Introduction
Primary hepatocellular carcinoma (HCC) is a common
malignancy with rapid progression and poor prognosis. HCC mortality
is still rising globally (1). The
treatment approaches include surgery, cryotherapy, hepatic arterial
chemoembolization, radiofrequency ablation, biological therapy,
radiotherapy and radioactive seed implantation (2). However, those methods have their
limitations, including incomplete treatment, recrudescence and
metastasis. Therefore, it is important to identify a reasonable and
effective treatment plan for advanced HCC.
Sorafenib is a diaryl urea derivative of a
multi-target receptor tyrosine kinase inhibitor, with the potential
to inhibit the tumor cell proliferation following binding to
specific targets (3). Sorafenib
has recently been approved by the US Food and Drug Administration
for the treatment of advanced HCC (4). However, the clinical efficacy of
sorafenib was limited due to low sensitivity and high drug
resistance (5). The first- and
second-line drugs for advanced HCC require further development
(6). Improving the efficacy of
sorafenib has been popular in the discussion of HCC
chemotherapy.
Activating transcription factor (ATF) 2 is a member
of the ATF family, members of this family have the basic leucine
zipper domain (7). ATF2 is
differentially expressed in different tissues, with the highest
expression in the brain tissue (8). Upon the stimulation from inflammatory
factors or extracellular stress, ATF2 is phosphorylated at the
Thr71 and/or Thr69 site by Jun N-terminal kinase (JNK) to activate
its transcriptional activity (9–11).
ATF2 functions in various cellular activities, such as chromatin
remodeling, transcription regulation and DNA damage response
(12–14). It is of note that ATF2 has a dual
role in cancer suppression and carcinogenesis. In non-small cell
lung cancer and melanoma, ATF2 has a carcinogenic role (15,16),
whereas it has been identified to function as a cancer suppressor
in breast and non-metastatic skin cancer (17,18).
The present study constructed a small interfering RNA (siRNA)-ATF2
sequence and investigated the associated effects of sorafenib
treatment combined with ATF2 silencing on HCC. This present study
aimed to provide experimental basis for clinical treatment of
HCC.
Materials and methods
Cell culture and transfection
The Huh-7 HCC cell line was purchased from Shanghai
Cell Bank of Chinese Academy of Science (Shanghai, China) and
cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Hyclone; GE Healthcare Life Sciences,
Logan, UT, USA) and 100 U/ml penicillin-streptomycin
(Sigma-Aldrich, Merck Millipore, Darmstadt, Germany) in 5%
CO2 at 37°C.
The present study used the following 6 treatment
groups: i) Control; ii) vector; iii) 6.8 µM sorafenib (cat. no.
S7397; Selleck Chemicals, Shanghai, China), iv) empty vector + 6.8
µM sorafenib, v) siRNA-ATF2; and vi) siRNA-ATF2 + 6.8 µM sorafenib.
Cells at 90% confluence were transfected with empty vector or
ATF2-siRNA (GenePharma Co. Ltd., Shanghai, China) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Sorafenib was applied 24 h after ATF2-siRNA
transfection. After 6 h, the medium was replaced with fresh DMEM
medium containing 10% FBS and cultured in an 5% CO2
incubator at 37°C for 24 h. Following this, subsequent experiments
were performed. The expression of ATF2 was verified using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting. The following siRNA sequence for ATF2 was used:
siRNA-ATF2 forward (F),
5′-GATCCGCGAAATCTGTGGTTGTAAATCTCGAGATTTACAACCACAGATTTCGCTTTTTG-3′
reverse (R),
5′-AATTCAAAAAGCGAAATCTGTGGTTGTAAATCTCGAGATTTACAACCACAGATTTCGCG-3′;
negative control F,
5′-GATCCTTCTCCGAACGTGTCACGTAATTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTG-3′
and R,
5′-AATTCAAAAAATTCTCCGAACGTGTCACGTAATCTCTTGAATTACGTGACACGTTCGGAGAAG-3′.
Cell Counting kit-8 (CCK-8) assay
HCC cells (3×103 cells/well) were seeded
in 96-well plates. Following transfection and/or sorafenib
treatment for 48 h, 10 µl DMEM with CCK-8 (Gibco; Thermo Fisher
Scientific, Inc.) was added. Following an additional 4 h incubation
in CO2 incubator at 37°C the absorbance was recorded at
560 nm using a microplate reader (Thermo Fisher Scientific, Inc.).
The optical density (OD) values represented cell viability.
Flow cytometry
HCC cells (3×103 cells/well) were seeded
in 6-well plates. Following transfection and/or sorafenib
treatment, cells were collected following digestion with trypsin
(Gibco; Thermo Fisher Scientific, Inc.). The cells were incubated
in the dark with Annexin V-fluorescein isothiocyanate and propidium
iodide (PI; Beyotime Institute of Biotechnology, Ningbo, China) for
30 min. Subsequently, apoptosis was detected using a flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA) within 1 h.
Transwell assay
HCC cells (3×103 cells/well) were seeded
in 6-well plates. Following transfection and/or sorafenib
treatment, the cells were digested and seeded into the upper
chamber (3×103 cells/well) of Transwell with 1-day
starvation of serum. The lower chamber contained DMEM medium with
10% FBS. The lower chamber contained DME 4% PFA for 20–30 min at
room temperature. Following staining with 0.1% crystal violet
(Amresco, LLC, Solon, OH, USA) for 5 min at room temperature, light
microscopy was used to obtain images in five random fields. Cell
numbers were counted and represented the capacity for cell
invasion.
Cell migration
HCC cells were seeded in 6-well plates
(3×103 cells/well). Following transfection and/or
sorafenib treatment for 48 h, the cell monolayer was scratched.
After 48 h incubation at 37°C and the images were obtained using a
light microscope (Olympus Corporation, Tokyo, Japan).
RT-qPCR
HCC cells (3×103 cells/well) were seeded
in 6-well plates. Following transfection and/or sorafenib treatment
for 48 h, mRNA expression levels in the different treatment groups
was extracted using a TRIzol assay kit (Baosheng Science &
Technology Innovation Co. Ltd., Shanghai, China). mRNA was
transcribed into cDNA using a reverse transcription kit (cat. no.
639522; Takara Biotechnology Co., Ltd., Dalian, China) and
performed at 25°C for 10 min, 37°C for 120 min and 85°C for 5 min,
qPCR was used to detect the expression level of the target genes by
using the SYBR-Green qPCR master mix (cat. no. HY-K0501;
MedChemExpress, Monmouth Junction, NJ, USA). The thermocycling
conditions were as follows: Initial denaturation at 95°C for 10
min, followed by 35 cycles of a two-step PCR at 95°C for 14 sec and
60°C for 1 min. The 2−ΔΔCq method (19) was used to quantify the results; the
relative expression level of JNK3, ATF2 and tumor necrosis factor
(TNF)-α was normalized to GAPDH. The primers (5′-3′) used were as
follows: JNK3, forward (F), GGAAAAGGACATCAGGGAAGA and reverse (R),
CATGGGCTACAAGGAAAACGT; ATF2, F, CCAGCAACATCCTCCAGT and R,
CTCTTCTCCGACGACCAC; TNF-α F, GCTGACCGACAAAGAAGGC and R,
TTTAGGGATGTGATGATGGG; GAPDH F, CAATGACCCCTTCATTGACC and R,
GAGAAGCTTCCCGTTCTCAG.
Western blot analysis
HCC cells were seeded in 6-well plates
(3×103 cells/well). Following transfection and/or
sorafenib treatment for 48 h, protein was extracted from cell lines
using for western blotting with the triplePrep kit (cat. no.
28-9425-44; ReadyPrep; GE Healthcare Life Sciences). Protein levels
were quantified with a bicinchoninic acid protein assay kit.
Protein (25 µg/lane) was separated via 12% SDS-PAGE and transferred
onto nitrocellulose membranes. The membranes were blocked in 5%
skim milk for 2 h at room temperature and incubated with the
following primary antibodies overnight at 4°C: JNK3 (cat. no.
ab76572; 1:1,000; Abcam, Cambridge, UK), ATF2 (cat. no. bs-0518R;
1:400; BIOSS, Beijing, China), GAPDH (cat. no. A007; 1:1,000;
ABclonal Biotech Co., Ltd., Woburn, MA, USA), TNF-α (cat. no.
BA14901; 1:800; BIOSS), phosphorylated (p)-JNK3 (cat. no. ab76572;
1:1,000; Abcam), p-ATF2 (cat. no. BS-84449R; 1:400, BIOSS)
overnight at 4°C. The secondary antibody (1:100; cat. nos.
ab131368; Abcam) was added and co-incubated for 2 h at room
temperature. Enhanced chemiluminescence exposure liquid droplet
(cat. no. RPN2133; GE Healthcare Life Sciences, Chalfont, UK) was
added to the membranes. The membranes were visualized using a gel
imaging system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Densitometry was performed using Quantity One version 1.4.6
(Bio-Rad Laboratories, Inc.) Experiments were repeated three
times.
Statistical analysis
Data were presented as the mean ± standard error of
the mean and analyzed using SPSS version 17.0 (SPSS, Inc., Chicago,
IL, USA). Significant differences were determined using one-way
analysis of variance followed by the Student-Newman-Keuls post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
SiRNA-ATF2 facilitates the
anti-proliferation effect of sorafenib
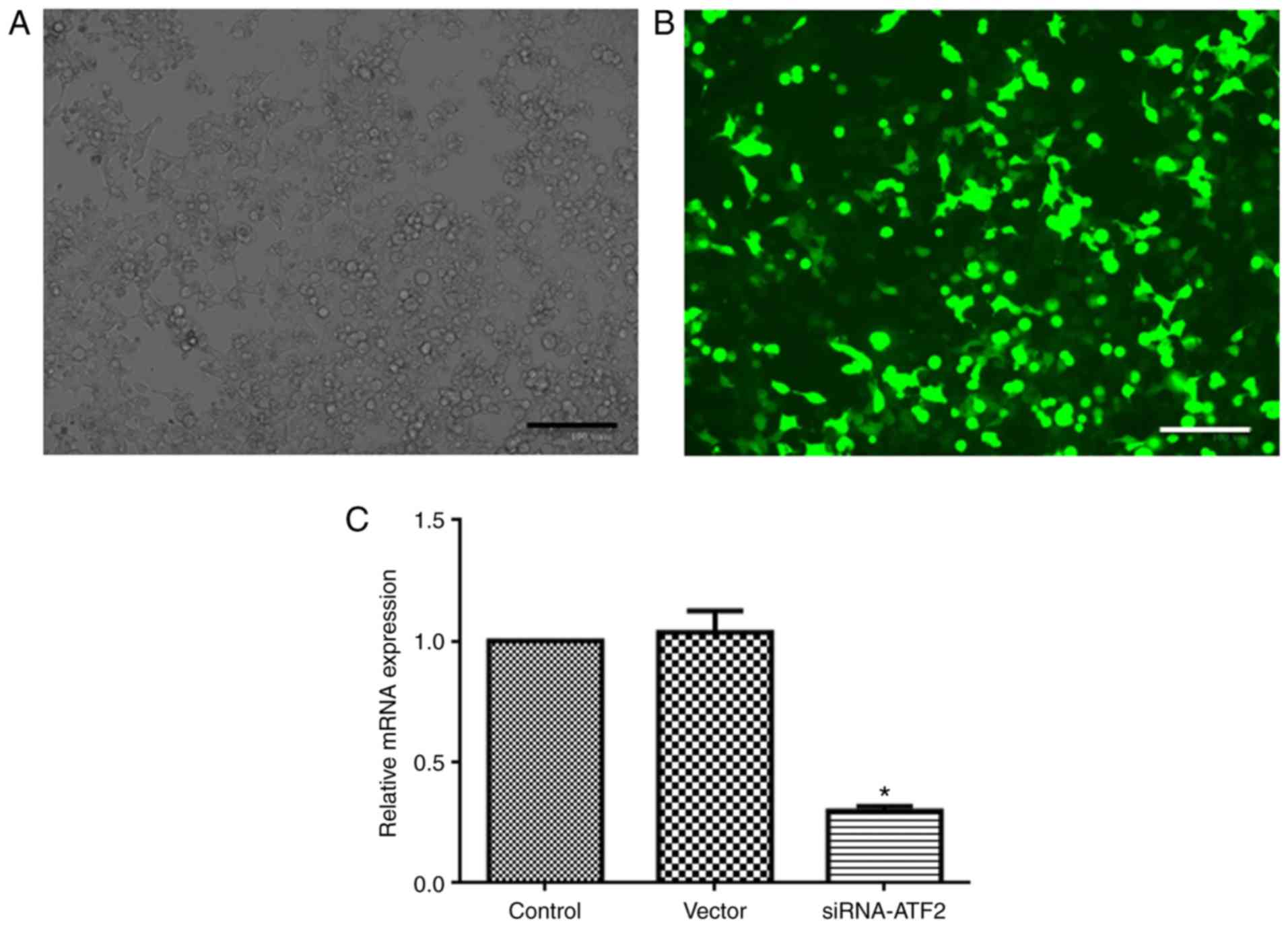
As presented in Fig.
1, Huh-7 cells achieved a good transfection efficiency and
normal morphology after following siRNA-ATF2 transfection. The
expression of ATF2 in the siRNA-ATF2 group was significantly
reduced (~80%, P<0.05) when compared with the control. ATF2
expression level in the control, vector and siRNA-ATF2 groups was
1, 1±0.09 and 0.30±0.02, respectively.
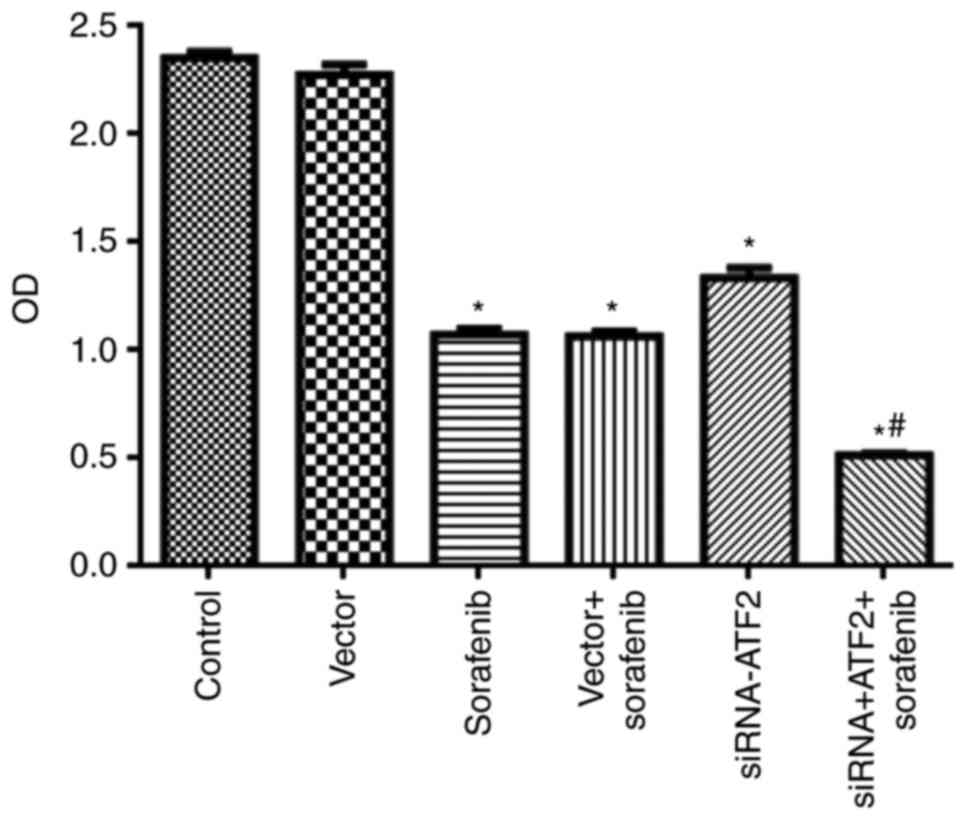
Sorafenib or siRNA-ATF2 only treatments
significantly reduced cell proliferation (F5,30=483.1;
P<0.05; Fig. 2) when compared
with the control group. It is of note, that sorafenib and
siRNA-ATF2 combination treatment significantly reduced cell
proliferation when compared with vector+sorafenib (P<0.05). The
OD values in each group were 2.35±0.03 in control, 2.27±0.05 in
vector, 1.07±0.03 in sorafenib, 1.06±0.02 in sorafenib+vector,
1.33±0.05 in siRNA-ATF2 and 0.51±0.01 in sorafenib+siRNA-ATF2.
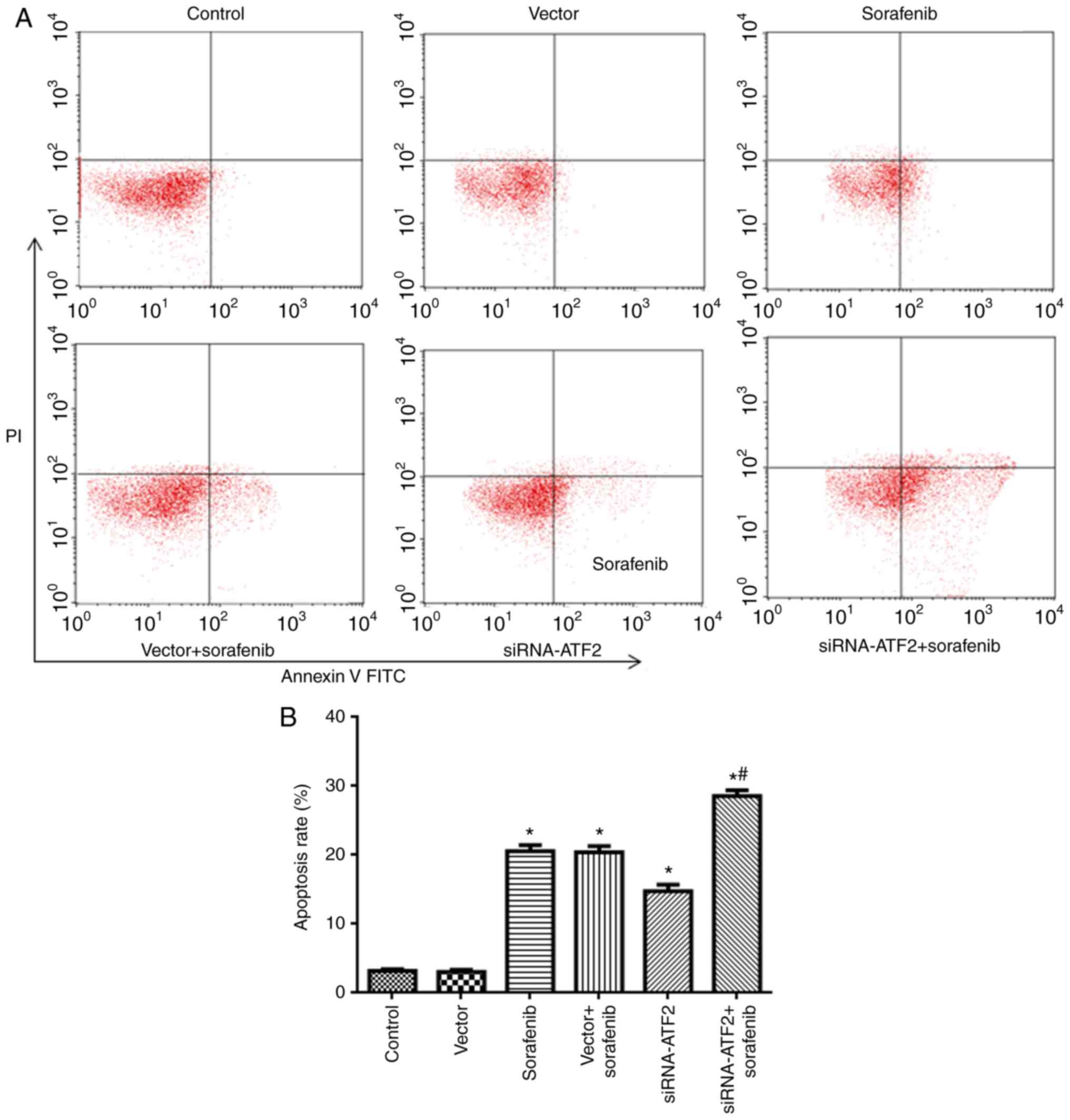
SiRNA-ATF2 facilitates apoptosis
induced by sorafenib
The flow cytometry findings are presented in
Fig. 3. Apoptosis in the sorafenib
and siRNA-ATF2 groups was significantly increased when compared
with the control (F5,30=189; P<0.05; Fig. 3). It is of note that sorafenib and
siRNA-ATF2 combined treatment additionally promoted apoptosis when
compared with the vector+sorafenib group (P<0.05). The apoptosis
rate in each group was 3.1±0.2% in control, 3.0±0.3% in vector,
20.5±0.9% in sorafenib, 20.3±0.9% in sorafenib+vector, 14.7±1.0% in
siRNA-ATF2 and 29.0±0.8% in sorafenib+siRNA-ATF2.
SiRNA-ATF2 facilitates the
anti-migration effect of sorafenib
Cell migration is presented in Fig. 4. Cell migration abilities of the
sorafenib or siRNA-ATF2 treatment groups were reduced when compared
with the control. It is of note that, sorafenib and siRNA-ATF2
combined treatment additionally inhibited cell migration.
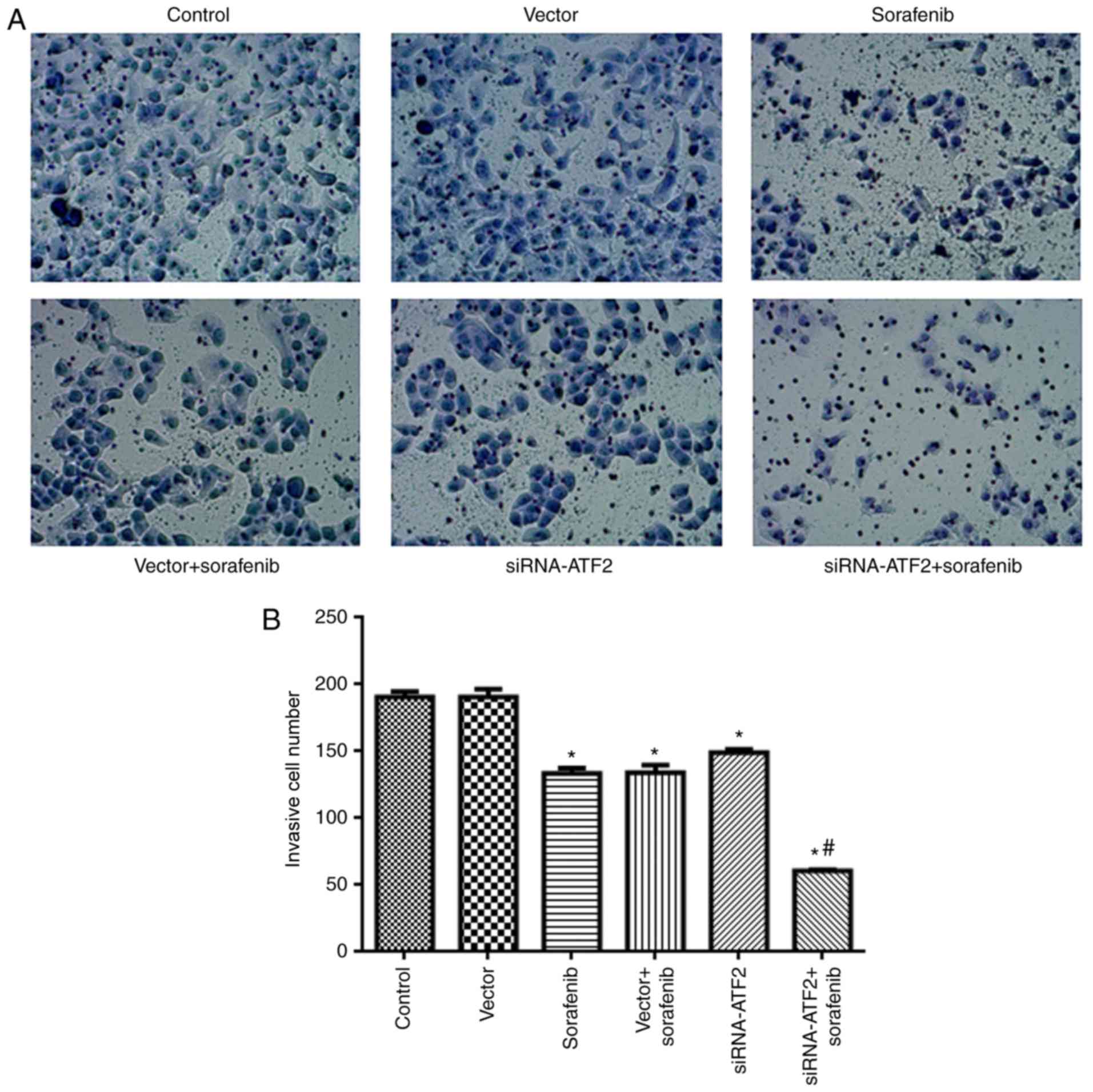
SiRNA-ATF2 facilitates the
anti-invasion effect of sorafenib
The findings of the cell invasion assay are
presented in Fig. 5. The cells in
the sorafenib or siRNA-ATF2 groups had significantly reduced
invasion ability when compared with the control
(F5,30=131; P<0.05; Fig.
5). It is of note that sorafenib and siRNA-ATF2 combined
treatment significantly inhibited the invasive abilities of cells
when compared with the vector+sorafenib treatment group
(P<0.05). The invasive cell number in each group was 190±4 in
control, 190±4 in vector, 133±4 in sorafenib, 134±6 in
sorafenib+vector, 149±3 in siRNA-ATF2 and 60±1 in
sorafenib+siRNA-ATF2.
Sorafenib and/or siRNA-ATF2 reduce
ATF2 and JNK3 expression and enhance TNF-α expression
ATF2, JNK3 and TNF-α mRNA and protein expression
levels were detected using RT-qPCR and western blotting. As
presented in Fig. 6, ATF2
expression was significantly different among groups (mRNA,
F5,30=149, P<0.05; protein, F5,30=239,
P<0.05). ATF2 mRNA and protein expression levels were
significantly reduced in the sorafenib and siRNA-ATF2 group when
compared with the control (Fig.
6A-C). It is of note that the sorafenib and siRNA-ATF2 combined
treatment significantly reduced ATF2 expression levels when
compared with the vector+sorafenib group (P<0.05). The mRNA
expression of ATF2 in each group was 1.0 in control, 1.04±0.1 in
vector, 0.59±0.03 in sorafenib, 0.59±0.01 in sorafenib+vector,
0.34±0.02 in siRNA-ATF2, and 0.23±0.02 in sorafenib+siRNA-ATF2. The
protein expression of ATF2 in each group was 0.90±0.03 in control,
0.90±0.03 in vector, 0.52±0.02 in sorafenib, 0.53±0.02 in
sorafenib+vector, 0.31±0.01 in siRNA-ATF2 and 0.24±0.03 in
sorafenib+siRNA-ATF2. The phosphorylation of ATF2 was also reduced
following sorafenib treatment and/or ATF2 silencing
(F5,30=239; P<0.05). The p-ATF2/GAPDH ratio in each
group was 0.95±0.03 in control, 0.96±0.03 in vector, 0.48±0.02 in
sorafenib, 0.54±0.02 in sorafenib+vector, 0.35±0.02 in siRNA-ATF2
and 0.28±0.02 in sorafenib+siRNA-ATF2. The JNK3 mRNA level was not
affected following sorafenib treatment and/or ATF2 silencing
(F5,30=89; P>0.05; Fig.
6A); however, JNK3 phosphorylation levels were significantly
greater when compared with the control (F5,30=319;
P<0.05; Fig. 6E). The JNK3 mRNA
expression was 1.0 in control, 1.0±0.08 in vector, 0.97±0.07 in
sorafenib, 1.0±0.1 in sorafenib+vector, 1.0±0.06 in siRNA-ATF2 and
1.0±0.08 in sorafenib+siRNA-ATF2. The p-JNK3/JNK3 ratio in each
group was 0.26±0.03 in control, 0.30±0.02 in vector, 0.56±0.04 in
sorafenib, 0.56±0.02 in sorafenib+vector, 0.73±0.01 in siRNA-ATF2
and 0.93±0.01 in sorafenib+siRNA-ATF2. Additionally, the present
study also detected TNF-α expression. As presented in Fig. 6A, B and D, TNF-α mRNA and protein
expression levels were increased following sorafenib treatment
and/or ATF2 silencing (mRNA, F5,30=229, P<0.05;
protein, F5,30=165, P<0.05). It is of note that
sorafenib and siRNA-ATF2 combined treatment further enhanced TNF-α
expression compared with the vector+sorafenib group (P<0.05).
The mRNA expression of TNF-α in each group was 1.0 in control,
1.0±0.06 in vector, 1.5±0.03 in sorafenib, 1.5±0.02 in
sorafenib+vector, 1.4±0.01 in siRNA-ATF2, and 1.7±0.02 in
sorafenib+siRNA-ATF2. The TNF-α protein expression in each group
was 0.28±0.02 in control, 0.30±0.01 in vector, 0.48±0.02 in
sorafenib, 0.48±0.02 in sorafenib+vector, 0.47±0.03 in siRNA-ATF2
and 0.89±0.02 in sorafenib+siRNA-ATF2.
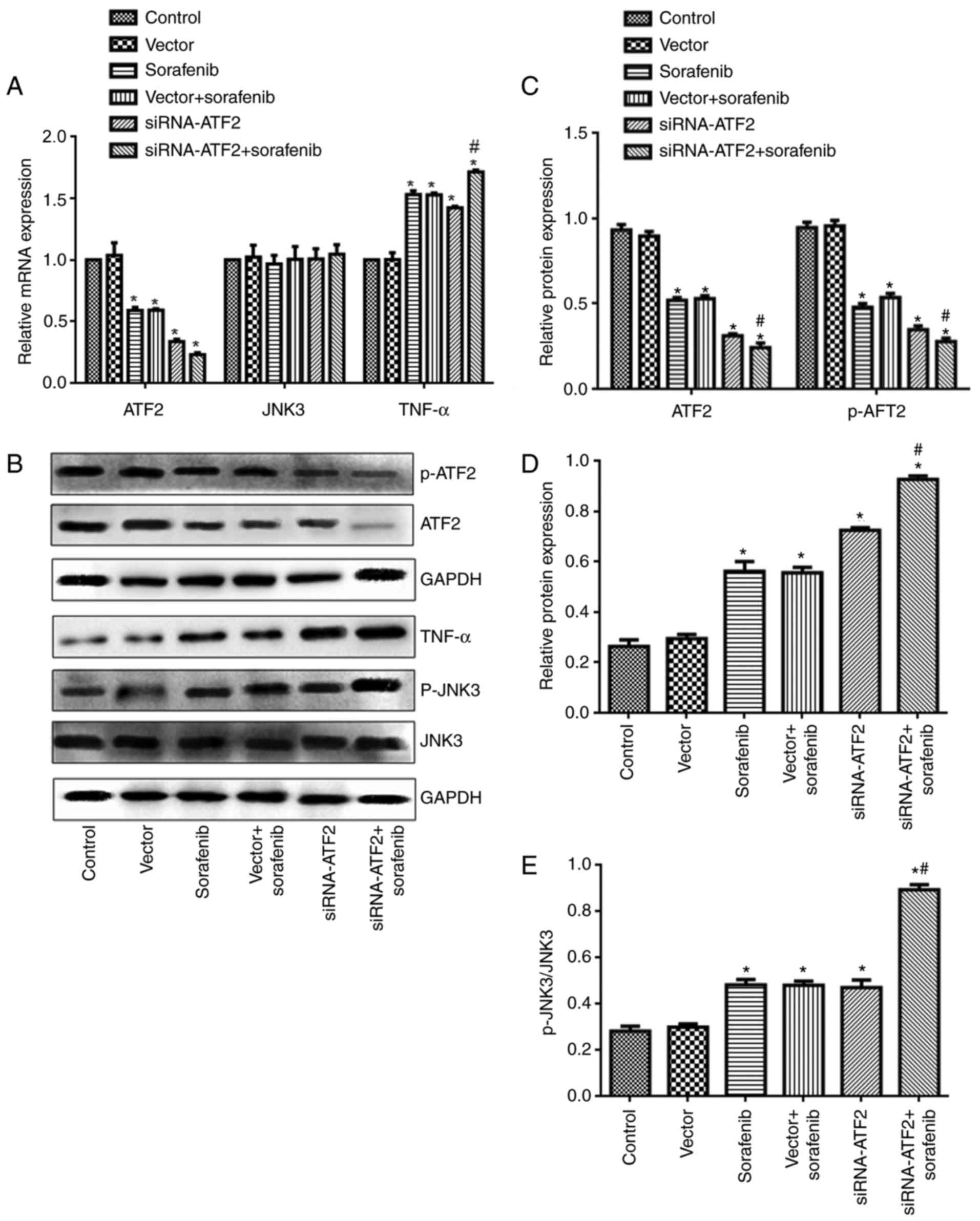 | Figure 6.Sorafenib and/or siRNA-ATF2 reduce
ATF2 and JNK3 expression and enhance TNF-α expression. mRNA
expression of (A) ATF2, JNK3 and TNF-α. (B) Representative western
blot images for ATF2, p-ATF2, TNF-α, JNK3 and p-JNK3.
Quantification of (C) ATF2 and p-ATF2, (D) TNF-α and (E)
p-JNK3/JNK3 protein expression levels. Data are presented as the
mean ± standard error of the mean of 6 repeats. *P<0.05 vs.
control, #P<0.05 vs. vector+sorafenib. p,
phosphorylated; ATF2, activating transcription factor-2; siRNA,
small interfering RNA; TNF-α, tumor necrosis factor-α; JNK3, c-Jun
N-terminal kinase. |
Discussion
Sorafenib is a candidate drug HCC treatment;
however, patients frequently develop drug resistance, thus reducing
the treatment effectivity (3–5). The
development of bio-engineering technology has improved gene
silencing technology over time (20). The use of siRNA fragments directly
in cells may promote specific degradation of target mRNAs. The
present study observed the effects of sorafenib combined with RNA
silencing technology on the biological characters of HCCs. The
present study demonstrated that sorafenib or ATF2 silencing alone
had an anticancer effect in HCC. It is of note that the combined
treatment of sorafenib and ATF2 silencing was able to additionally
facilitate the anticancer effects.
In addition to its role in normal tissues, ATF2 has
a dual role in cancer (21). ATF2
expression is required for tumor cell growth in non-small cell lung
cancer and melanoma (15,16). Conversely, ATF2 expression also
limited tumor cell growth in breast and non-metastatic skin cancer
(17,18). The present study used an siRNA
sequence for ATF2 and RT-qPCR to confirm the silencing effect.
Subsequent experiments confirmed that ATF2 silencing had an
anticancer effect in HCC as cell proliferation, migration and
invasion were inhibited. These findings revealed that ATF2 acts as
an oncogene in HCC similar to breast and non-metastatic skin cancer
(17,18).
Sorafenib is a candidate drug for HCC treatment
(3). However, drug resistance
restricted its clinical application. Therefore, sensitizers which
promote the effect of sorafenib have been widely investigated. For
example, amentoflavone enhances sorafenib-induced apoptosis in
sorafenib-resistant SK-Hep1 cell line (20). Aspirin also overcomes sorafenib
resistance in HCC (21).
Genetically, phosphoprotein enriched in diabetes may reduce the
antitumor effect of sorafenib in HCC (22). The present study confirmed that
sorafenib inhibited cell proliferation, migration and invasion and
promoted apoptosis in HCCs. It is of note that ATF2 silencing
promoted anticancer of sorafenib in HCC. These results implicate
that siRNA-ATF2 could promote the anticancer effect of sorafenib on
Huh-7 HCC cells.
TNF-α is a type II membrane protein, which is
secreted primarily by activated monocyte macrophages (23). TNF-α has two forms: Membrane
associated TNF-α and soluble TNF-α. TNF-α exerts its biological
function by binding to its TNF receptor (TNFR). There are two TNFR
types: TNFRl and TNFR2, which are type I membrane proteins. Soluble
TNF-α has an important role in regulating cell survival and death
and the inflammatory response (24). Following binding with TNF-α, TNF,
receptor-associated, death, domain is recruited to bind to the
intracellular portion of the TNF-α receptor. Subsequently, the
remaining 2 activated receptor-associated proteins, TRAF2 and
Fas-associated death domain, which activates multiple signaling
pathways, including JNK, nuclear factor-κB and mitogen-activated
protein kinase (MAPK) to promote the apoptotic pathway (25). The present study also detected
TNF-α expression levels in HCC. Following siRNA-ATF2 and/or
sorafenib treatment, TNF-α mRNA and protein expression was
increased. These findings suggest that sorafenib regulated TNF-α
expression via ATF2.
JNK is a member of the MAPK superfamily. JNK may be
activated by various extracellular stimuli at the Tyrl8 and Thrl83
sites. Following phosphorylation, JNK participated in the
initiation of apoptosis, which has a crucial role in various
diseases, such as autoimmune hepatitis (26,27).
JNK-mediated apoptosis primarily involves two pathways (28). One pathway acts through promoting
apoptosis-associated gene transcription activity, by regulating
apoptosis-associated proteins and mediating cell apoptosis,
including death receptors, B cell leukemia/lymphoma 2 family
proteins and tumor suppressor genes. The other pathway is directly
through promoting Bim protein phosphorylation in mitochondria,
activating caspase-mediated apoptosis (29). The present study used siRNA-ATF2
and sorafenib in Huh-7 cells. However, total JNK expression was not
significantly different following siRNA-ATF2 and/or sorafenib
treatment, whereas the p-JNK expression was significantly
increased. These findings suggest that siRNA-ATF2 and sorafenib may
induce the HCC apoptosis by activating the JNK signaling pathway.
Combined with the upregulation of TNF-α expression level,
siRNA-ATF2 and sorafenib exert anticancer activity in HCC via the
TNF-α/JNK3 pathway (30).
In conclusion, the current findings revealed that
siRNA-ATF2 or sorafenib treatment not only reduces cell
proliferation, migration and invasion, but also facilitated
apoptosis of HCC. Additionally, the combined treatment of
siRNA-ATF2 and sorafenib increased the anticancer effect. The
possible mechanism may be associated with the activation of the
TNF-α/JNK signaling pathway.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LifL, LC, LaiL and ZT performed the experiments and
analyzed the data. LifL and XM designed the study and wrote the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bai DS, Zhang C, Chen P, Jin SJ and Jiang
GQ: The prognostic correlation of AFP level at diagnosis with
pathological grade, progression, and survival of patients with
hepatocellular carcinoma. Sci Rep. 7:128702017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thein HH, Qiao Y, Zaheen A, Jembere N,
Sapisochin G, Chan KKW, Yoshida EM and Earle CC: Cost-effectiveness
analysis of treatment with non-curative or palliative intent for
hepatocellular carcinoma in the real-world setting. PLoS One.
12:e01851982017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cainap C, Qin S, Huang WT, Chung IJ, Pan
H, Cheng Y, Kudo M, Kang YK, Chen PJ, Toh HC, et al: Linifanib
versus Sorafenib in patients with advanced hepatocellular
carcinoma: Results of a randomized phase III trial. J Clin Oncol.
33:172–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cervello M, Bachvarov D, Lampiasi N,
Cusimano A, Azzolina A, McCubrey JA and Montalto G: Molecular
mechanisms of sorafenib action in liver cancer cells. Cell Cycle.
11:2843–2855. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chow AK, Ng L, Lam CS, Wong SK, Wan TM,
Cheng NS, Yau TC, Poon RT and Pang RW: The Enhanced metastatic
potential of hepatocellular carcinoma (HCC) cells with sorafenib
resistance. PLoS One. 8:e786752013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fouladi F, Jehn LB, Metzelder SK, Hub F,
Henkenius K, Burchert A, Brendel C, Stiewe T and Neubauer A:
Sorafenib induces paradoxical phosphorylation of the extracellular
signal-regulated kinase pathway in acute myeloid leukemia cells
lacking FLT3-ITD mutation. Leuk Lymphoma. 56:2690–2698. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Endo M, Su L and Nielsen TO: Activating
transcription factor 2 in mesenchymal tumors. Hum Pathol.
45:276–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goetz J, Chatton B, Mattei MG and Kedinger
C: Structure and expression of the ATFa gene. J Biol Chem.
271:29589–29598. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lo Iacono M, Monica V, Vavala T, Gisabella
M, Saviozzi S, Bracco E, Novello S, Papotti M and Scagliotti GV:
ATF2 contributes to cisplatin resistance in non-small cell lung
cancer and celastrol induces cisplatin resensitization through
inhibition of JNK/ATF2 pathway. Int J Cancer. 136:2598–2609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shatanawi A, Lemtalsi T, Yao L, Patel C,
Caldwell RB and Caldwell RW: Angiotensin II limits NO production by
upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur J
Pharmacol. 746:106–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sreekanth GP, Chuncharunee A,
Sirimontaporn A, Panaampon J, Noisakran S, Yenchitsomanus PT and
Limjindaporn T: SB203580 modulates p38 MAPK signaling and dengue
virus-induced liver injury by reducing MAPKAPK2, HSP27, and ATF2
phosphorylation. PLoS One. 11:e01494862016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan Z, Duan X, Cai H, Wang L, Li M, Qu J,
Li W, Wang Y and Wang J: Curcumin inhibits the invasion of lung
cancer cells by modulating the PKCα/Nox-2/ROS/ATF-2/MMP-9 signaling
pathway. Oncol Rep. 34:691–698. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song S, Fajol A, Tu X, Ren B and Shi S:
miR-204 suppresses the development and progression of human
glioblastoma by targeting ATF2. Oncotarget. 7:70058–70065. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HS, Choi ES, Shin JA, Jang YK and Park
SD: Regulation of Swi6/HP1-dependent heterochromatin assembly by
cooperation of components of the mitogen-activated protein kinase
pathway and a histone deacetylase Clr6. J Biol Chem.
279:42850–42859. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shah M, Bhoumik A, Goel V, Dewing A,
Breitwieser W, Kluger H, Krajewski S, Krajewska M, Dehart J, Lau E,
et al: A role for ATF2 in regulating MITF and melanoma development.
PLoS Genet. 6:e10012582010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
You Z, Zhou Y, Guo Y, Chen W, Chen S and
Wang X: Activating transcription factor 2 expression mediates cell
proliferation and is associated with poor prognosis in human
non-small cell lung carcinoma. Oncol Lett. 11:760–766. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maekawa T, Shinagawa T, Sano Y, Sakuma T,
Nomura S, Nagasaki K, Miki Y, Saito-Ohara F, Inazawa J and Kohno T:
Reduced levels of ATF-2 predispose mice to mammary tumors. Mol Cell
Biol. 27:1730–1744. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bhoumik A, Fichtman B, Derossi C,
Breitwieser W, Kluger HM, Davis S, Subtil A, Meltzer P, Krajewski
S, Jones N and Ronai Z: Suppressor role of activating transcription
factor 2 (ATF2) in skin cancer. Proc Natl Acad Sci USA.
105:1674–1679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen WL, Hsieh CL, Chen JH, Huang CS, Chen
WT, Kuo YC, Chen CY and Hsu FT: Amentoflavone enhances
sorafenib-induced apoptosis through extrinsic and intrinsic
pathways in sorafenib-resistant hepatocellular carcinoma SK-Hep1
cells in vitro. Oncol Lett. 14:3229–3234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li S, Dai W, Mo W, Li J, Feng J, Wu L, Liu
T, Yu Q, Xu S, Wang W, et al: By inhibiting PFKFB3, aspirin
overcomes sorafenib resistance in hepatocellular carcinoma. Int J
Cancer. 141:2571–2584. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quintavalle C, Hindupur SK, Quagliata L,
Pallante P, Nigro C, Condorelli G, Andersen JB, Tagscherer KE, Roth
W, Beguinot F, et al: Phosphoprotein enriched in diabetes
(PED/PEA15) promotes migration in hepatocellular carcinoma and
confers resistance to sorafenib. Cell Death Dis. 8:e31382017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shivashankar R and Pardi DS: Use of
anti-tumor necrosis factors and anti-integrins in the treatment of
crohn's disease. Gastroenterol Clin North Am. 46:589–601. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sedger LM and McDermott MF: TNF and
TNF-receptors: From mediators of cell death and inflammation to
therapeutic giants-past, present and future. Cytokine Growth Factor
Rev. 25:453–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X and Lin Y: Tumor necrosis factor
and cancer, buddies or foes? Acta Pharmacol Sin. 29:1275–1288.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Xia Y, Liu T, Wang J, Dai W, Wang F,
Zheng Y, Chen K, Li S, Abudumijiti H, et al: Protective effects of
astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK
pathway-mediated inhibition of autophagy and apoptosis. PLoS One.
10:e01204402015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251, 0000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yi J, Yang X, Zheng L, Yang G, Sun L, Bao
Y, Wu Y, Huang Y, Yu C, Yang SN and Li Y: Photoactivation of
hypericin decreases the viability of RINm5F insulinoma cells
through reduction in JNK/ERK phosphorylation and elevation of
caspase-9/caspase-3 cleavage and Bax-to-Bcl-2 ratio. Biosci Rep.
35:pii: e00195. 352015
|
|
30
|
Deng Y, Ren X, Yang L, Lin Y and Wu X: A
JNK-dependent pathway is required for TNFalpha-induced apoptosis.
Cell. 115:61–70. 2003. View Article : Google Scholar : PubMed/NCBI
|