Introduction
Neuroblastoma, the most common extracranial solid
tumor, is the third most common cause of cancer-associated
mortality in childhood, causing 7% of all cancer cases in this age
group (1). Neuroblastoma develops
from undifferentiated neural crest cells in the embryo and is able
to spread to other parts of the body through the blood and lymph
prior to the manifestation of any apparent symptoms, resulting in a
majority of diagnosed cases involving metastasis (2). Current therapies include surgery,
radiation and chemotherapy, and their use depends on the cancer
stage. Low and intermediate risk neuroblastoma tends to have a
favorable outcome and maybe curable with surgery only, whereas high
risk neuroblastoma is difficult to treat successfully even using
intensive multi-modal therapies (3) which risk severe complications,
including cardiac toxicity, infertility, hearing loss and secondary
cancers resulting from high-dose chemotherapy (4). As a result, survival and prognoses
remain poor for patients with high risk neuroblastoma.
The Wnt/β-catenin signaling pathway, which is
critical during embryonic development, is one of the fundamental
regulatory mechanisms of cellular proliferation, differentiation,
apoptosis, polarity and pluripotency (5,6).
Dysregulation of the pathway is associated with birth defects and
multiple human diseases, including liver and colon cancer, and
certain types of brain cancer (7).
The stability of β-catenin is used to evaluate the activity of
Wnt/β-catenin pathway. β-catenin is modulated by a destruction
complex consisting of glycogen synthase kinase-3, casein kinase 1,
adenomatous polyposis coli and the scaffolding protein axin. In the
absence of Wnt stimulation, β-catenin is phosphorylated and
degraded by the ubiquitin proteasome pathway mediated by the
destruction complex, resulting in the attenuation of Wnt signaling.
Wnt stimulation contributes to the accumulation of cytoplasmic
β-catenin and its translocation into the nucleus, where β-catenin
associates with transcription factor 4/lymphoid enhancer-binding
factor to activate the transcription of its target genes, cyclin D1
and c-Myc, which control the G1 to S phase transition in the cell
cycle (8–10), resulting in abnormal cellular
proliferation. High β-catenin expression levels or activation of
its downstream signaling cascades are associated with tumor grade
and poor prognosis in glioma (11–15).
Blocking Wnt signaling has attracted attention as a therapeutic
strategy for the treatment of cancer (16).
N-Myc, a member of the Myc family, is a
proto-oncogene protein encoded by the v-myc avial myelocytomatosis
viral oncogene neuroblastoma derived homolog (MYCN) gene.
N-Myc is highly expressed in the fetal brain and is important for
normal brain development (17).
MYCN amplification is presented in ~25% of cases and is
hypothesized to be associated with high-risk neuroblastoma and poor
prognosis (18). MYCN
amplification is a genetic marker that is used to stratify grade in
neuroblastoma. Previous studies have demonstrated that N-Myc is
involved in cell growth, apoptosis, metastasis, angiogenesis and
tumorigenesis in neuroblastoma (19). N-Myc primarily functions as a
downstream target of Wnt signaling (20). However, it remains unclear if N-Myc
regulates Wnt signaling to affect cellular biological functions.
N-Myc has previously been reported to directly reduce dickkopf Wnt
signaling pathway inhibitor 1(DKK1) expression, and DKK1 regulates
neuroblastoma cell proliferation (21). Furthermore, DKK1 was demonstrated
to disrupt the Wnt/β-catenin signaling pathway (22,23).
Therefore, the present study hypothesized and confirmed that N-Myc
regulates neuroblastoma cell growth through affecting the
Wnt/β-catenin pathway.
Materials and methods
Cell culture
Human neuroblastoma cell lines CHP134 [isolated from
a patient at The Children's Hospital of Philadelphia (Philadelphia,
PA, USA) and purchased from the European Collection of
Authenticated Cell Cultures (Public Health England, Salisbury, UK)
and BE-2C (provided by The Children's Hospital of Philadelphia)
were maintained in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.), 1%
penicillin/streptomycin (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and 2 mM glutamine. Cells were cultured in a 37°C
incubator with 5% CO2.
RNA interference
MYCN-specific small interfering RNA (siRNA;
siN-Myc) and control siRNA (siControl) were purchased from
Sigma-Aldrich; Merck KGaA. The siN-Myc sequences used were as
previously described (24). The si
Control sequence used was 5′-ACGTGACACGTTCGGAGAATT-3′, and does not
match with any known human cDNA. The synthesized oligonucleotides
were annealed and cloned into AgeI/EcoRI (Fermentas;
Thermo Fisher Scientific, Inc.) double digested pLKO.1-puro
lentivirus plasmids (preserved in our laboratory, The Research
Center for Vascular Biology, College of Medicine, Yangzhou
University, Yangzhou, China). Green fluorescent protein was used as
a reporter gene. Lentiviruses containing pLKO.1-puro vectors were
produced by cotransfection with Δ8.2 and vesicular stomatitis virus
(VSV-G) plasmids (preserved in our laboratory) into HEK293T packing
cells purchased from the Cell Bank of Type Culture Collection of
the Chinese Academy of Sciences (Shanghai, China) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. Cells
were further grown for 48 h after transfection, virus supernatants
were collected and used to transfect CHP134 and BE-2C neuroblastoma
cells by spin inoculation in the presence of 8 µl/ml Polybrene
(Sigma-Aldrich; Merck KGaA) for 72 h. After 48 h of incubation with
2 mg/ml puromycin (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), cells were selected for further analysis. The efficacy of
interference was assessed by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting.
RT-qPCR
Total RNA was extracted from cells and purified
using TRIzol reagent (cat. no. 15596-026; Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
total RNA isolated using TRIzol was free of protein and DNA
contamination. The isolated RNA was then treated with amplification
grade DNase I (cat. no. 18068-015; Invitrogen; Thermo Fisher
Scientific, Inc.). cDNA was obtained by reverse transcription using
RevertAid™ First Strand cDNA synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc.) and was amplified using
TaqMan®Gene Expression assays (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with 6-carboxyfluorescein-labeled
probes according to the manufacturer's protocols. The primers used
for MYCN gene amplification were as follows: Forward
5′-CCCCTGGGTCTGCCCCGTTT-3′, reverse, 5′-GCCGAAGTAGAAGTCATCTT-3′.
The sequence of the TaqMan fluorogenic probe was
5′-CCCACCCTCTCCGGTGTGTCTGTCGGTT-3′. For the β-actin gene, the
primers were as follows: β-actin, forward
5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse
5′-CAGCGGAACCGCTCATTGCCAATGG-3′; fluorogenic probe, forward
5′-ATGCCCTCCCCCATGCCATCCTGCGT-3′. Both genes were amplified with
the following thermocycler protocol: A first step of 120 sec at
95°C, followed by 45 cycles of 30 sec at 95°C, 30 sec at 60°C, and
30 sec at 72°C. Fluorescence detection was performed using the ABI
PRISM 7700 Sequence Detector (PerkinElmer, Inc. Waltham, MA, USA).
N-Myc expression was normalized to β-actin expression and
was calculated using the 2−ΔΔCq formula (25). The relative N-Myc mRNA expression
levels were presented as a percentage of the control.
Western blotting
Cells were washed twice with PBS and were lysed in
lysis buffer [50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% NP40, 150 mM
NaCl, 10 mM NaF, 1 mM Na3VO4] containing a
protease inhibitor cocktail (Roche Diagnostics, Basel,
Switzerland). Following centrifugation at 12,000 × g for 10 min,
the supernatant was collected and quantified using a bicinchoninic
acid quantification kit (Beyotime Institute of Biotechnology,
Haimen, China). Proteins (50 µg) were separated on 10% SDS-PAGE gel
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and transferred to
Immobilon-P membranes (Merck KGaA). The membranes were blocked with
5% non-fat dried milk in tris-buffered saline with 0.1% Tween-20
for 1 h, and incubated with specific primary antibodies overnight
at 4°C. Rabbit polyclonal immunoglobulin G (IgG) specific to N-Myc
(1:500; cat. no. ab24193; Abcam, Cambridge, UK), mouse monoclonal
IgG specific to β-catenin (1:1,000; cat. no. sc7963; Santa Cruz
Biotechnology, Inc.), rabbit polyclonal IgG specific to Cyclin D1
(1:1,000; cat no. sc753; Santa Cruz Biotechnology, Inc.), mouse
monoclonal IgG specific to c-Myc (1:1,000; cat. no. sc40; Santa
Cruz Biotechnology, Inc.), rabbit polyclonal IgG specific to DKK1
(1:1,000; cat. no. 4687S; Cell Signaling Technology, Inc., Danvers,
MA, USA), goat polyclonal IgG specific to N-Myc and STAT interactor
(Nmi; 1:1,000; cat. no. sc9483; Santa Cruz Biotechnology, Inc.),
mouse monoclonal IgG specific to B cell lymphoma 2 (Bcl-2; 1:500;
cat. no. sc7382; Santa Cruz Biotechnology, Inc.), rabbit polyclonal
IgG specific to Bcl-2 associated X apoptosis regulator (Bax; 1:500;
cat. no. sc493; Santa Cruz Biotechnology, Inc.), mouse monoclonal
IgG specific to β-actin (1:1,000; cat. no. sc47778; Santa Cruz
Biotechnology, Inc.), rabbit polyclonal IgG specific to
cleaved-caspase-3 (1:1,000; cat. no. 9661S; Cell Signaling
Technology, Inc.) and rabbit monoclonal IgG specific to
cleaved-poly ADP ribose polymerase (PARP; 1:1,000; cat. no. 5625P;
Cell Signaling Technology, Inc.) were used for detection by an
enhanced chemiluminescence detecting reagent (GE Healthcare Life
Sciences, Chalfont, UK). Horseradish peroxidase-conjugated
secondary antibodies: goat anti-mouse (1:2,000; cat. no. sc-2005;
Santa Cruz Biotechnology, Inc.) and goat anti-rabbit IgG (1:2,000;
cat. no. sc-2004; Santa Cruz Biotechnology, Inc.) were used to
incubation for 1 h at room temperature. The protein blots were
quantified by densitometry using Quantity One software version
4.6.2 (Bio-Rad, Hercules, CA, USA), and the amounts were expressed
relative to the internal reference β-actin.
MTT assay
Cell viability was evaluated by MTT assay. CHP134
and BE-2C cells (1×104 cells/well) were plated in a 96
well plate, and were cultured for 48 h at 37°C. MTT (20 µl, 5
mg/ml; Sigma-Aldrich, Merck KGaA) was added to the medium and
incubated for 4 h, and the medium was then replaced with 150 µl
dimethyl sulfoxide for 10 min at room temperature to dissolve the
cells. Cell viability was measured using an enzyme-linked
immunosorbent assay (ELISA) spectrophotometer (JK-UVS-760CRT;
Shanghai Jingke Scientific Instrument Co., Ltd., Shanghai, China)
at 490 nm.
Apoptosis assay
Apoptosis was assessed by ELISA, using the Cell
Death Detection ELISAPLUS kit (cat. no. 11774425001;
Roche Applied Science, Penzberg, Germany). CHP134 and BE-2C cells
(4×103 cells/well) were plated in a 96-well plate
(Thermo Fisher Scientific, Inc.). Following incubation for 9 h,
samples were collected and analyzed according to the manufacturer's
protocol. Results were expressed as the fold induction relative to
control. In addition, caspase-3 activation was detected to verify
the effect on apoptosis using the active caspase-3 ELISA kit (cat.
no. K106-100; R&D Systems, Inc., Minneapolis, MN, USA). A total
of 1×106 cells/well transfected with siN-Myc and
siControl were grown in 6-well plates (Thermo Fisher Scientific,
Inc.). Following 8 h incubation, the cells were lysed and ELISA was
performed according to the manufacturer's protocol.
Morphological observations of
neuroblastoma cells
Following transfection, 1×105 cells were
cultured in 60 mm culture dishes (Thermo Fisher Scientific, Inc.)
for 48 h. The medium was then replaced with fresh medium. Cell
morphology was observed and photographed using a light vertical
microscope (magnification, ×400) (Olympus Corporation, Tokyo,
Japan).
Statistical analysis
All experiments were repeated ≥3 times. Statistical
analysis was preformed using SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA). Values are expressed as the mean ± standard error of the
mean. One-way analysis of variance was used to assess differences
between groups. Duncan method was employed for pairwise comparison
and followed by Bonferroni correction. P<0.05 was considered to
indicate a statistically significant difference.
Results
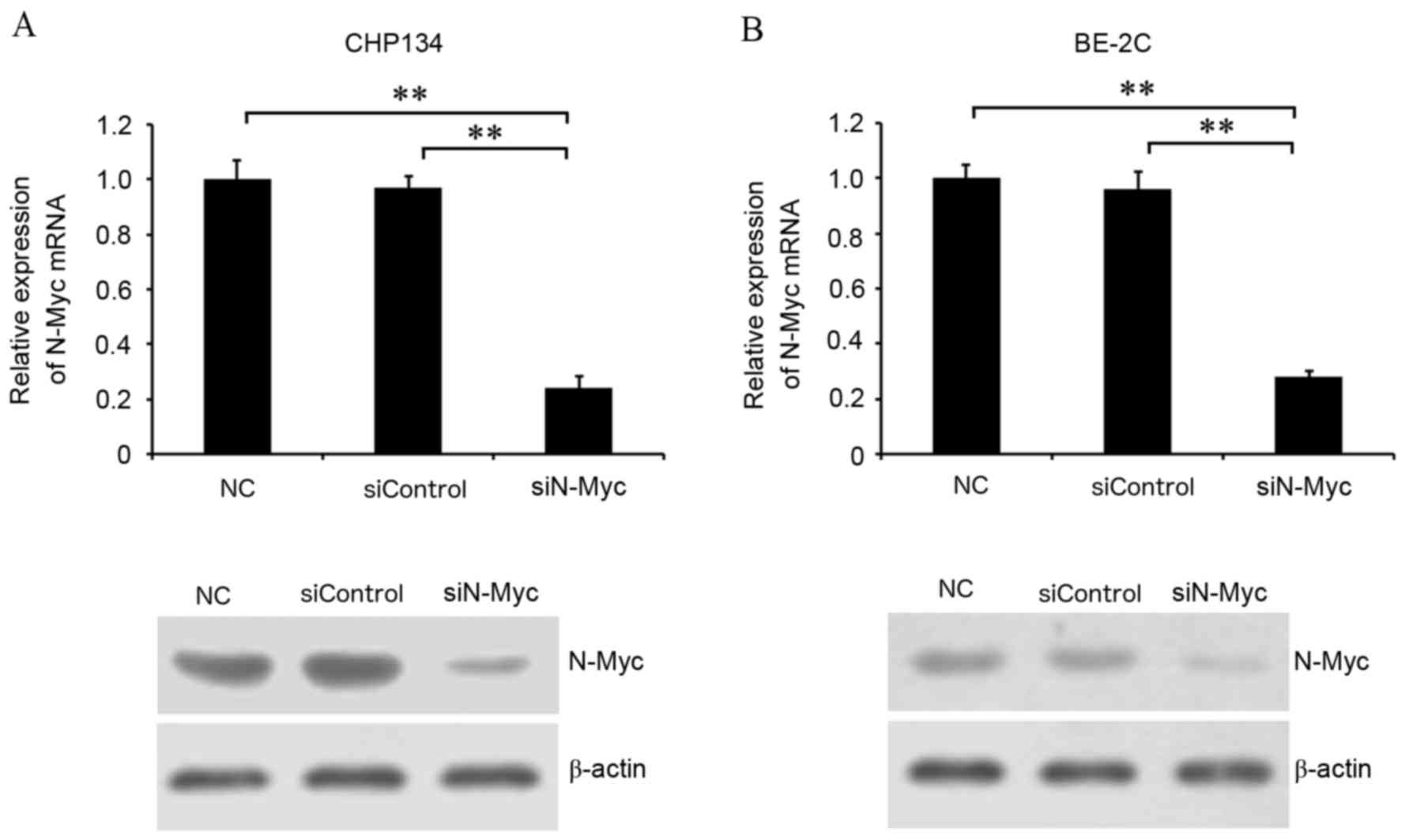
Transfection with siN-Myc silences
N-Myc expression in CHP134 and BE-2C neuroblastoma cells
To determine the function of N-Myc in neuroblastoma
cells, N-Myc was knocked down by transfection with N-Myc-specific
siRNA technology in neuroblastoma cells expressing MYCN.
N-Myc mRNA and protein expression levels were detected by RT-qPCR
and western blotting, respectively. N-Myc mRNA expression levels
were significantly decreased in cells transfected with siN-Myc
compared with negative control and siControl cells (P<0.01;
CHP134 cells, Fig. 1A; BE-2C
cells, Fig. 1B). Residual N-Myc
protein levels in cells transfected with siN-Myc were also visibly
inhibited compared with normal control and siControl cells (CHP134
cells, Fig. 1A; BE-2C cells,
Fig. 1B). Therefore, transfection
with siN-Myc successfully silenced N-Myc expression in CHP134 and
BE-2C cells, enabling the identification of the biological function
of N-Myc in neuroblastoma cells.
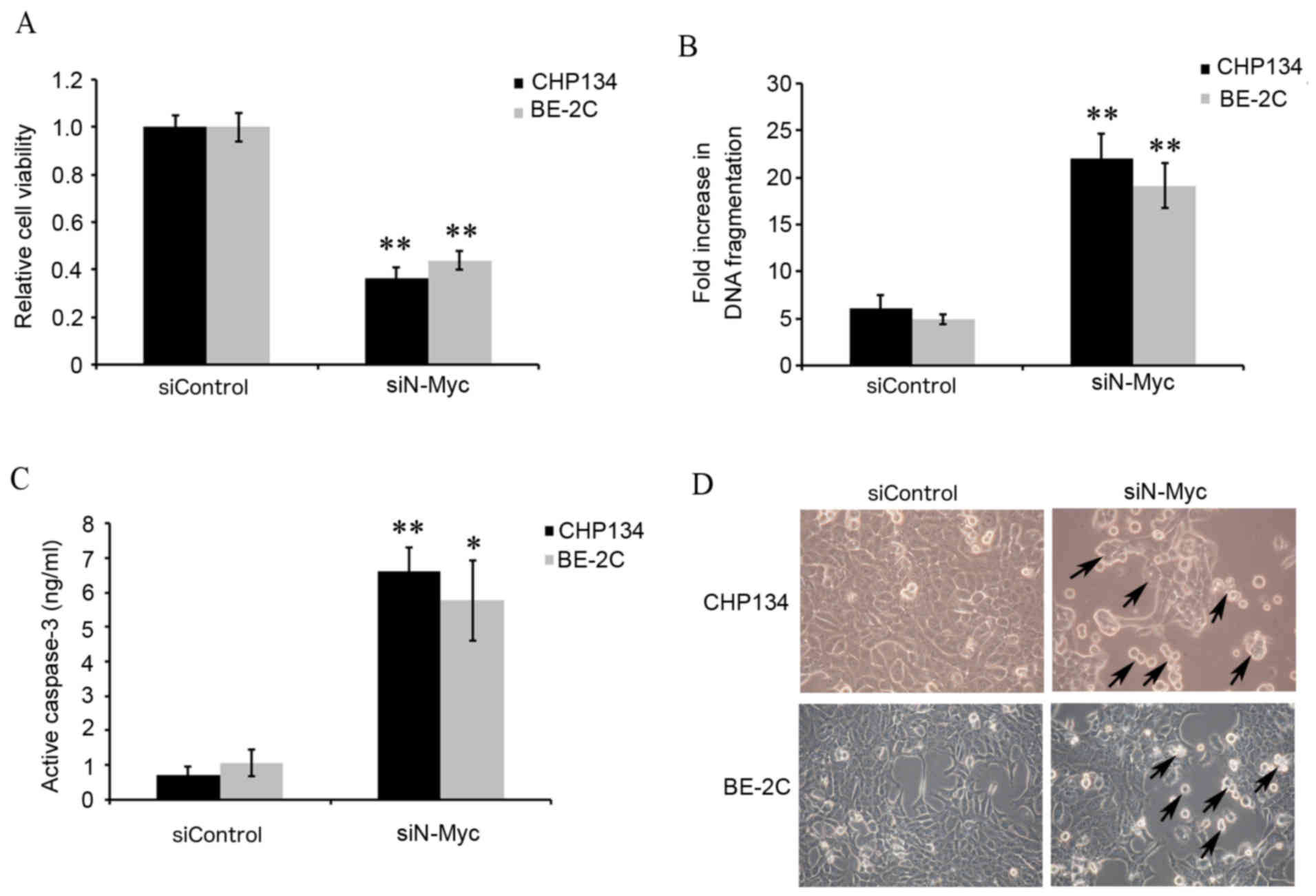
MYCN silencing inhibits cellular
growth and promotes apoptosis
The functional effects of N-Myc knockdown were
investigated by measuring cell viability and apoptosis. The MTT
assay results demonstrated that CHP134 and BE-2C cell viability was
significantly inhibited in CHP134 and BE-2C cells transfected with
siN-Myc compared with siControl cells (P<0.01; Fig. 2A). Apoptosis was analyzed using
ELISA to detect histone release, which is an epigenetic marker of
early apoptosis. CHP134 and BE-2C cells transfected with siN-Myc
demonstrated significantly increased DNA fragmentation compared
with siControl cells (P<0.01; Fig.
2B), indicating that MYCN silencing induced cell
apoptosis. Caspase-3 is activated by intrinsic mitochondrial and
extrinsic death ligand pathways in apoptotic cells (26,27).
Therefore, to confirm the involvement of N-Myc in the regulation of
apoptosis, caspase-3 activity was also detected. Caspase-3 activity
was significantly higher in CHP134 and BE-2C cells transfected with
siN-Myc compared with siControl cells (P<0.01 and P<0.05,
respectively; Fig. 2C), suggesting
that MYCN silencing promoted apoptosis. Cellular growth
capabilities were also compared by visualizing cell morphology.
CHP134 and BE-2C cells transfected with siN-Myc appeared to grow
more slowly and exhibit morphological changes, including shrinkage
of cell volume and membrane-bound apoptotic bodies in comparison
with siControl cells (Fig.
2D).
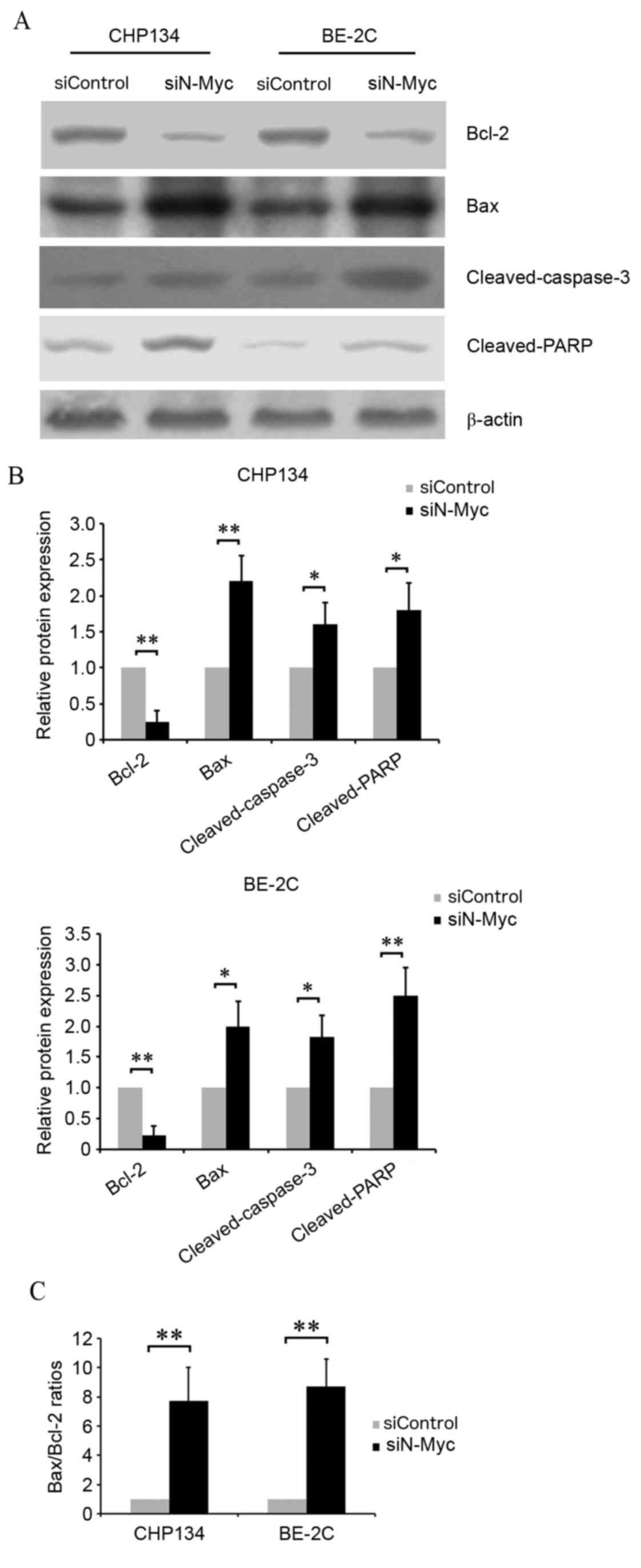
Effects of N-Myc on
apoptosis-associated protein expression levels in neuroblastoma
cells
To further confirm the involvement of N-Myc in
neuroblastoma cell apoptosis, western blotting was performed to
determine the expression levels of the following
apoptosis-associated proteins: Bcl-2, Bax, cleaved-caspase-3 and
cleaved-PARP (Fig. 3A and B).
Protein expression levels of Bcl-2, an anti-apoptosis protein, were
significantly decreased in CHP134 and BE-2C cells transfected with
siN-Myc compared with siControl cells (P<0.01). In addition,
transfection with siN-Myc significantly increased pro-apoptosis
protein expression levels compared with siControl cells, including
Bax (CHP134 cells, P<0.01; BE-2C cells, P<0.05)
cleaved-caspase-3 (P<0.05) and cleaved-PARP (CHP134 cells,
P<0.05, BE-2C cells, P<0.01). The Bax/Bcl-2 ratio was
significantly increased in CHP134 and BE-2C cells transfected with
siN-Myc compared with siControl cells (P<0.01; Fig. 3C), and was a key factor in
determining the occurrence and level of apoptosis. These data
indicated that N-Myc regulated cell apoptosis in CHP134 and BE-2C
cells, potentially through modulation of Bcl-2, Bax,
cleaved-caspase-3 and cleaved-PARP.
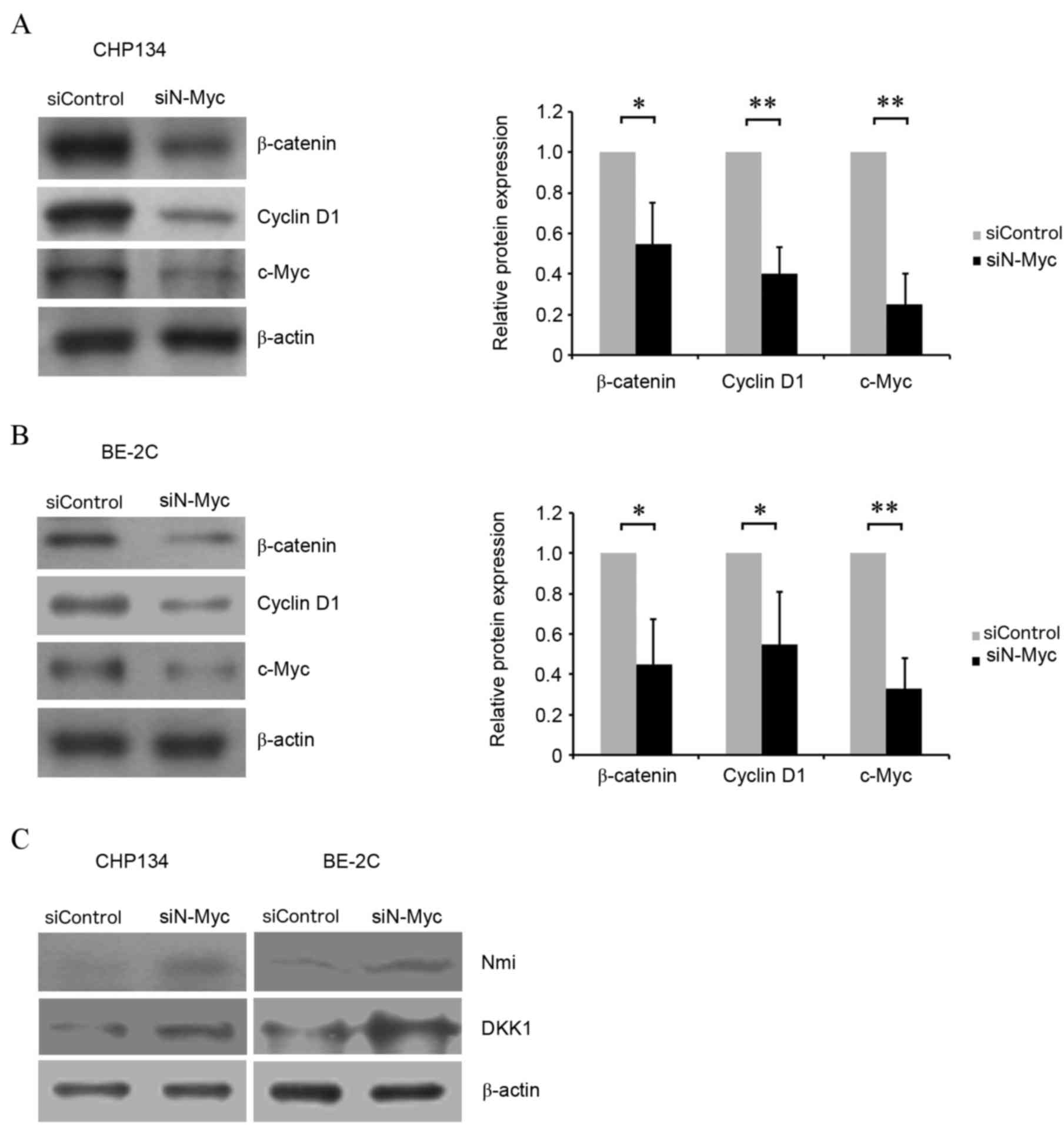
MYCN silencing inhibits the
Wnt/β-catenin signaling pathway
To explore the molecular mechanisms underlying N-Myc
regulation of neuroblastoma cell viability and apoptosis, the
expression levels of key proteins in the Wnt/β-catenin signaling
pathway were evaluated. Western blotting revealed that β-catenin
(P<0.05), cyclin D1 (P<0.01, CHP134 cells; P<0.05, BE-2C
cells) and c-Myc (P<0.01) protein expression levels were
significantly decreased in CHP134 and BE-2C cells transfected with
siN-Myc compared with siControl cells (Fig. 4A and B), suggesting that N-Myc may
regulate cell viability and apoptosis through Wnt signaling. In
order to clarify the mechanisms involved in N-Myc induced
modulation of the Wnt/β-catenin pathway, DKK1, an inhibitor of the
Wnt/β-catenin signaling cascade, and Nmi, an upstream regulator of
DKK1, were detected by western blotting. DKK1 and Nmi protein
levels were increased in cells transfected with siN-Myc compared
with siControl cells (Fig. 4C).
This suggested that MYCN silencing may affect the
Wnt/β-catenin pathway in CHP134 and BE-2C neuroblastoma cells
through Nmi-mediated upregulation of DKK1 expression levels.
Discussion
High risk neuroblastoma is an aggressive cancer that
is often accompanied by metastasis, resulting in poor prognosis
even with coordinated surgical, radiation and chemotherapy
treatment (28). Therefore,
exploring the molecular mechanisms underlying neuroblastoma is
important for the identification of novel therapeutic targets.
The Myc proto-oncogene family is associated with the
establishment of multiple types of human cancer, functioning as
transcription factors that trigger cell immortalization, continued
cell-cycle progression and inhibition of differentiation in various
cell lines. N-Myc is a member of the Myc family that has a basic
helix-loop-helix (bHLH) domain. N-Myc is located in the nucleus and
binds DNA by dimerizing with another bHLH protein (29). N-Myc expression has been previously
reported to be elevated in several types of cancer, including
prostate, colorectal, lung and breast cancer, and is associated
with malignancy and a poor prognosis (30–34).
In addition, amplification of MYCN has previously been
observed in human neuroblastoma and is correlated with the
malignant progression of neuroblastoma (35,36).
Therefore, N-Myc may be a potential therapeutic target for the
treatment of malignant neuroblastoma that overexpresses
MYCN. In the present study, downregulation of MYCN by
siN-Myc inhibited the cellular growth of CHP134 and BE-2C
MYCN-amplified neuroblastoma cell lines, potentially through
the induction of apoptosis.
To explore the molecular mechanisms underlying the
function of N-Myc in neuroblastoma cells, the present study
detected apoptosis-associated protein (Bcl-2, Bax,
cleaved-caspase-3 and cleaved-PARP) expression levels by western
blotting. The anti-apoptosis protein Bcl-2 and the pro-apoptosis
protein Bax are members of the Bcl-2 protein family, and regulate
mitochondrial permeability and apoptosis through the intrinsic
pathway (37). N-Myc inhibition
significantly decreased Bcl-2 protein expression levels and
significantly increased Bax protein expression levels in CHP134 and
BE-2C cells, and significantly elevated the Bax/Bcl-2 ratio, which
is a standard used to measure the occurrence and level of
apoptosis. Cleaved-caspase-3 is an activated form of caspase-3 that
is an important mediator of cell apoptosis (38). All caspases require cleavage
adjacent to aspartates to liberate one large and one small subunit,
which associate into an a2b2 tetramer to form the active enzyme.
Caspase-3 cleaves PARP to create the specific 85 kDa form observed
during apoptosis. In the present study, inhibition of N-Myc
increased cleaved-caspase-3 and cleaved-PARP protein expression
levels, which was consistent with the promotion of apoptosis.
Wnt/β-catenin signaling has previously been
demonstrated to regulate cellular proliferation and apoptosis
during embryonic development, and is involved in advanced disease
stages of several human cancers, including liver, colon and brain
cancer (4,5,39).
It has previously been reported that the Wnt/β-catenin signaling is
involved in neuroblastoma proliferation (40). β-catenin stability may reflect the
activity levels of this pathway. In the present study, MYCN
silencing induced a decrease of β-catenin protein levels in CHP134
and BE-2C cells, suggesting that N-Myc inhibition contributes to
the degradation of β-catenin. The present study also demonstrated
that protein expression levels of cyclin D1 and c-Myc, the
downstream target proteins of β-catenin, were reduced following
MYCN silencing, indicating that the Wnt/β-catenin pathway
was inhibited. β-catenin has been demonstrated to bind T-cell
factor (TCF) to stimulate cellular growth and proliferation in
tumorigenesis by triggering the cell-cycle regulator cyclin D1
(41), and furthermore, c-Myc is
involved in neuroblastoma prognosis (8). In addition, N-Myc has previously been
demonstrated to mediate Wnt signaling functions as the downstream
target. Wnt signaling has been demonstrated to promote neuronal
fate commitment and the proliferation of neural precursor cells via
N-Myc (19). Wnt/β-catenin
signaling is a key upstream regulator of N-Myc and modulates
proximal-distal patterning in the lung, in part, through the
interaction of β-catenin and lymphoid enhancer binding factor/TCF
transcription factors to activate the promoter of MYCN
(42). However, the present study
revealed that N-Myc was an upstream modulator of Wnt signaling.
These results appear inconsistent but they were not contradictory,
which presents the cross-talk among intracellular signal pathways.
Additionally, MYCN silencing was demonstrated to upregulate
DKK1 protein expression levels, revealing the potential mechanism
underlying MYCN silencing-induced Wnt signaling
downregulation. DKK1 has previously been reported to be a secreted
protein that interacts with the Wnt receptor LDL receptor related
protein (LRP)5/6, resulting in the rapid removal of the receptor
through transmembrane protein Kremen 1/2-mediated endocytosis,
followed by blocking of Wnt signaling (43). However, there are several
mechanisms involved in the modulation of N-Myc on the Wnt/β-catenin
pathway. N-Myc downstream regulated gene 1 (NDRG1) has been
observed to modulate Wnt/β-catenin signaling via interaction with
the Wnt receptor LRP6, and pleiotropically suppresses metastasis in
prostate and breast cancers (44).
Nmi has also been previously demonstrated to inhibit Wnt/β-catenin
signaling by upregulating DKK1 expression in MDA-MB-231 breast
cancer and MDA-MB-435 melanoma cells, and retard tumor growth
(45). In the present study, Nmi
protein expression levels were demonstrated to increase in
MYCN-silenced cells. The interaction of Nmi with N-Myc has
also been observed in neuroblastoma cells (46). Therefore, MYCN silencing may
inhibit Wnt signaling through Nmi-mediated DKK1 upregulation in
neuroblastoma cells. However, the effect of NDRG1 or other
molecules on the modulation of N-Myc on Wnt signaling in
neuroblastoma remains to be established.
In conclusion, these data indicated that N-Myc
downregulation suppressed cell viability and promoted apoptosis in
CHP134 and BE-2C neuroblastoma cells, potentially via the
Wnt/β-catenin signaling pathway. However, these results require
further validation in vivo and in vitro to improve
understanding of the mechanism and the potential relevance of N-Myc
for neuroblastoma therapy. The present study lays a theoretical
foundation for novel, targeted neuroblastoma therapies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
Research and Development Program of China (grant no.
2016YFE0126000), China National Natural Science Foundation (grant
no. 81570392), China Postdoctoral Science Foundation funded project
(grant no. 2016M591937), Natural Science Fund for Colleges and
Universities in Jiangsu Province (grant no. 16KJB320017), and High
Level Talent Support Program of Yangzhou University (grant no.
137080077).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JL and SG made substantial contributions to the
conception and design of the present study. YW, WW and YX made
substantial contributions of data analysis and performed the
experiments. YW and JL were major contributors in drafting the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lanzkowsky P: Chapter
22-NeuroblastomaManual of Pediatric Hematology and Oncology. 5th
edition. Academic Press; pp. 671–694. 2011, View Article : Google Scholar
|
|
2
|
John J and Gregory Jr: Neuroblastoma:
Pediatric Cancers: Merck Manual Professional. Retrieved. Apr
17–2008.
|
|
3
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brodeur GM, Hogarty MD, Mosse YP and Maris
JM: NeuroblastomaPizzo PA, Adamson PC and Poplack DG: Principles
and Practice of Pediatric Oncology. 6th edition. Philadelphia, PA:
Wolters Kluwer Health/Lippincott Williams & Wilkins; pp.
886–922. 2011
|
|
5
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu C, Tu Y, Sun X, Jiang J, Jin X, Bo X,
Li Z, Bian A, Wang X, Liu D, et al: Wnt/beta-Catenin pathway in
human glioma: Expression pattern and clinical/prognostic
correlations. Clin Exp Med. 11:105–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu X, Wang L, Zhao S, Ji X, Luo Y and
Ling F: beta-Catenin overexpression in malignant glioma and its
role in proliferation and apoptosis in glioblastma cells. Med
Oncol. 28:608–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pu P, Zhang Z, Kang C, Jiang R, Jia Z,
Wang G and Jiang H: Downregulation of Wnt2 and beta-catenin by
siRNA suppresses malignant glioma cell growth. Cancer Gene Ther.
16:351–561. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sareddy GR, Panigrahi M, Challa S,
Mahadevan A and Babu PP: Activation of Wnt/beta-catenin/Tcf
signaling pathway in human astrocytomas. Neurochem Int. 55:307–317.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Utsuki S, Sato Y, Oka H, Tsuchiya B,
Suzuki S and Fujii K: Relationship between the expression of E-,
N-cadherins and beta-catenin and tumor grade in astrocytomas. J
Neurooncol. 57:187–192. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rossi M, Magnoni L, Miracco C, Mori E,
Tosi P, Pirtoli L, Tini P, Oliveri G, Cosci E and Bakker A:
β-catenin and Gli1 are prognostic markers in glioblastoma. Cancer
Biol Ther. 11:753–761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Knoepfler PS, Cheng PF and Eisenman RN:
N-myc is essential during neurogenesis for the rapid expansion of
progenitor cell populations and the inhibition of neuronal
differentiation. Genes Dev. 16:2699–2712. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mueller S and Matthay KK: Neuroblastoma:
Biology and staging. Curr Oncol Rep. 11:431–438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang M and Weiss WA: Neuroblastoma and
MYCN. Cold Spring Harb Perspect Med. 3:a0144152013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barker N and Clevers H: Mining the Wnt
pathway for cancer therapeutics. Nat Rev Drug Discov. 5:997–1014.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuwahara A, Hirabayashi Y, Knoepfler PS,
Taketo MM, Sakai J, Kodama T and Gotoh Y: Wnt signaling and its
downstream target N-myc regulate basal progenitors in the
developing neocortex. Development. 137:1035–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koppen A, Ait-Aissa R, Hopman S, Koster J,
Haneveld F, Versteeg R and Valentijn LJ: Dickkopf-1 is
down-regulated by MYCN and inhibits neuroblastoma cell
proliferation. Cancer Lett. 256:218–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou XL, Qin XR, Zhang XD and Ye LH:
Downregulation of Dickkopf-1 is responsible for high proliferation
of breast cancer cells via losing control of Wnt/beta-catenin
signaling. Acta Pharmacol Sin. 31:202–210. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mao B, Wu W, Davidson G, Marhold J, Li M,
Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, et al: Kremen
proteins are Dickkopf receptors that regulate Wnt/beta-catenin
signalling. Nature. 417:664–667. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bell E, Premkumar R, Carr J, Lu X, Lovat
PE, Kees UR, Lunec J and Tweddle DA: The role of MYCN in the
failure of MYCN amplified neuroblastoma cell lines to G1 arrest
after DNA damage. Cell Cycle. 5:2639–2647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salvesen GS: Caspases: Opening the boxes
and interpreting the arrows. Cell Death Differ. 9:3–5. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ and
Los M: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maris JM: Recent advances in
neuroblastoma. N Engl J Med. 362:2202–2211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
MYCN v-myc myelocytomatosis viral related
oncogene, neuroblastoma derived [Mus musculus (house mouse)]. Gene
ID: 18109. https://www.ncbi.nlm.nih.gov/gene/18109Updated.
Apr 8–2018.
|
|
30
|
Song Y, Oda Y, Hori M, Kuroiwa K, Ono M,
Hosoi F, Basaki Y, Tokunaga S, Kuwano M, Naito S and Tsuneyoshi M:
N-myc downstream regulated gene-1/Cap43 may play an important role
in malignant progression ofprostate cancer, in its close
association with E-cadherin. Hum Pathol. 41:214–222. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song Y, Lv L, Du J, Yue L and Cao L:
Correlation of N-mycdownstream-regulated gene 1 subcellular
localization and lymph node metastases ofcolorectal neoplasms.
Biochem Biophys Res Commun. 439:241–246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saksela K, Bergh J and Nilsson K:
Amplification of the N-myconcogene in an adenocarcinoma of the
lung. J Cell Biochem. 31:297–304. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nau MM, Carney DN, Battey J, Johnson B,
Little C, Gazdar A and Minna JD: Amplification, expression and
rearrangement of c-myc and N-myconcogenes in human lung cancer.
Curr Top Microbiol Immunol. 113:172–177. 1984.PubMed/NCBI
|
|
34
|
Metge BJ, Mitra A, Chen D, Shevde LA and
Samant RS: N-Mycand STAT Interactor regulates autophagy and
chemosensitivity in breast cancer cells. Sci Rep. 5:119952015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-mycin untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schwab M, Ellison J, Busch M, Rosenau W,
Varmus HE and Bishop JM: Enhanced expression of the human gene
N-mycconsequent to amplification of DNA may contribute to malignant
progression of neuroblastoma. Proc Natl Acad Sci USA. 81:4940–4944.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lopez J and Tait SW: Mitochondrial
apoptosis: Killing cancer using the enemy within. Br J Cancer.
112:957–962. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun H, Gao Y, Lu K, Zhao G, Li X, Li Z and
Chang H: Overexpression of Klotho suppresses liver cancer
progression and induces cell apoptosis by negatively regulating
wnt/β-catenin signaling pathway. World J Surg Oncol. 13:3072015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vieira GC, Chockalingam S, Melegh Z,
Greenhough A, Malik S, Szemes M, Park JH, Kaidi A, Zhou L,
Catchpoole D, et al: LGR5 regulates pro-survival MEK/ERK and
proliferative Wnt/β-catenin signalling in neuroblastoma.
Oncotarget. 6:40053–40067. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Damalas A, Ben-Ze'ev A, Simcha I, Shtutman
M, Leal JF, Zhurinsky J, Geiger B and Oren M: Excess beta-catenin
promotes accumulation of transcriptionally active p53. EMBO J.
18:3054–3063. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shu W, Guttentag S, Wang Z, Andl T,
Ballard P, Lu MM, Piccolo S, Birchmeier W, Whitsett JA, Millar SE
and Morrisey EE: Wnt/beta-catenin signaling acts upstream of N-myc,
BMP4, and FGF signaling to regulate proximal-distal patterning in
the lung. Dev Biol. 283:226–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mao B and Nichrs C: Kremen2 modulates
Dickkopf2 activity during Wnt/LRP6 signaling. Gene. 302:179–183.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu W, Xing F, Iiizumi-Gairani M, Okuda H,
Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, et al:
N-myc downstream regulated gene 1 modulates Wnt-β-catenin
signalling and pleiotropically suppresses metastasis. EMBO Mol Med.
4:93–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fillmore RA, Mitra A, Xi Y, Ju J, Scammell
J, Shevde LA and Samant RS: Nmi (N-Myc interactor) inhibits
Wnt/beta-catenin signaling and retards tumor growth. Int J Cancer.
125:556–564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bannasch D, Weis I and Schwab M: Nmi
protein interacts with regions that differ between MycN and Myc and
is localized in the cytoplasm of neuroblastoma cells in contrast to
nuclear MycN. Oncogene. 18:6810–6817. 1999. View Article : Google Scholar : PubMed/NCBI
|