Introduction
Pulmonary thromboembolism is the most common cause
of mortality in hospitalized patients, accounting for ~60% of
in-hospital mortality at present (1). A massive pulmonary embolism can lead
to cardiac arrest (CA) in 41% of patients, with a mortality ranging
from 65–95% (2). Pneumocyte
apoptosis is the most significant pathological observation in the
lung following an incidence of acute pulmonary embolism (APE). Li
et al (3) have reported the
presence of apoptotic cells as well as decreased expression of the
anti-apoptotic proteins B-cell lymphoma (Bcl)-2 and Bcl-extra large
in lung tissues following an APE, as compared with the controls.
This cellular damage in the lung might be responsible for a
subsequent severe lung dysfunction (4,5).
The renin-angiotensin system serves an important
role in the regulation of apoptosis in alveolar epithelial cells
(AECs). Recently, several research groups have identified a novel
pathway called angiotensin-converting enzyme2 (ACE2)-angiotensin
(Ang)-1-7-Mas1 proto-oncogene, G protein-coupled receptor (Mas)
axis that acts contrary to the classic ACE-Ang II-Ang II type 1
(AT1) receptor axis (6–9). The pro-apoptotic role of the classic
ACE axis in AECs in response to Fas activation has been well
studied (6). Ang II is known to
chemically induce apoptosis in AECs (7). Wang et al (8) demonstrated that ACE2 can attenuate
bleomycin-induced lung fibrosis by inhibiting the apoptosis in
pulmonary epithelial cells. Similarly, Ji et al (9) demonstrated that ACE2 inhibits
apoptosis in pulmonary endothelial cells during acute lung injury.
Our previous study demonstrated a reduction in post-resuscitation
pulmonary vascular resistance following captopril treatment, which
activated the ACE2-Ang-(1–7)-Mas axis (10). However, the role of ACE2/ACE
balance on pneumocyte apoptosis in massive APE-CA remains unclear.
Thus, in the present study, it was aimed to examine the correlation
between ACE2/ACE balance and apoptotic factors in the lung and to
investigate the effect of captopril on pulmonary apoptosis in an
APE-CA pig model.
Materials and methods
Animals
As described previously (10), twenty-nine land race pigs (both
sexes, aged 3 months, 28±2 kg) were obtained from the Beijing
Experimental Animal Center (license no. SCXK 11-00-002). All
animals were housed in a cage of size 80×80×90 cm and had free
access to water and standard chow. Room temperature was adjusted to
26°C, and humidity was 60%. The study procedures were approved by
The Capital Medical University Institutional Animal Care Committee
(permit no. 2010-D-013). Experimental protocols were designed in
strict compliance with the guidelines of the Animal Care and Use
Committee of Capital Medical University (Beijing, China).
Experimental preparation
All animals received an intramuscular premedication
with 0.2 mg/kg midazolam followed by an ear vein injection of 1.0
mg/kg propofol. Animals were maintained under general anesthesia by
a continuous administration of 3% pentobarbital (8 mg/kg/h). An
endotracheal tube (internal diameter 6.5 mm) was fixed into trachea
and a ventilator (Evita 4; Draeger Medical UK Ltd., Hertfordshire,
UK) was used to assist ventilation. The ventilation mode was
synchronized to a tidal volume of 8 ml/kg and a respiratory
frequency of 12–20 breaths per minute on room air. An end-tidal
pressure of carbon dioxide was maintained between 30 and 40 mmHg
and monitored using an infrared CO2 analyzer
(CO2SMO Plus monitor; Respironics, Inc., Murrysville,
PA).
A7-F Swan-Ganz catheter (Edwards Lifesciences Corp.,
Irvine, CA, USA) was flowed into the pulmonary artery from the
right external jugular vein to monitor mean pulmonary artery
pressure. An arterial catheter was inserted into the left femoral
artery to measure aortic pressure. A triple lumen central venous
catheter was inserted into the left femoral vein to monitor central
venous pressure. A large-bore catheter (internal diameter 1.0 cm)
was inserted into the left external jugular vein with its tip
touching the opening of the pulmonary artery for infusion of blood
clots, under the guidance of a computed tomography. Normal saline
and 5% glucose saline (8 ml/kg/h) were infused via the right
femoral vein to compensate for the fluid loss and to maintain a
constant central venous pressure (5–12 mmHg). Electrocardiographic
and hemodynamic parameters were also monitored (M8001A; Philips
Medizin Systeme Böblingen GmbH, Böblingen, Germany).
Experimental protocol
An APE-CA model was established as described
previously (10). Briefly, 100 ml
of blood was drawn out from the femoral vein and allowed to
self-coagulate at room temperature (2–3 h). The clots (10–15 ml)
were cut into pieces (1.5×1×1 cm) and were suspended in normal
saline in a large catheter tip syringe. Subsequently, the clot
pieces were injected into the left external jugular vein >2 min
until a mean arterial pressure <30 mmHg was reached, yielding a
porcine model of APE-CA (11). To
establish an APE-return of spontaneous circulation (ROSC) model,
urokinase (15,000 U/kg) was infused into the pulmonary artery using
a Swan-Ganz catheter and cardiopulmonary resuscitation was
immediately initiated (following the 2010 American Heart
Association guidelines) (12).
ROSC was verified based on a systolic blood pressure >50 mmHg
for 10 min (13). If ROSC was not
restored within 30 min, the animals were considered dead.
Pigs (n=29) were randomly assigned into four groups.
Group 1 (control group, n=5) was injected with 10 ml potassium
chloride (15%) intravenously followed by a bolus of propofol (100
mg) at 6 h following experimental preparation. All animals in this
group were sacrificed due to ventricular fibrillation. Group 2
(APE-CA group, n=5) underwent the procedure outlined above. All
animals in this group were sacrificed due to ventricular
fibrillation or electromechanical diastolic arrest. Nineteen
animals received a thrombus injection as described above for the
establishment of APE-ROSC model. Ten out of nineteen animals were
successfully resuscitated and were randomly assigned into two
subgroups: ROSC-Cap (n=5), receiving an injection of captopril
(22.22 mg/kg; cat. no. C8856-1G, Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) 30 min after ROSC, and ROSC-SA (saline; n=5),
receiving an injection of equivalent normal saline 30 min after
ROSC. Subsequently, all animals in both subgroups (~6 h following
ROSC) were euthanized with an intravenous injection of 10 ml
potassium chloride (15%) following a bolus of 100 mg propofol. Lung
tissue was isolated and was immediately frozen in liquid nitrogen
and stored at −80°C until further analysis.
Histological evaluation
Lung tissues were fixed in 10% buffered formalin for
48 h, dehydrated with alcohol solutions of gradient concentration
(100, 95, 80 and 75%), sliced at 4 µm thickness, and sections were
stained with hematoxylin (15 sec)-eosin (30 sec) and dried 20 min
at 55°C. The lung pathological changes were observed under a light
Nikon Eclipse Ci-Emicroscope (Olympus Corporation, Tokyo, Japan)
under the low (10×10) and high magnification (10×40).
Western blot analysis
Each lung tissue sample (20 mg) was chopped into
fragments and homogenized in 200 µl radioimmunoprecipitation lysis
buffer (Roche Diagnostics, Basel, Switzerland). Tissue lysates were
centrifuged (13,000 × g, 20 min, 4°C) and immediately harvested and
stored at −80°C until further use. A bicinchoninic acid protein
assay kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) was used to measure protein concentrations in the supernatant.
The tissue lysate was mixed with the same volume of 5X sodium
lauryl sulfate (SDS) buffer [2.5 ml 0.5 mol/l Tris-HCl (pH 6.8),
0.39 g DTT, 0.5 g SDS, 0.025 g bromophenol blue, and 2.5 ml
glycerol] to obtain a mixture at a final concentration of 3 µg/µl.
The homogenates were heated at 95°C for 5 min. Using 8, 10 and 15%
SDS-polyacrylamide gels, total proteins (40 µg) were separated and
transferred onto a 0.45-µm polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA) for 40–100 min. Following blocking
with 5% nonfat dry milk for 1 h at room temperature, the membranes
were incubated, first, with 1:500 dilution of ACE2 (cat. no.
SC-390851), ACE (cat. no. SC-2079), AT1 receptor (cat. no.
SC-81671), Bcl-2 (cat. no. SC-492) and Bcl-2-associated X protein
(Bax) antibodies (cat. no. SC-6236; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), 1:200 dilution of Mas receptor antibody (cat. no.
AAR-013; Alomone Labs, Jerusalem, Israel), 1:1,000 dilution of
cleaved caspase-3 (cat. no. 9664s; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and caspase-3 antibodies (cat. no. SC-7272; Santa
Cruz Biotechnology, Inc.), and 1:1,000 dilution of β-actin antibody
(Abcam, Cambridge, UK), overnight at 4°C, and subsequently with
peroxidase-conjugated anti-rabbit or anti-mouse IgG-HRP linked
secondary antibody (1:10,000 dilution; cat. no. 111-035-003;
Jackson Immuno Research Laboratories, Inc., West Grove, PA, USA),
for 40 min at room temperature. Bound antibodies were detected via
enhanced chemiluminescence (EMD Millipore) and analyzed using a Gel
Imaging System version 4.00 (Tanon Science & Technology Co.,
Ltd., Shanghai. China).
Immunohistochemical staining
For the detection of cleaved caspase-3 in lung
tissues, immunohistochemical staining was used. Lung tissues were
fixed in 10% buffered formalin for 48 h at room temperature,
dehydrated with alcohol solutions of gradient concentration (100,
95, 80 and 75%), sliced at 4 µm thickness, and embedded in
paraffin. The sections were placed in histosol for the removal of
paraffin, rehydrated in graded ethanol, and blocked in 5% bovine
serum albumin (Sigma-Aldrich; Merck KGaA, Darmstadt, USA) for 4 h.
Slides were incubated with cleaved-caspase3 antibody (1:1,000
dilution; cat. no. 9664s; CST Biological Reagents Co., Ltd.,
Shanghai, China) overnight at 4°C. Sections incubated in normal
rabbit serum served as negative controls. Following primary
antibody incubation, sections were washed with phosphate buffer
saline and were incubated with avidin-biotin peroxidase-conjugated
secondary antibody (1:200; cat. no. PK-4001; ZSGB-Biotechnology;
OriGene Technologies, Inc., Beijing, China) for 2 h at room
temperature. For the development of peroxidase activity, 0.05%
diaminobenzidinetetrahydrochloride (Sigma-Aldrich; Merck KGaA) was
used for 10 min at room temperature. Sections were counterstained
with hematoxylin for 20 sec and viewed under an inverted phase
microscope (IX80 microscopy; Olympus Corporation) and analyzed
using Image Pro Insight 6.0 (Media Cybernetics, Inc., Rockville,
MD, USA).
Electron microscopy
Within 1–2 min following isolation, lung tissues
were sliced (~1 mm thick) on ice, fixed with 2.5% (v/v)
glutaraldehyde for 30 min at 4°C, postfixed with 1% (v/v) osmic
acid for 1 h at 4°C, and were dehydrated and embedded with Epon812
at 40°C for 4 h, 50°C for 2 h, and 90°C for 12 h. Thinner sections
(0.5–1 µm) were sliced, stained with 0.5% (w/v) toluidine blue, and
viewed under a Nikon Eclipse Ci-E microscope (Nikon Corporation,
Tokyo, Japan). Ultrathin sections (~60 nm) were cut, double-stained
with 2.0% (w/v) uranyl acetate for 20 min and 2.0% (w/v) lead
citrate for 15 min at room temperature, and viewed under a
transmission electron microscope (HITACHI HT7700; Hitachi Ltd.,
Tokyo, Japan).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
To obtain evidence of apoptosis in lung cells, TUNEL
assay was performed according to the manufacturer's protocol (Roche
Molecular Diagnostics, Pleasanton, CA, USA). Briefly, slices of
lung tissues (4–5 µm) were embedded in paraffin. Cells were counted
based on positive nuclei staining from five microscopic fields
(magnification, ×200) under a light microscope (Olympus
Corporation) by an experienced pathologist. The apoptotic index of
each experimental group was expressed as the ratio of the number of
apoptotic cells to the total number of lung cells.
Statistical analysis
Data were expressed as the mean ± standard
deviation. Comparisons between groups were made using one-way
analysis of variance with Bonferroni correction for post hoc
comparison. Continuous variables were fixed to normal distribution
by Kolmogorov-Smirnov test and equal variances by the homogeneity
of variance test. Pearson's correlation coefficient was used to
determine correlations between the ratios of ACE2/ACE, Bax, cleaved
caspase-3, and Bcl-2 and the ratio of Bcl-2/Bax. P<0.05 was
considered to indicate a statistically significant difference.
Results
Histological analysis of the APE-CA
lung
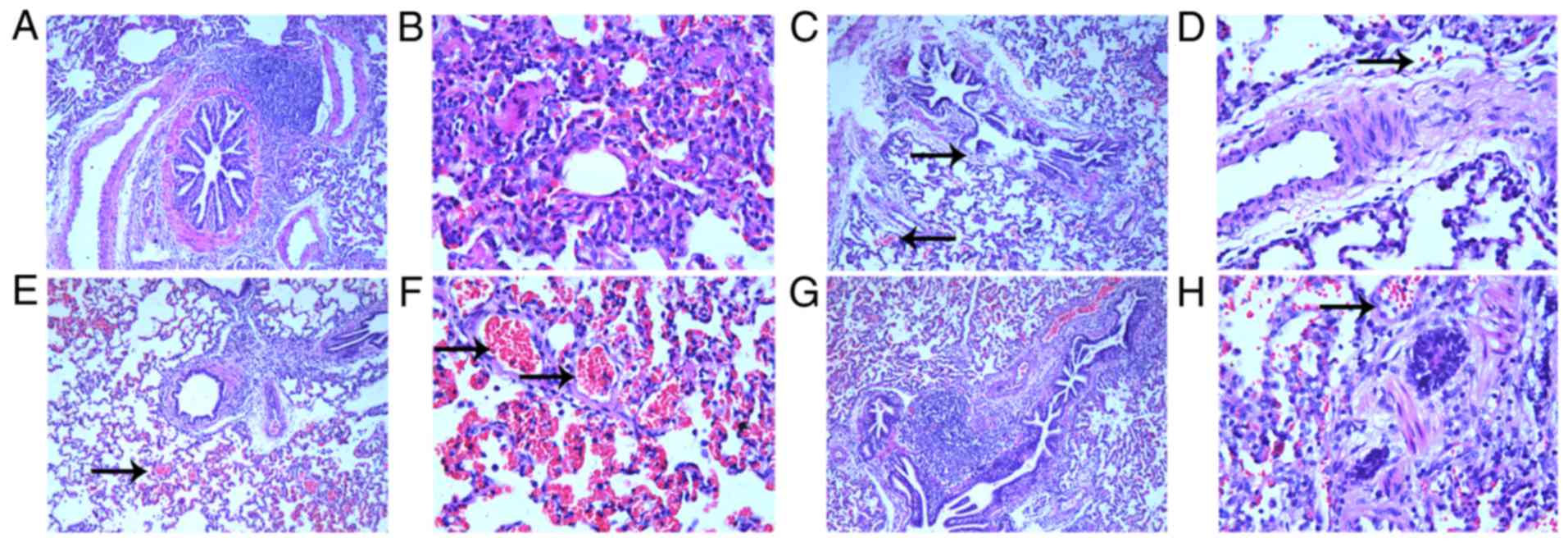
Histological analysis of the control animals
demonstrated regular bronchiolar and alveolar walls, with no
congestion in the alveolus (Fig. 1A
and B), whereas APE-CA animals had damaged bronchiolar and
alveolar walls and mildly congestive alveolar space (Fig. 1C and D). For the ROSC-SA group,
histological analysis demonstrated increased alveolar space
congestion (Fig. 1E and F) with a
large number of exudative cells and pulmonary interstitial edema.
However, captopril treatment alleviated the alveolar space
congestion following ROSC (Fig. 1G and
H).
ACE2/ACE imbalance in the APE-CA
group
The expression of ACE, AT1, ACE2, and Mas proteins
in porcine lung tissues was measured by western blotting in the CA
and ROSC groups following induction of APE (Fig. 2A-C). As depicted in Fig. 2C, the levels of ACE and AT1 (ACE,
0.24±0.02 to 0.48±0.03; AT1, 0.27±0.01 to 0.69±0.02) in the CA
group were significantly higher than those of the control group
(P<0.001), but lower in the ROSC-SA (0.31±0.14 and 0.33±0.09)
compared with the CA group (P<0.05). The ACE2/ACE ratio declined
sharply in CA (from 1.61±0.03 to 0.72±0.04) following induction of
APE. Notably, the protein levels of Mas receptor were decreased in
the ROSC-SA group following induction of APE (0.19±0.04) compared
with the APE-CA group (0.30±0.01) (Fig. 2C). The expression of AT1 receptor
increased, but ACE2 and Mas receptor expression levels were
unchanged in the ROSC-Cap compared with the ROSC-SA group.
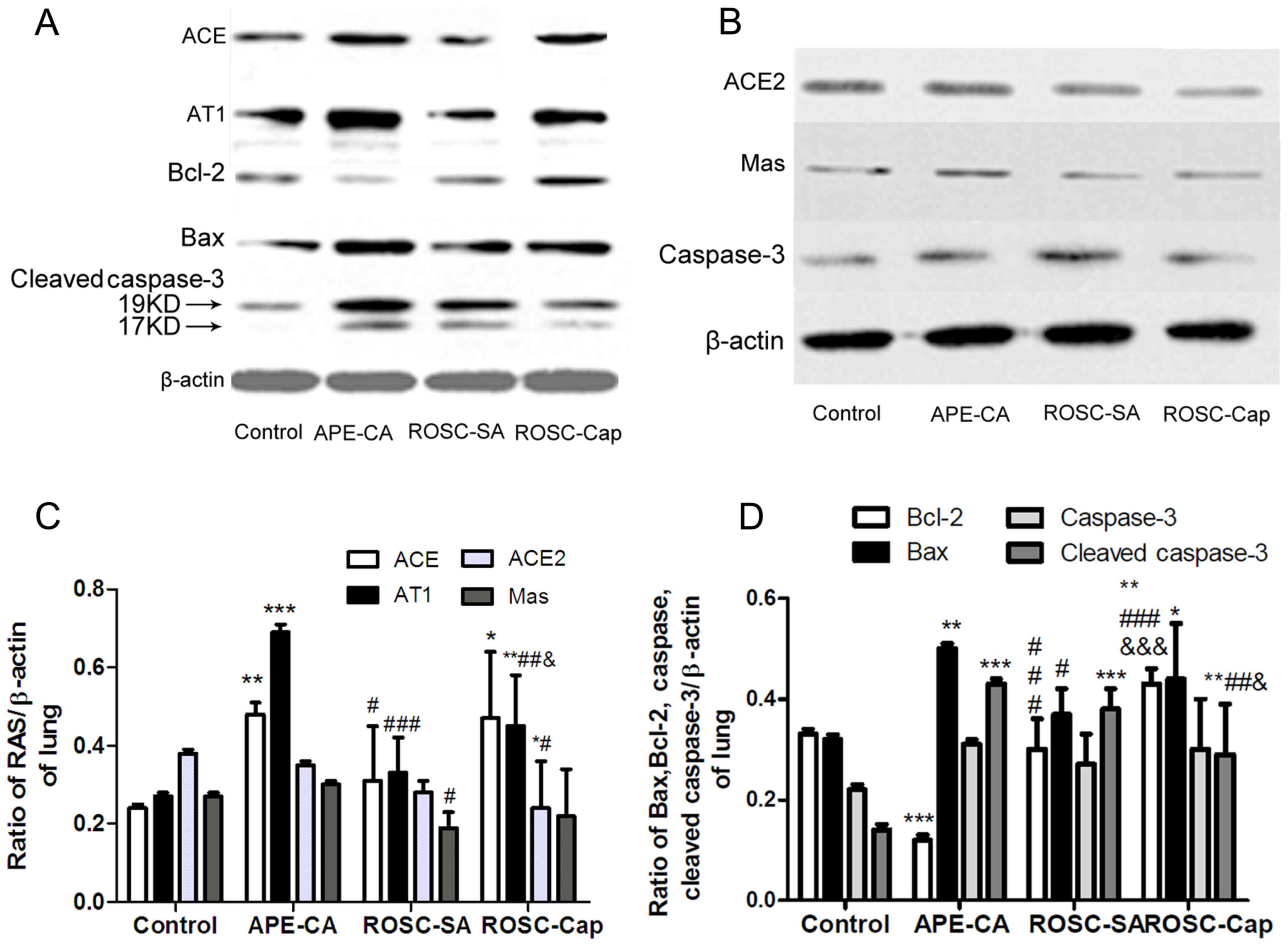 | Figure 2.Representative western blots of (A)
ACE, AT1, Bax, Bcl-2, cleaved caspase-3 and (B) ACE2, Mas and
caspase-3, in the lung and (C) quantification of ACE2, ACE, AT1 and
Mas expression levels and (D) quantification of apoptosis factors
expression. Data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01 and ***P<0.001 vs. control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. APE-CA group;
&P<0.05 and &&&P<0.001
vs. ROSC-SA group. APE, acute pulmonary embolism; CA, cardiac
arrest; ROSC, return of spontaneous circulation; SA, saline; Cap,
captopril; ACE, angiotensin-converting enzyme; AT1, angiotensin
receptor type-1; Mas, Mas1 proto-oncogene G protein-coupled
receptor. ACE, angiotensin-converting enzyme; AT1, ACE-Ang II-Ang
II type1; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X
protein. |
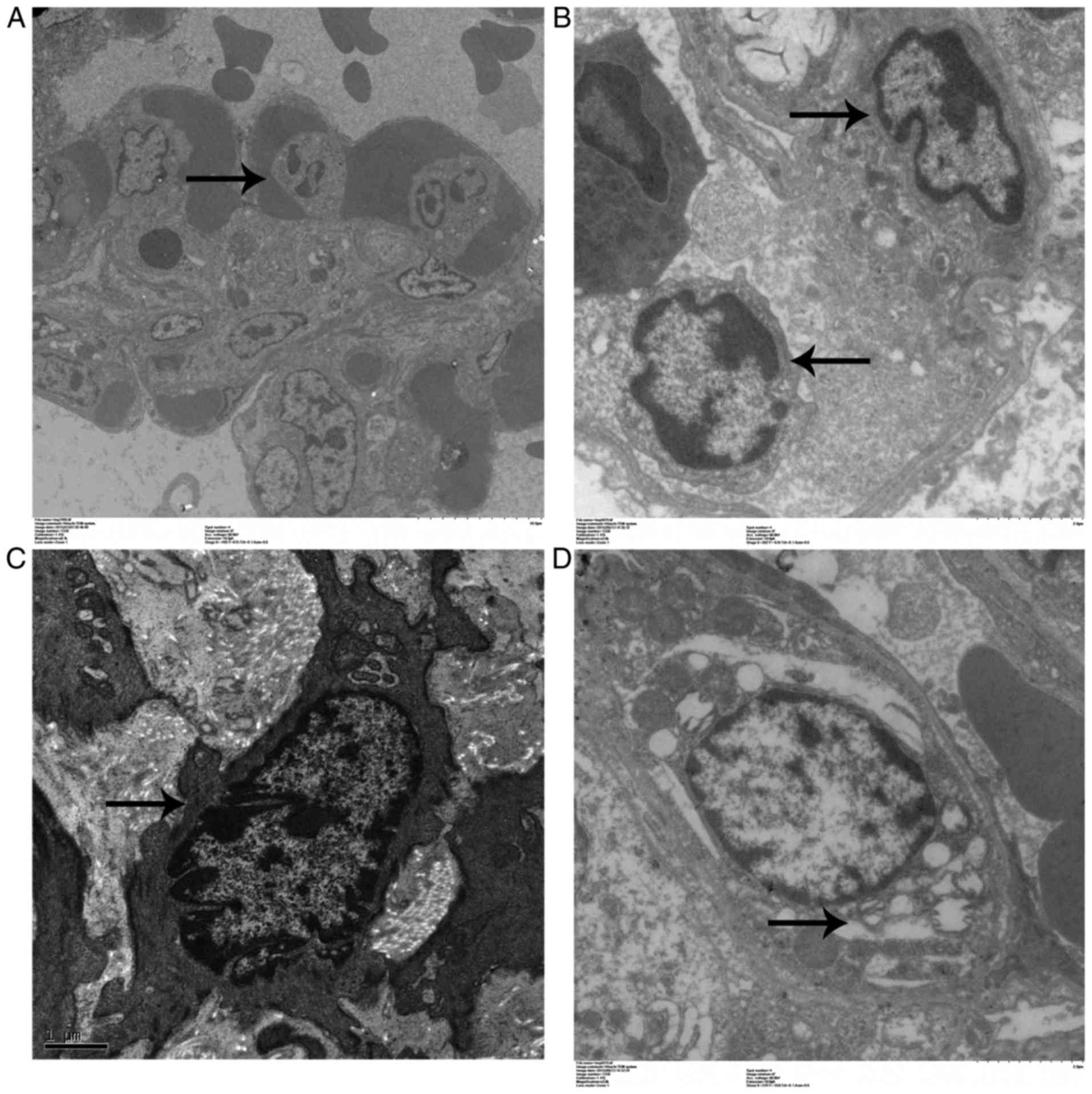
Ultrastructural changes in lung
cells
Pulmonary endothelial cell apoptosis and endothelial
matrix proliferation in the APE-CA and ROSC-SA groups are
illustrated in Fig. 3A and B.
Electron microscopy demonstrated loose connections between
endothelial cells, extensive nuclear shrinkage, chromatin
condensation and margination, cytoplasmic vacuoles (Fig. 3C), as well as type II pneumocyte
with lamellar body swelling and partial desquamation, in the APE-CA
group (Fig. 3D).
Pulmonary apoptosis and the effect of
captopril on the APE-CA group
To measure pulmonary apoptosis, the expression of
Bax, Bcl-2, cleaved caspase-3 (Fig.
2A) and caspase-3 (Fig. 2B)
proteins in the lung tissues was evaluated by western blot
analysis. The pro-apoptotic protein Bax was higher in the pulmonary
tissue of the APE-CA and ROSC-SA groups following induction of APE
compared with the control group (P<0.05; Fig. 2D). However, Bax protein levels were
lower in the ROSC-SA group than in the APE-CA group (P<0.05).
Compared with the control group, although the expression of
anti-apoptotic protein Bcl-2 (and the Bcl-2/Bax ratio) were lower
in the APE-CA group (P<0.001), these returned to normal levels
during ROSC following APE. Cleaved caspase-3 or cleaved
caspase-3/caspase-3 ratio was upregulated in the CA and ROSC-SA
groups compared with the control group (P<0.001; Fig. 2D). Captopril treatment inhibited
pulmonary cleaved caspase-3 expression and promoted Bcl-2
expression compared with the saline administration following ROSC
(Fig. 2A, D). The expression of
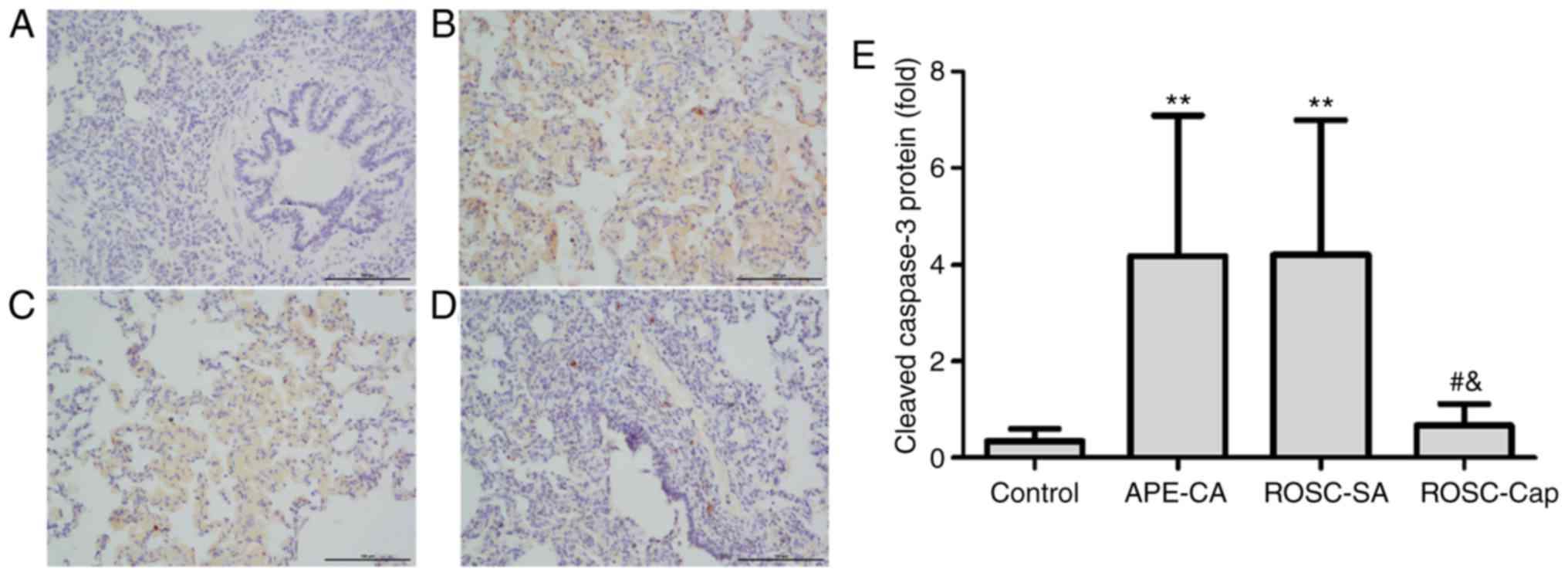
cleaved caspase-3 was analyzed by conventional
immunohistochemistry. Cleaved caspase-3 expression was not detected
in the control group (Fig. 4A) but
was markedly observable in the nucleus and cytoplasm of pulmonary
epithelial cells of the APE-CA and ROSC groups (Fig. 4B-E). Additionally, captopril
reduced cleaved caspase-3 staining in lung epithelial cells
following ROSC (Fig. 4D and E).
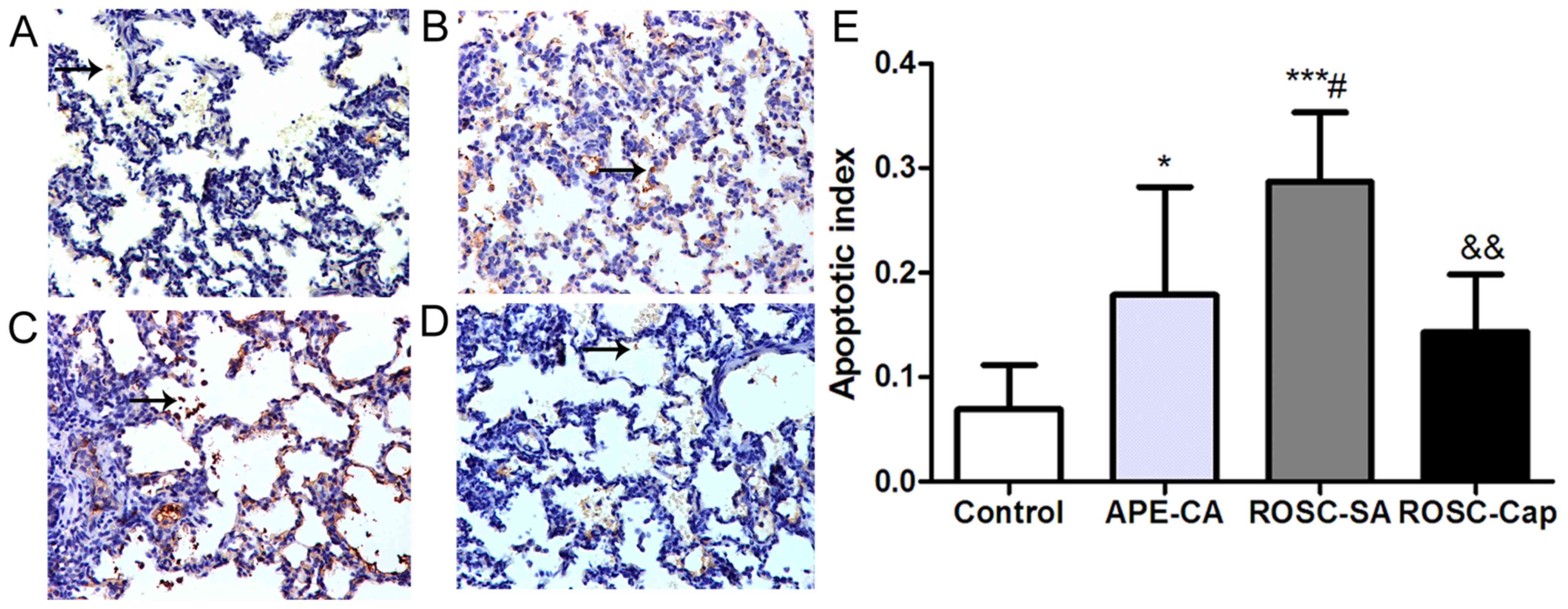
Furthermore, based on the results of TUNEL assay, the apoptotic
index in APE-CA and ROSC-SA groups increased compared with the
control group, and after ROSC, captopril treatment partially
attenuated pulmonary apoptotic index (Fig. 5).
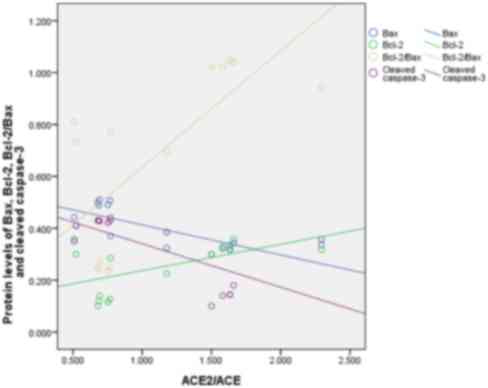
Correlation between ACE2/ACE and
apoptotic factors during an APE
A negative correlation between ACE2/ACE ratio and
Bax (r=−0.772, P<0.01) was detected. The protein levels between
ACE2/ACE ratio and cleaved caspase-3 were negative correlative
factors (r=−0.689, P<0.05) and the Bcl-2 (r=0.559, P<0.05)
and Bcl-2/Bax ratio (r=0.716, P<0.01) were positive correlative
factors of ACE2/ACE ratio (Fig.
6).
Discussion
Pneumocyte apoptosis is the most significant
pathological observation in the lung tissue following an APE. The
association between ACE2/ACE axis and the pathological changes in
the lung during ischemia and reperfusion induced by massive APE has
not been established. The present study aimed to investigate this
association by inducing CA (ischemia) and ROSC (reperfusion) in an
APE pig model. The present study observed congested alveolar space
and apoptosis in endothelial cells, which may be due to the loss of
ACE2/ACE balance in the lung tissue and reduced ACE2/ACE ratio. The
apoptosis in lung tissue was aggravated at the APE-CA group, but
was attenuated following ROSC. Following captopril treatment,
apoptosis in the lung tissue was alleviated. Thus, it can be
concluded that the ACE2/ACE balance may be positively correlated
with the expression of anti-apoptotic factors during APE-induced
lung injury in the pig model.
Apoptosis is the main mechanism of histological
pulmonary injury in APE-CA (3).
Deng et al (14)
established a canine model of pulmonary thromboembolism by
selectively embolizing blood clots to the right lower lobar
pulmonary artery. After 2 weeks, embolectomy of the reperfusion and
ischemia groups revealed similar pulmonary pathology to the present
study, including collapsed alveolar structures and the presence of
few exudative cells in the alveolar space in the ischemia group
(comparable to the APE-CA group) and a large number of exudative
cells and exudation within the alveolar spaces in the reperfusion
group (comparable to the ROSC groups). In the present study, higher
congestion in the alveolar spaces was present in the ROSC group
compared with the APE-CA group. However, captopril treatment
inhibited alveolar congestion. Electron microscopy visualization
revealed apoptosis in pulmonary endothelial cells and type II
pneumocytes with lamellar bodies partially desquamated in the
APE-CA model. Thus, alveolar congestion may have been caused by
injury to the alveolar epithelial barriers resulting from pulmonary
endothelial cell apoptosis.
The role of apoptosis in the pathogenesis of
interstitial pulmonary fibrosis, acute respiratory distress
syndrome, and chronic obstructive pulmonary disease has been
described previously (15–18). Recent studies suggested that
apoptosis serves an important role in chronic pulmonary embolism
(19–22). In a mouse model of chronic
pulmonary thromboembolism, lung parenchyma had markedly elevated
levels of proliferating cells and apoptotic cells, and activity of
pro-apoptotic caspase-3 compared with normal lungs (19). Mercier et al (20) investigated vessel alterations
induced by high flow through the creation of an aortopulmonary
shunt and reported increased smooth muscle cell proliferation, but
following 1 week of shunt closure, smooth muscle cells demonstrated
increased apoptosis without proliferation. Dolkart et al
(21) reported that the rate of
apoptosis in the bronchoalveolar lavage peaks at 12 and 48 h
following ischemia-reperfusion. When investigating the mechanisms
associated with lung ischemia-reperfusion injury in pulmonary
thromboembolism, Deng et al (22) demonstrated that the number of
apoptotic pneumocytes had a negative correlation with the ratio of
arterial oxygen partial pressure to fractional inspired oxygen,
whereas it had a positive correlation with alveolar
polymorphonuclear neutrophils in the reperfusion group. In the
present study, APE-CA and ROSC groups had increased levels of the
pro-apoptotic factors Bax and cleaved caspase-3, and inhibition of
the anti-apoptotic factor Bcl-2 compared with the control group.
Following ROSC, the reperfusion may have alleviated apoptosis in
the lung cells as indicated by the reduction in Bax and the
increase of Bcl-2 expression compared with the APE-CA group.
Previous studies have reported that the classical
ACE-AngII-AT1 axis of the renin-Ang system serves an important role
in the apoptosis of AECs induced by either Fas activation, or
chemically by antiarrhythmic agents benzofuran amiodarone (6) or fibrogenic agent bleomycin (7). A study reported that an AT1 receptor
blocker, losartan, reduced the protein levels of caspase-3 in
lipopolysaccharide (LPS)-induced lung injury (23). Additionally, evidence of inhibition
of pneumocyte apoptosis via activation of ACE2/Ang-(1–7)/Mas axis
has been reported. Yang et al (24) proved that Ang-(1–7) treatment is
effective in ameliorating Ang II-induced apoptosis in human
umbilical vein endothelial cells. Li et al (25) illustrated that LPS induced
apoptosis in pulmonary microvascular endothelial cells and reduced
the ratio of ACE2/ACE, while ACE2 treatment alleviated LPS-induced
apoptosis by reversing the ACE2/ACE imbalance and increasing
Ang-(1–7) levels. Other studies have demonstrated that the ACE2/ACE
imbalance is an important pathological mechanism not only in
LPS-induced acute lung injury but also in many diseases such as in
spontaneous hypertension (26),
acute pancreatitis (27,28), hepatic fibrogenesis (29), and in renal injury (30). In agreement with these studies, the
present reported that there was a positive correlation between
ACE2/ACE ratio and anti-apoptotic factors in APE-CA pigs,
suggesting that restoring the ACE2/ACE balance may be a therapeutic
strategy for resolving pneumonocyte apoptosis.
Captopril treatment has been widely investigated in
several studies on pulmonary and cardiac diseases. Fan et al
(31) demonstrated that captopril
treatment resulted in lower expression of ACE and AT1 receptors and
higher expression of ACE2 and AT2 receptors in mouse Lewis lung
carcinoma cells under hypoxia conditions. Li et al (32) in an in vivo study using rat
pulmonary microvascular endothelial cells demonstrated that
captopril pretreatment significantly reversed the LPS-induced
pathophysiological changes in the lung, reduced the ratio of Ang II
to Ang-(1–7), and restored the ACE/ACE2 ratio to normal levels. Our
previous study demonstrated that captopril treatment lowered
post-resuscitation pulmonary vascular resistance in pulmonary
embolism by activating the serum ACE2/Ang-(1–7)/Mas axis (10). In the present study, the
anti-apoptotic action of captopril in lung epithelial cells
following ROSC was demonstrated by increased Bcl-2 protein levels,
as assessed by western blot analysis and decreased cleaved
caspase-3 protein levels, as assessed by immunohistochemical
analysis. In addition, the TUNEL assay showed that the apoptotic
index was decreased following captopril treatment.
However, the present study has some limitations.
Firstly, although the porcine model is expected to simulate the
human model of APE-CA injury, the results may be hindered by
species and organ-dependent differences. Additionally, the present
experiments did not measure lung injury biomarkers in parallel, and
thus the correlation between ACE2/ACE axis and lung injury could
not be validated. Therefore, the molecular signaling pathway(s)
mediating the effects of the ACE2 axis on post-resuscitation lung
apoptosis in APE require further investigation.
In conclusion, the present study confirmed that a
loss of ACE2/ACE balance may be a key mechanism of pathogenesis in
APE-CA. Levels of apoptotic proteins were elevated in APE-CA
animals, but this effect was reversed following ROSC. Treatment
with captopril had anti-apoptotic effects in the lung tissue
following ROSC. Taken together, the present study indicated that
restoring the ACE/ACE2 balance may be an important therapeutic
strategy in the management of APE-CA.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81372025), the 2015
Annual Special Cultivation and Development Project for Technology
Innovation Base of Beijing Key Laboratory of Cardiopulmonary
Cerebral Resuscitation (grant no. Z151100001615056) and the Beijing
Natural Science Foundation (grant no. 7173253).
References
|
1
|
Chesnutt MS, Prendergast TJ, McPhee SJ,
Papadakis MA and Tierney LM Jr: Pulmonary venous
thromboembolismCurrent Medical Diagnosis and Treatment. 46th. New
York McGraw Hill; 28. pp. 284–294. 2007
|
|
2
|
Kuisma M and Alaspää A: Out-of-hospital
cardiac arrests of non-cardiac origin. Epidemiology and outcome.
Eur Heart J. 18:1122–1128. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li SQ, Jian W, Liu AR, Zhao F, Ti XY and
Ouyang HF: Expression of apoptosis related protein in transforming
growth factors-beta signaling pathway and its effects on the cell
apoptosis in the lung tissues after acute pulmonary embolism.
Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 20:353–356. 2008.(In
Chinese). PubMed/NCBI
|
|
4
|
Fischer S, Cassivi SD, Xavier AM, Cardella
JA, Cutz E, Xavier AM, Edwards V, Liu M and Keshavjee S: Cell death
in human lung transplantation: Apoptosis induction in human lungs
during ischemia and after transplantation. Ann Surg. 231:424–431.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Putte BP, Kesecioglu J, Hendriks JM,
Persy VP, van Marck E, van Schil P and Broe ME: Cellular
infiltrates and injury evaluation in a rat model of warm pulmonary
ischemia-reperfusion. Crit Care. 9:R1–R8. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bargout R, Jankov A, Dincer E,
Ibarra-Sunga O, Komodromos T, Filippatos G and Uhal BD: Amiodarone
induces apoptosis in human and rat alveolar epithelial cells in
vitro. Am J Physiol Lung Cell MolPhysiol. 278:L1039–L1044. 2000.
View Article : Google Scholar
|
|
7
|
Wang R, Ibarra-Sunga O, Verlinski L, Pick
R and Uhal BD: Abrogation of bleomycin-induced epithelial apoptosis
and lung fibrosis by captopril or by a caspase inhibitor. Am J
Physiol Lung Cell MolPhysiol. 279:L143–L151. 2000. View Article : Google Scholar
|
|
8
|
Wang L, Wang Y, Yang T, Guo Y and Sun T:
Angiotensin-Converting Enzyme 2 attenuates Bleomycin-induced lung
fibrosis in mice. Cell Physiol Biochem. 36:697–711. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ji Y, Gao F, Sun B, Hao J and Liu Z:
Angiotensin-converting enzyme 2 inhibits apoptosis of pulmonary
endothelial cells during acute lung injury through suppressing
SMAD2 phosphorylation. Cell Physiol Biochem. 35:2203–2212. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiao HL, Li CS, Zhao LX, Yang J, Tong N,
An L and Liu QT: Captopril improves postresuscitation hemodynamics
protective against pulmonary embolism by activating the
ACE2/Ang-(1–7)/Mas axis. Naunyn Schmiedebergs Arch Pharmacol.
389:1159–1169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pantazopoulos IN, Xanthos TT, Vlachos I,
Troupis G, Kotsiomitis E, Johnson E, Papalois A and Skandalakis P:
Use of the impedance threshold device improves survival rate and
neurological outcome in a swine model of asphyxial cardiac arrest.
Crit Care Med. 40:861–868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Travers AH, Rea TD, Bobrow BJ, Edelson DP,
Berg RA, Sayre MR, Berg MD, Chameides L, O'Connor RE and Swor RA:
Part 4: CPR overview: 2010 American heart association guidelines
for cardiopulmonary resuscitation and emergency cardiovascular
care. Circulation. 122 18 Suppl 3:S676–S684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stadlbauer KH, Rheinberger K, Wenzel V,
Raedler C, Krismer AC, Strohmenger HU, Augenstein S, Wagner-Berger
HG, Voelckel WG, Lindner KH and Amann A: The effects of nifedipine
on ventricular fibrillation mean frequency in a porcine model of
prolonged cardiopulmonary resuscitation. Anesth Analg. 97:226–230.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng C, Yang M, Lin Q, Yang Y, Zhai Z, Liu
K, Ding H, Cao X, Huang Z, Zhang L and Zhao J: Beneficial effects
of inhaled NO on apoptotic pneumocytes in pulmonary thromboembolism
model. Theor Biol Med Model. 11:362014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hagimoto N, Kuwano K, Miyazaki H, Kunitake
R, Fujita M, Kawasaki M, Kanika Y and Hara N: Induction of
apoptosis and pulmonary fibrosis in mice in response to ligation of
FAS antigen. Am J Respir Cell MolBiol. 17:272–278. 1997. View Article : Google Scholar
|
|
16
|
Kuwano K, Kunitake R, Kawasaki M, Nomoto
Y, Hagimoto N, Nakanishi Y and Hara N: P21Waf1/Cip1/Sdi1 and p53
expression in association with DNA strand breaks in idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med. 154:477–483. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matute-Bello G, Liles WC, Steinberg KP,
Kiener PA, Mongovin S, Chi EY, Jonas M and Martin TR: Soluble Fas
ligand induces epithelial cell apoptosis in humans with acute lung
injury (ARDS). J Immunol. 163:2217–2225. 1999.PubMed/NCBI
|
|
18
|
Segura-Valdez L, Pardo A, Gaxiola M, Uhal
BD, Becerril C and Selman M: Upregulation of gelatinases A and B,
collagenases 1 and 2, and increased parenchymal cell death in COPD.
Chest. 117:684–694. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wagner EM, Petrache I, Schofield B and
Mitzner W: Pulmonary ischemia induces lung remodeling and
angiogenesis. J Appl Physiol (1985). 100:587–593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mercier O, Sage E, de Perrot M, Tu L,
Marcos E, Decante B, Baudet B, Hervé P, Dartevelle P, Eddahibi S
and Fadel E: Regression of flow-induced pulmonary arterial
vasculopathy after flow correction in piglets. J Thorac Cardiovasc
Surg. 137:1538–1546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dolkart O.E.A.S.S.S.M.P.G..Aa W: Temporal
determination of lung NO system and COX-2 upregulation following
ischemia-reperfusion injury. Exp Lung Res. 40:22–29. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng C, Zhai Z, Wu D, Lin Q, Yang Y, Yang
M, Ding H, Cao X, Zhang Q and Wang C: Inflammatory response and
pneumocyte apoptosis during lung ischemia-reperfusion injury in an
experimental pulmonary thromboembolism model. J Thromb
Thrombolysis. 40:42–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Deng W, Deng Y, Deng J, Wang DX and Zhang
T: Losartan attenuated lipopolysaccharide-induced lung injury by
suppression of lectin-like oxidized low-density lipoprotein
receptor-1. Int J Clin Exp Pathol. 8:15670–15676. 2015.PubMed/NCBI
|
|
24
|
Yang HY, Bian YF, Zhang HP, Gao F, Xiao
CS, Liang B, Li J, Zhang NN and Yang ZM: Angiotensin-(1–7)
treatment ameliorates angiotensin II-induced apoptosis of human
umbilical vein endothelial cells. Clin Exp Pharmacol Physiol.
39:1004–1010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Y, Cao Y, Zeng Z, Liang M, Xue Y, Xi C,
Zhou M and Jiang W: Angiotensin-converting enzyme
2/angiotensin-(1–7)/Mas axis prevents lipopolysaccharide-induced
apoptosis of pulmonary microvascular endothelial cells by
inhibiting JNK/NF-κB pathways. Sci Rep. 5:82092015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gowrisankar YV and Clark MA: Angiotensin
II regulation of angiotensin-converting enzymes in spontaneously
hypertensive rat primary astrocyte cultures. J Neurochem.
138:74–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gaddam RR, Ang AD, Badiei A, Chambers ST
and Bhatia M: Alteration of the renin-angiotensin system in
caerulein induced acute pancreatitis in the mouse. Pancreatology.
15:647–653. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu R, Qi H, Wang J, Wang Y, Cui L, Wen Y
and Yin C: Angiotensin-converting enzyme (ACE and ACE2) imbalance
correlates with the severity of cerulein-induced acute pancreatitis
in mice. Exp Physiol. 99:651–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moreira de Macêdo S, Guimarães TA,
Feltenberger JD and Sousa Santos SH: The role of renin-angiotensin
system modulation on treatment and prevention of liver diseases.
Peptides. 62:189–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang XH, Wang YH, Wang JJ, Liu YC, Deng W,
Qin C, Gao JL and Zhang LY: Role of angiotensin-converting enzyme
(ACE and ACE2) imbalance on tourniquet-induced remote kidney injury
in a mouse hindlimb ischemia-reperfusion model. Peptides. 36:60–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fan L, Feng Y, Wan HY, Ni L, Qian YR, Guo
Y, Xiang Y and Li QY: Hypoxia induces dysregulation of local
renin-angiotensin system in mouse Lewis lung carcinoma cells. Genet
Mol Res. 13:10562–10573. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang
X and Jiang W: Angiotensin-converting enzyme inhibition attenuates
lipopolysaccharide-induced lung injury by regulating the balance
between angiotensin-converting enzyme and angiotensin-converting
enzyme 2 and inhibiting mitogen-activated protein kinase
activation. Shock. 43:395–404. 2015. View Article : Google Scholar : PubMed/NCBI
|