated that
PD-1-targeted combination therapy approaches may be useful for
augmenting CAR-T-cell efficacy and persistence in patients. The
phase 1 CARPETS trial (registration no. ACTRN12613000198729)
utilized GD2-iCAR consisting of CD3ζ, CD28 and OX40 signaling
domains coupled to a 14g2a scFv and an inducible caspase-9 suicide
gene, with PD-1 blocked using pembrolizumab. In a protocol
amendment for the GRAIN trial of GD2-specific CAR-T-cells in
neuroblastoma patients, concurrent treatment with anti-PD-1 mAb was
used (58). The researchers
applied their understanding of the in vitro results to an
analysis of peripheral blood samples derived from patients enrolled
in the ongoing CARPETS clinical trial. During the investigations,
it was observed that PD-1 blockade restored CAR-T-cell cytokine
production and promoted GD2-iCAR T-cell survival and the killing of
GD2+PD-L1+ tumor cells (58). However, the limited number of
patients enrolled means that the results that were presented were
descriptive and may not be used to form definitive conclusions
until more patients are enrolled in the study.
Preclinical studies have illustrated the synergistic
efficacy of the combination of CAR-T and PD-1 blockade. By
contrast, fewer clinical trials have been performed to evaluate the
effects of the combination approach; therefore, whether CAR-T in
combination with PD-1 blockade is a rational strategy for clinical
trials requires further elucidation. However, it may be
hypothesized that if translated to the clinic, PD-1 blockade and
CAR-T may be an efficacious treatment, since preclinical evidence
supports the synergistic combination of CAR-T and PD-1
blockade.
Conclusions and future perspectives
The immune system serves an important role in
controlling and eradicating malignant cells. Based on the rapid
development of immunotherapy, combination therapy using CAR-T and
PD-1 blockade has become a novel research area. Preclinical studies
have demonstrated that CAR-T and PD-1 blockade are synergistic,
leading to long-term survival without causing any signs of
pathology in vivo. Moon et al (55) reported that the combination
strategy was able to slow tumor growth, although it did not result
in regression or cure. Despite recent progress, the field remains
at the preclinical phase. However, previous data have suggested
that combination therapy may enhance therapeutic efficacy and
broaden the range of antitumor treatments (43,51,55–57,59).
It may be hypothesized that the combination strategy may be a
rational approach for future clinical trials, although further
research is required. Prior to wide adoption of the CAR-T and PD-1
blockade combination in clinical practice, a number of challenges
must be addressed, including low response rates, toxicity,
relatively short response duration, inability to achieve curative
effects, and lack of effective and specific tumor-associated
antigen targets. Trial-and-error approaches may be used to optimize
the strategy in order to provide more rational principles for
future clinical practice. At present, further research is required
to improve the efficacy and decrease the toxicity of the
combination treatment.
Future strategies may improve the efficacy of the
combination therapy of CAR-T and PD-1 blockade. Immunotherapy for
cancer is primarily dependent on T-cells, particularly
CD8+ CTL and CD4+ T-helper cells (10). The ability to identify important
T-cell characteristics and systematically optimize CAR-T-cell
preparation has the potential to markedly improve the efficacy of
adoptive T-cell therapy. A previous study demonstrated that
CAR-T-cells are enriched in the central memory (TCM) phenotype and
that TCM-derived CAR-T-cells are functionally superior to those
generated using bulk CD8+ T-cells (60). Methods to increase the persistence
of CAR-T-cells to promote treatment efficacy include using
allogeneic virus-specific T-cells and a combination of
CD8+ TCM cells and CD4+ T-cells (61,62).
Strategies to increase the efficacy of CAR-T-cells through the
modification of CAR constructs, including the use of 3rd generation
and 4th generation armored constructs, are being evaluated
(63). An additional approach is
to infuse patients with polyspecific CAR-T-cells that target
multiple cell surface proteins to prevent immune evasion.
The toxicity of the combined therapy requires
further investigation. Due to previous studies of toxicity in
certain CAR-T-cell (64),
anti-PD-1 (34) and combination
CAR-T and PD-1 blockade trials (51,56,57),
future studies are required to further optimize the dose and timing
regimens of CAR-T-cells with PD-1 blockade in self-antigen mouse
models prior to phase I clinical trials. However, identifying an
ideal dose of CAR-T-cells to use in combination with PD-1 blockade
is difficult as the in vivo expansion of the cells is
variable, potentially resulting in inconsistent responses and
unpredictable toxicity. Novel methods to increase therapeutic
safety are being evaluated and include the introduction of a
suicide gene via herpes simplex virus thymidine kinase or inducible
caspase-9, in addition to the use of targetable cell-surface
proteins, including truncated epidermal growth factor receptor or
CD20 (65,66). Sadelain et al (5) reported the cotransfection of two
different CARs that recognize two different tumor surface antigens,
one providing TCR-like signals and the other co-stimulation. The
need for simultaneous recognition of two antigens may provide
increased specificity and safety.
The combination therapy of CAR-T and PD-1 blockade
may be promising for patients with cancer as the research continues
and the techniques improve. The combination strategy requires
optimization through repeated preclinical and clinical trials in
order to minimize toxicity and maximize treatment efficacy for
patients with malignancies. The results of ongoing and future
studies may facilitate understanding of the differential use of
these treatments as a single or a combined modality that improves
patient prognosis. It may be hypothesized that immunotherapies will
be increasingly applied in the clinic due to the rapid development
of cellular immunology and molecular biology, and that an era of
novel immunotherapies for malignancy may be approaching.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant no. 81301946), the Natural
Science Foundation of Jiangsu Province (grant no. BK2012146) and
the Postdoctoral Science Foundation of China (grant nos. 2015T80588
and 2013M540467).
References
|
1
|
Yang JC and Rosenberg SA: Adoptive T-Cell
therapy for cancer. Adv Immunol. 130:279–294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maus MV and June CH: Making better
chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer
Res. 22:1875–1884. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Whilding LM and Maher J: CA0R T-cell
immunotherapy: The path from the by-road to the freeway? Mol Oncol.
9:1994–2018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haji-Fatahaliha M, Hosseini M, Akbarian A,
Sadreddini S, Jadidi-Niaragh F and Yousefi M: CAR-modified T-cell
therapy for cancer: An updated review. Artif Cells Nanomed
Biotechnol. 44:1339–1349. 2016.PubMed/NCBI
|
|
5
|
Sadelain M, Brentjens R and Rivière I: The
basic principles of chimeric antigen receptor design. Cancer
Discov. 3:388–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schreiber RD, Old LJ and Smyth MJ: Cancer
immunoediting: Integrating immunity's roles in cancer suppression
and promotion. Science. 331:1565–1570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee DW, Kochenderfer JN, Stetler-Stevenson
M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M,
Shah NN, et al: T-cells expressing CD19 chimeric antigen receptors
for acute lymphoblastic leukaemia in children and young adults: A
phase 1 dose-escalation trial. Lancet. 385:517–528. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davenport AJ, Jenkins MR, Cross RS, Yong
CS, Prince HM, Ritchie DS, Trapani JA, Kershaw MH, Darcy PK and
Neeson PJ: CAR-T-cells inflict sequential killing of multiple tumor
targeT-cells. Cancer Immunol Res. 3:483–494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruella M and Kalos M: Adoptive
immunotherapy for cancer. Immunol Rev. 257:14–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riaz IB, Zahid U, Kamal MU, Husnain M,
McBride A, Hua A, Hamadani AA, George L, Zeeshan A, Sipra QR, et
al: Anti-CD 19 and anti-CD 20 CAR-modified T cells for B-cell
malignancies: A systematic review and meta-analysis. Immunotherapy.
9:979–993. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor A, Rothstein D and Rudd C: Small
molecule inhibition of PD-1 transcription is an effective
alternative to antibody blockade in cancer therapy. Cancer Res.
2017. View Article : Google Scholar
|
|
14
|
Sathyanarayanan V and Neelapu SS: Cancer
immunotherapy: Strategies for personalization and combinatorial
approaches. Mol Oncol. 9:2043–2053. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang WL: CAR T-cell therapy:
Opportunities and challenges. Immunotherapy. 8:245–247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim MG, Kim D, Suh SK, Park Z, Choi MJ and
Oh YK: Current status and regulatory perspective of chimeric
antigen receptor-modified T-cell therapeutics. Arch Pharm Res.
39:437–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Restifo NP, Dudley ME and Rosenberg SA:
Adoptive immunotherapy for cancer: Harnessing the T-cell response.
Nat Rev Immunol. 12:269–281. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
June CH: Principles of adoptive T-cell
cancer therapy. J Clin Invest. 117:1204–1212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
June CH: Adoptive T-cell therapy for
cancer in the clinic. J Clin Invest. 117:1466–1476. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Till BG, Jensen MC, Wang J, Chen EY, Wood
BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, et al:
Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle
cell lymphoma using genetically modified autologous CD20-specific
T-cells. Blood. 112:2261–2271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Louis CU, Savoldo B, Dotti G, Pule M, Yvon
E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al: Antitumor
activity and long-term fate of chimeric antigen receptor-positive
T-cells in patients with neuroblastoma. Blood. 118:6050–6056. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brentjens RJ, Riviere I, Park JH, Davila
ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda
O, et al: Safety and persistence of adoptively transferred
autologous CD19-targeted T-cells in patients with relapsed or
chemotherapy refractory B-cell leukemias. Blood. 118:4817–4828.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kakarla S and Gottschalk S: CAR T-cells
for solid tumors: Armed and ready to go? Cancer J. 20:151–155.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Figueroa JA, Reidy A, Mirandola L, Trotter
K, Suvorava N, Figueroa A, Konala V, Aulakh A, Littlefield L,
Grizzi F, et al: Chimeric antigen receptor engineering: A right
step in the evolution of adoptive cellular immunotherapy. Int Rev
Immunol. 34:154–187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bedognetti D, Maccalli C, Bader SB,
Marincola FM and Seliger B: Checkpoint inhibitors and their
application in breast cancer. Breast care (Basel). 11:108–115.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thommen DS, Schreiner J, Muller P, Herzig
P, Roller A, Belousov A, Umana P, Pisa P, Klein C, Bacac M, et al:
Progression of lung cancer is associated with increased dysfunction
of T-cells defined by coexpression of multiple inhibitory
receptors. Cancer Immunol Res. 3:1344–1355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blank C and Mackensen A: Contribution of
the PD-L1/PD-1 pathway to T-cell exhaustion: An update on
implications for chronic infections and tumor evasion. Cancer
Immunol Immunother. 56:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kedmi M, Avigdor A and Nagler A:
Anti-PD-1-targeted therapies focusing on lymphatic malignancies:
Biological rationale, clinical challenges and opportunities. Acta
Haematol. 133:129–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Keir ME, Butte MJ, Freeman GJ and Sharpel
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao S and Chen L: PD-1 as an immune
modulatory receptor. Cancer J. 20:262–264. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen DS, Irving BA and Hodi FS: Molecular
pathways: Next-generation immunotherapy-inhibiting programmed
death-ligand 1 and programmed death-1. Clin Cancer Res.
18:6580–6587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Okazaki T, Chikuma S, Iwai Y, Fagarasan S
and Honjo T: A rheostat for immune responses: The unique properties
of PD-1 and their advantages for clinical application. Nat Immunol.
14:1212–1218. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Massari F, Santoni M, Ciccarese C, Santini
D, Alfieri S, Martignoni G, Brunelli M, Piva F, Berardi R,
Montironi R, et al: PD-1 blockade therapy in renal cell carcinoma:
Current studies and future promises. Cancer Treat Rev. 41:114–121.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Topalian SL, Hodi FS, Brahmer JR,
Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD,
Sosman JA, Atkins MB, et al: Safety, activity and immune correlates
of anti-PD-1 antibody in cancer. N Engl J Med. 366:2443–2454. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ratta R, Zappasodi R, Raggi D, Grassi P,
Verzoni E, Necchi A, Di Nicola M, Salvioni R, de Braud F and
Procopio G: Immunotherapy advances in uro-genital malignancies.
Crit Rev Oncol Hematol. 105:52–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bracarda S, Altavilla A, Hamzaj A, Sisani
M, Marrocolo F, Del Buono S and Danielli R: Immunologic checkpoints
blockade in renal cell, prostate and urothelial malignancies. Semin
Oncol. 42:495–505. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Joshi M, Pal SK and Drabick JJ: Novel
approaches in cancer immunotherapy-a light at the end of the
tunnel. Discov Med. 21:479–487. 2016.PubMed/NCBI
|
|
39
|
Thanarajasingam G, Thanarajasingam U and
Ansell SM: Immune checkpoint blockade in lymphoid malignancies.
FEBS J. 283:2233–2244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weber JS: Practical management of
immune-related adverse events from immune checkpoint protein
antibodies for the oncologist. Am Soc Clin Oncol Educ Book. 1–177.
2012.
|
|
41
|
Zhu Y, Tan Y, Ou R, Zhong Q, Zheng L, Du
Y, Zhang Q and Huang J: Anti-CD19 chimeric antigen
receptor-modified T cells for B-cell malignancies: A systematic
review of efficacy and safety in clinical trials. Eur J Haematol.
96:389–396. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Srivastava S and Riddell SR: Engineering
CAR-T-cells: Design concepts. Trends Immunol. 36:494–502. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu X, Ranganathan R, Jiang S, Fang C, Sun
J, Kim S, Newick K, Lo A, June CH, Zhao Y and Moon EK: A chimeric
switch-receptor targeting PD1 augments the efficacy of
second-generation car T-cells in advanced solid tumors. Cancer Res.
76:1578–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Adachi K and Tamada K: Immune checkpoint
blockade opens an avenue of cancer immunotherapy with a potent
clinical efficacy. Cancer Sci. 106:945–950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chicaybam L, Sodré AL and Bonamino M:
Chimeric antigen receptors in cancer immuno-gene therapy: Current
status and future directions. Int Rev Immunol. 30:294–311. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morales-Kastresana A, Labiano S, Quetglas
JI and Melero I: Better performance of CARs deprived of the PD-1
brake. Clin Cancer Res. 19:5546–5548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Postow MA, Callahan MK and Wolchok JD:
Immune checkpoint blockade in cancer therapy. J Clin Oncol.
33:1974–1982. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shirasu N and Kuroki M: Functional design
of chimeric T-Cell antigen receptors for adoptive immunotherapy of
cancer: Architecture and outcomes. Anticancer Res. 32:2377–2383.
2012.PubMed/NCBI
|
|
49
|
Cheadle EJ, Gornall H, Baldan V, Hanson V,
Hawkins RE and Gilham DE: CAR T-cells: Driving the road from the
laboratory to the clinic. Immunol Rev. 257:91–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Blank C, Gajewski TF and Mackensen A:
Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific
T-cells as a mechanism of immune evasion: Implications for tumor
immunotherapy. Cancer Immunol Immunother. 54:307–314. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
John LB, Devaud C, Duong CP, Yong CS,
Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH and Darcy PK:
Anti-PD-1 antibody therapy potently enhances the eradication of
established tumors by gene-modified T-cells. Clin Cancer Res.
19:5636–5646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu J, Wang Y, Yan F, Li H and Ren X:
Response to comment on ‘Myeloid-derived suppressor cells suppress
antitumor immune responses through IDO expression and correlate
with lymph node metastasis in patients with breast cancer’. J
Immunol. 190:5341–5342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mazzoni A, Bronte V, Visintin A, Spitzer
JH, Apolloni E, Serafini P, Zanovello P and Segal DM: Myeloid
suppressor lines inhibit T-cell responses by an NO-dependent
mechanism. J Immunol. 168:689–695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mauldin IS, Tung KS and Lorenz UM: The
tyrosine phosphatase SHP-1 dampens murine Th17 development. Blood.
119:4419–4429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Moon EK, Wang LC, Dolfi DV, Wilson CB,
Ranganathan R, Sun J, Kapoor V, Scholler J, Puré E, Milone MC, et
al: Multifactorial T-cell hypofunction that is reversible can limit
the efficacy of chimeric antigen receptor-transduced human T-cells
in solid tumors. Clin Cancer Res. 20:4262–4273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Burga RA, Thorn M, Point GR, Guha P,
Nguyen CT, Licata LA, DeMatteo RP, Ayala A, Joseph Espat N,
Junghans RP and Katz SC: Liver myeloid-derived suppressor cells
expand in response to liver metastases in mice and inhibit the
anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother.
64:817–829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Suarez ER, Chang de K, Sun J, Sui J,
Freeman GJ, Signoretti S, Zhu Q and Marasco WA: Chimeric antigen
receptor T-cells secreting anti-PD-L1 antibodies more effectively
regress renal cell carcinoma in a humanized mouse model.
Oncotarget. 7:34341–34355. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gargett T, Yu W, Dotti G, Yvon ES, Christo
SN, Hayball JD, Lewis ID, Brenner MK and Brown MP: GD2-specific CAR
T-cells undergo potent activation and deletion following antigen
encounter but can be protected from activation-induced cell death
by PD-1 blockade. Mol Ther. 24:1135–1149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Prosser ME, Brown CE, Shami AF, Forman SJ
and Jensen MC: Tumor PD-L1 co-stimulates primary human CD8(+)
cytotoxic T-cells modified to express a PD1: CD28 chimeric
receptor. Mol Immunol. 51:263–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chang ZL, Silver PA and Chen YY:
Identification and selective expansion of functionally superior
T-cells expressing chimeric antigen receptors. J Transl Med.
13:1612015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Terakura S, Yamamoto TN, Gardner RA,
Turtle CJ, Jensen MC and Riddell SR: Generation of CD19-chimeric
antigen receptor modified CD8+ T-cells derived from virus-specific
central memory T-cells. Blood. 119:72–82. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cruz CR, Micklethwaite KP, Savoldo B,
Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, et al:
Infusion of donor-derived CD19-redirected virus-specific T-cells
for B-cell malignancies relapsed after allogeneic stem cell
transplant: A phase 1 study. Blood. 122:2965–2973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pegram HJ, Park JH and Brentjens RJ: CD28z
CARs and Armored CARs. Cancer J. 20:127–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shi H, Liu L and Wang Z: Improving the
efficacy and safety of engineered T-cell therapy for cancer. Cancer
Lett. 328:191–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jones BS, Lamb LS, Goldman F and Di Stasi
A: Improving the safety of cell therapy products by suicide gene
transfer. Front Pharmacol. 5:2542014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang X, Chang WC, Wong CW, Colcher D,
Sherman M, Ostberg JR, Forman SJ, Riddell SR and Jensen MC: A
transgene-encoded cell surface polypeptide for selection, in vivo
tracking, and ablation of engineered cells. Blood. 118:1255–1263.
2011. View Article : Google Scholar : PubMed/NCBI
|