Introduction
Familial adenomatous polyposis (FAP) is an autosomal
dominant inherited disorder with an incidence of approximately
3–10/100,000 (1). FAP is
characterized by the presence of hundreds to thousands of
adenomatous polyps in the colon and rectum, and is a disease
predisposing individuals to colorectal cancer (CRC) (2). Affected FAP subjects without early
diagnosis and treatment subsequently develop CRC in the late
childhood to the sixties, with a mean age at diagnosis of 40 years
old (3). CRC is the third most
common malignant disease in both males and females in Asia, and is
the second leading cause of cancer death in the past ten years
(4). Patients who suffer from FAP
also have increased risk of extra-colonic manifestations, including
duodenal polyposis, sebaceous cysts, congenital hypertrophy of the
retinal pigment epithelium (CHRPE) and tumors in the upper
gastrointestinal tract, thyroid gland and brain (5,6).
Germline mutations in the tumor suppressor
adenomatous polyposis coli gene (APC) on chromosome 5q22.2 are
responsible for the most cases of FAP. The APC gene comprises of 16
exons (NM_000038.5), including1 upstream non-coding exon and 15
coding exons. The last coding exon encompasses the majority
(>75%) of the total coding region. Most of the mutations causing
FAP are nonsense or frameshift mutations, and can result in
premature stop codons thus produce truncated APC proteins (7). The APC protein, which comprises of
2843 amino acids, plays an important role in the β-catenin nuclear
localization (8). Loss of APC
function results in increased level of β-catenin and activation of
growth-promoting genes via the increased β-catenin/Tcf-4
transcription complexes, subsequently leading to the development of
adenomatous colorectal polyps at a young age (9). Adenomatous polyposis will
consequently progress to CRC if left untreated.
Genetic screening along with other preventative
strategies can conspicuously improve the overall survival of FAP
patients. In the present study, we screened for novel mutations
that might cause FAP in a four-generation Chinese family with FAP
using targeted next-generation sequencing method. A novel mutation
in the exon 10 of APC (c.1317delA) was identified in all of the
affected members, but not in the unaffected members or the 600
normal controls. In addition, this mutation was correlated to
extra-colonic phenotypes featuring by duodenal polyposis and
sebaceous cysts in this pedigree. This novel mutation can hopefully
be utilized as a biomarker for molecular diagnosis and precise
treatment in the foreseeable future.
Materials and methods
Subjects
The family with FAP was recruited from Hospital of
University of Electronic Science and Technology of China and
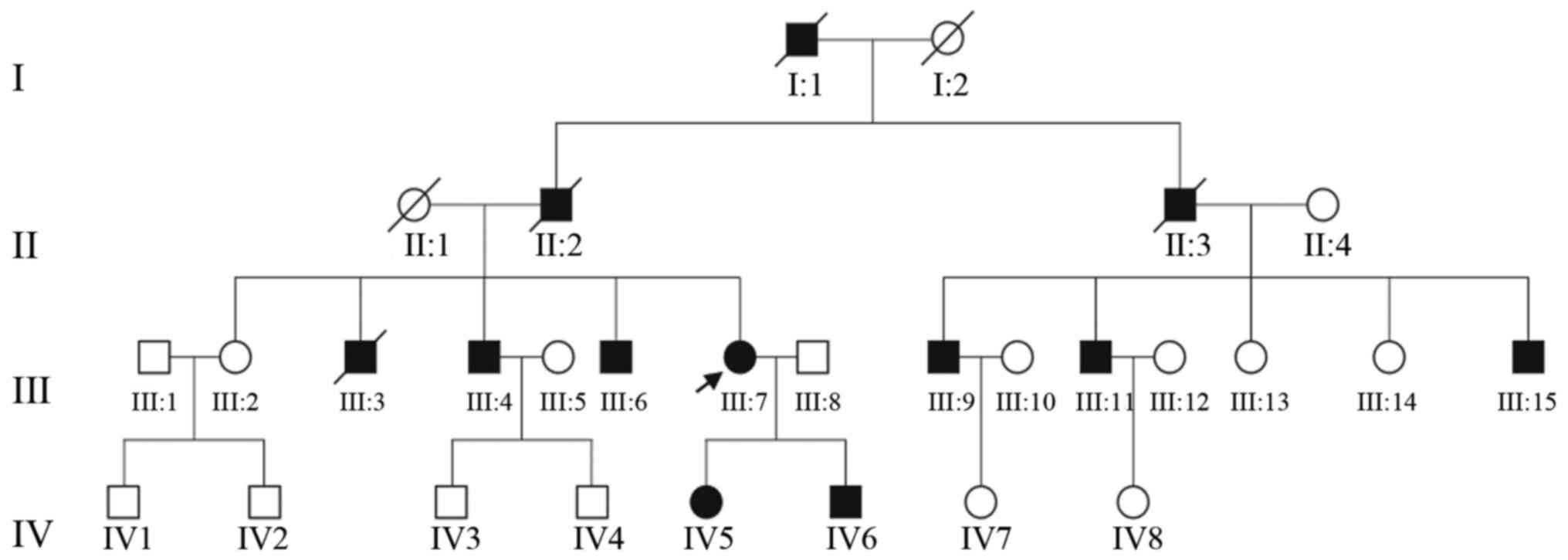
Sichuan Provincial People's Hospital (Fig. 1). This study was conducted in
accordance to the tenets of the Declaration of Helsinki and
approved by the Institutional Review Boards of Sichuan Academy of
Medical Sciences and Sichuan Provincial People's Hospital. Written
informed consents were obtained from the family prior to the study.
600 unrelated normal control subjects were recruited from
participants who attended to the annual physical examination in
Sichuan Provincial People's Hospital. They underwent comprehensive
examinations and were free from any related diseases.
Clinical diagnosis
Twelve of the family members were diagnosed with
FAP. Non-consanguineous marriages were found in the four-generation
family and their clinical information is summarized in Table I. All the available members in this
family were enrolled in our study and underwent complete clinical
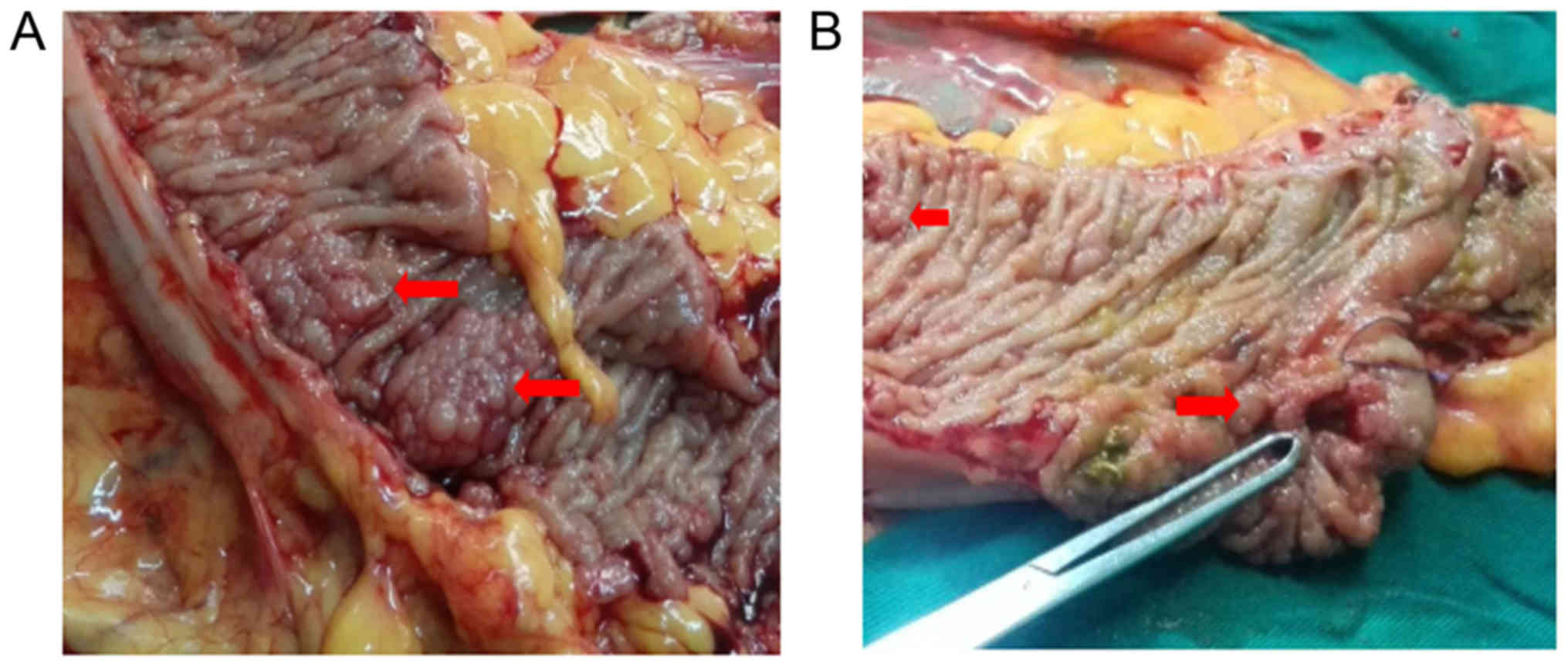
examination. Patient III: 7, diagnosed and treated in Sichuan
Provincial People's Hospital, was the proband of this family
(Fig. 1). FAP was confirmed in the
family by endoscopic screening. The diagnostic criteria were as
follows: (1) more than 100
colorectal polyps in total, and (2) at least 20 synchronous adenomatous
polyps (Fig. 2). All patients'
clinical information, family history, and the results of
colonoscopic, laboratory, and pathologic examinations were
collected. We obtained 5–10 ml of peripheral blood from as many
families members as possible, with full informed consent, and
reviewed pathologic slides whenever available.
 | Table I.Clinical information of the
participants in the family. |
Table I.
Clinical information of the
participants in the family.
| Family ID | Gender | Age (years) | Diagnosis | Age at diagnosis | Mutation | Polyp count | CRC (age of
onset) | Extra-colonic
features |
| I:1a | M | Deceased | Patient | Unspecified | Unspecified | Unspecified | Unspecified | Unspecified |
| I:2a | F | Deceased | Normal | / | / | / | / | / |
| II:1a | F | Deceased | Normal | / | / | / | / | / |
| II:2a | M | Deceased | Patient | Unspecified | Unspecified | Unspecified | Unspecified | Unspecified |
| II:3b | M | Deceased | Patient | Unspecified | Unspecified | Unspecified | Unspecified | Unspecified |
| II:4 | F | 75 | Normal | / | / | / | / | / |
| III:1 | M | 64 | Normal | / | / | / | / | / |
| III:2 | F | 62 | Normal | / | / | / | / | / |
| III:3b | M | Deceased | Patient | 34 | c.1317delA | >100 | Unspecified | None |
| III:4 | M | 47 | Patient | 37 | c.1317delA | >100 | None | None |
| III:5 | F | 46 | Normal | / | / | / | / | / |
| III:6 | M | 45 | Patient | 38 | c.1317delA | >100 | None | None |
| III:7 | F | 43 | Patient | 36 | c.1317delA | >100 | 42 | None |
| III:8 | M | 45 | Normal | / | / | / | / | / |
| III:9 | M | 53 | Patient | 44 | c.1317delA | >100 | None | None |
| III:10 | F | 50 | Normal | / | / | / | / | / |
| III:11 | M | 50 | Patient | 49 | c.1317delA | >100 | None | None |
| III:12 | F | 48 | Normal | / | / | / | / | / |
| III:13 | F | 48 | Normal | / | / | / | / | / |
| III:14 | F | 47 | Normal | / | / | / | / | / |
| III:15 | M | 43 | Patient | 40 | c.1317delA | >100 | 43 | None |
| IV:1 | M | 39 | Normal | / | / | / | / | / |
| IV:2 | M | 37 | Normal | / | / | / | / | / |
| IV:3 | M | 24 | Normal | / | / | / | / | / |
| IV:4 | M | 12 | Normal | / | / | / | / | / |
| IV:5 | F | 24 | Patient | 21 | c.1317delA | >100 | None | None |
| IV:6 | M | 19 | Patient | 16 | c.1317delA | >100 | None | None |
| IV:7 | F | 20 | Normal | / | / | / | / | / |
| IV:8 | F | 22 | Normal | / | / | / | / | / |
DNA extraction
All genomic DNA was extracted from peripheral blood
using a blood DNA extraction kit (QIAamp DNA Blood Midi kit; Qiagen
GmbH, Hilden, Germany) according to the manufacturer's protocol.
DNA samples were stored at −20°C until used. DNA integrity was
evaluated by 1% agarose gel electrophoresis and NanoDrop 2000
(Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Targeted next-generation sequencing
and variant detection
In accordance with the literatures searched within
online databases, a total of 200 candidate genes associated with
colorectal cancer and ID/DD were selected as the genes of interest.
We used a custom-designed gene panel, synthesized using the Agilent
Sure-Select Target Enrichment technique (Zhongguancun Huakang Gene
Institute, China), to capture the coding regions from the 200
genes, including their exons and exon-intron boundaries (1.285M bp
in total). The following targeted next-generation sequencing (NGS)
was performed on an Illumina GAIIx platform (Illumina, Inc., San
Diego, CA, USA) using paired-end sequencing of 110 bp for two
patients (III:7 and III:9). Bioinformatics analysis of the raw data
included the following steps: i) image analysis using RTA software
version 1.9 (real-time analysis, Illumina); ii) base calling using
CASAVA software v1.8.2 (Illumina); iii) filtered out duplicate and
low base quality score reads using the Genome Analysis Tool kit
(GATK; Broad Institute) version 4; iv) aligned clean paired-end
reads to the human reference genome build hg19 using BWA software
(Pittsburgh Supercomputing Center, Pittsburgh, PA, U.S.A.) and v)
identified insertion-deletions (indels) and single-nucleotide
polymorphisms (SNPs) using the GATK and annotated using ANNOVAR
(version 2017, July 16). The sequencing depth was more than 5X
(range of 5X-185X; average of 136X), and the mean coverage was
98.56%. The detected variants were annotated and filtered based on
public and in-house databases: i) Variants within intergenic,
intronic, and UTR regions and synonymous mutations were excluded
from downstream analysis and ii) variants in dbSNP138 (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000
Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), YH Database
(http://yh.genomics.org.cn/), and HapMap
Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap) were excluded.
Heterozygous variations of genes with autosomal dominant heredity
were regarded as likely causative variations. We performed
validation and parental origin analyses for these variations by
conventional Sanger sequencing, and confirmed causative mutations
according to parental origin of the variations and clinical
features of the patients.
Mutation validation
The heterozygous APC (NM_000038.5) mutation
identified by targeted NGS was further confirmed in all the family
members and 600 normal controls by direct sequencing. Primers
flanking the mutation were designed based on genomic sequences of
Human Genome database and synthesized by Invitrogen; Thermo Fisher
Scientific, Inc.: APC-F: GGGTTATATTAGTGATCCCTGCA; APC-R:
ACACAGAGGAAGCAGCTGAT. Amplified PCR products were purified with
spin columns (QIAquick; Qiagen, Inc., Valencia, CA, USA) and
sequenced directly (BigDye Terminators Sequencing kit; Applied
Biosystems; Thermo Fisher Scientific, Inc.) in both directions with
an automated genetic analysis system (3730; ABI; Thermo Fisher
Scientific, Inc.). Multiple sequence alignment of the human APC
protein was performed along with other APC protein across different
species, to check for the conservation of the residues. The
homology modelling server SWISS-MODEL (www.swissmodel.expasy.org/) was used to predict the
tertiary structure of APC protein.
Brief Literature review
A systematic literature search was conducted using
the PubMed, Embase, Web of Science, and the Chinese Biomedical
Database, in order to identify all published studies on the APC
mutations of Han Chinese FAP subjects from their starting date to
December 31, 2017. Two observers (YL and MP) independently piled up
data from all eligible publications onto paper data collection
forms. Disagreements were resolved by discussion until a consensus
was achieved. Otherwise, two other investigators (BG and ZY) were
consulted to resolve the dispute. The search strategy was based on
the following search term/phase, ‘Familial adenomatous polyposis or
FAP’, ‘adenomatous polyposis coli gene or APC’ and ‘Chinese’. The
eligible articles were considered if they: i) evaluated germline
mutations of APC gene in Chinese FAP families and ii) were original
research articles, not reviews or comments. The articles selected
were limited to studies in humans and in Han Chinese, but without
restriction on sample size or time period. Excluded were abstracts
from conferences, full texts without raw data available for
retrieval, republished data, duplicate studies, association studies
and reviews. A total of 79 Han Chinese families from 20 independent
studies (including the current study) were included (some of the
studies reported more than one family). All the mutations in the
APC gene were coordinated using the same transcript reference
NM_000038.5.
Results
Clinical findings
A four-generation family composed of 29 members from
Sichuan Province of China was recruited in this study. Among them,
I1, I2, II1, II2, and II3 were deceased before this study, their
information were collected from their medical records. III3 was
deceased after he participated in this study and donated his blood
sample. In total, 12 family members were diagnosed with FAP
according to their clinical features, family history, medical
history, colonoscopy, and pathology examinations. The disease
exhibited a pattern of autosomal dominant inheritance, as indicated
by the familial pedigree (Fig. 1).
Detailed clinical information was presented in Table I.
Extracolonic manifestations featuring by duodenal
polyposis and sebaceous cysts were observed in all the available
patients of this family, indicating a correlation with this novel
mutation. The proband (III:7) had obvious duodenal polyposis, and
was diagnosed with CRC at the age of 42 year old; while his cousin
(III:15) had an onset of CRC at 43 year old. Two grape beaded
polyps and multiple small polyps were present in the transverse
colon and descending colon of the proband (Fig. 2). The mean age of diagnosis of
polyposis was 30.5 years old among the patients in this family. The
daughter (IV:5) and son (IV:6) of the proband had an aggressive
phenotype with early onset of the polyposis at younger age of 16
and 21 years old, respectively.
Targeted next-generation sequencing
and mutation validation
To ensure complete sequencing coverage of all coding
regions in APC, the quality and reliability of NGS data were
evaluated based on the percentage of readable bases and the
coverage depth in the targeted region. In the APC gene, the
coverage depth was up to 200×, with 100% of bases being readable in
coding regions. This suggests high capacity for variant
identification in most of the exons. Additionally, the mean depth
was close to the median depth in each exon, indicating a good
randomicity. On average, 295 variations within the 200 genes were
found in the two patients (III:7 and III:9). Under the autosomal
dominant model, 8 novel variations were filtered out. Because
previously unclassified variations and pathogenic mutations
detected by NGS were considered as causative candidate, the
filtered data was narrowed down to a pathogenic heterozygous
variant (c.1317delA in the APC gene). This variant was further
validated using Sanger sequencing on other family members and 600
normal controls. Finally, we confirmed the novel heterozygous
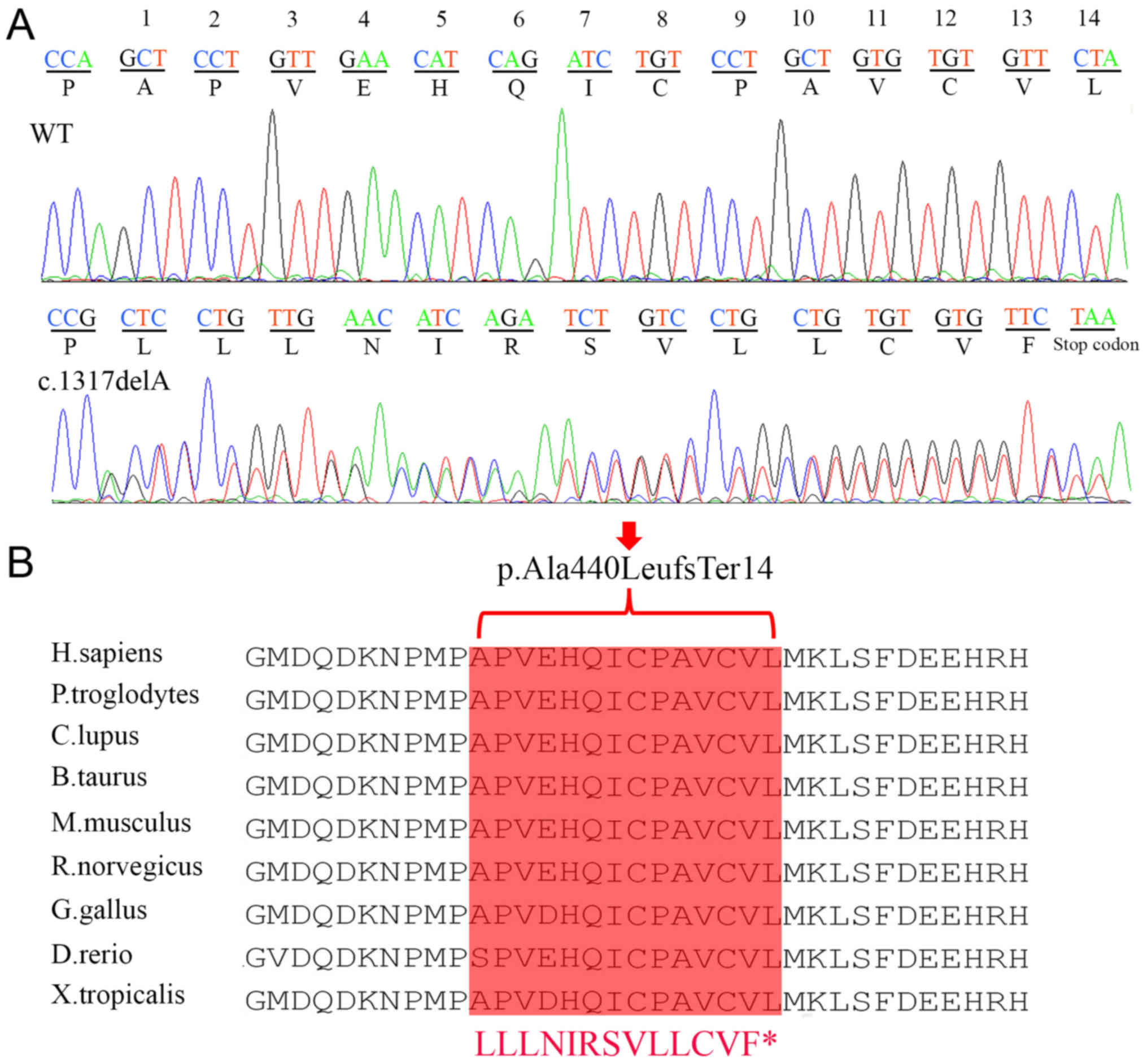
deletion mutation c.1317delA (p.Ala440LeufsTer14; Fig. 3A) in exon 10 of APC in the all of
the affected members, and this mutation was absent in the
unaffected member and the other 600 normal controls. Genotypes and
phenotypes for the patients with APC mutations were described in
Table I. Therefore, this mutation
was co-segregated with the phenotype in this family. Comparative
amino acid sequence alignment of other APC protein across different
species revealed that this novel mutation occurred at highly
conserved positions (Fig. 3B).
This mutation is a 1-bp deletion of A at exon 10 (c.1317delA),
leading to a subsequent reading frame-shift and producing a
premature termination codon located at the 454th amino acid
downstream (p.Ala440LeufsTer14). This premature termination
consequently resulted in a truncation of 2,389 amino acids
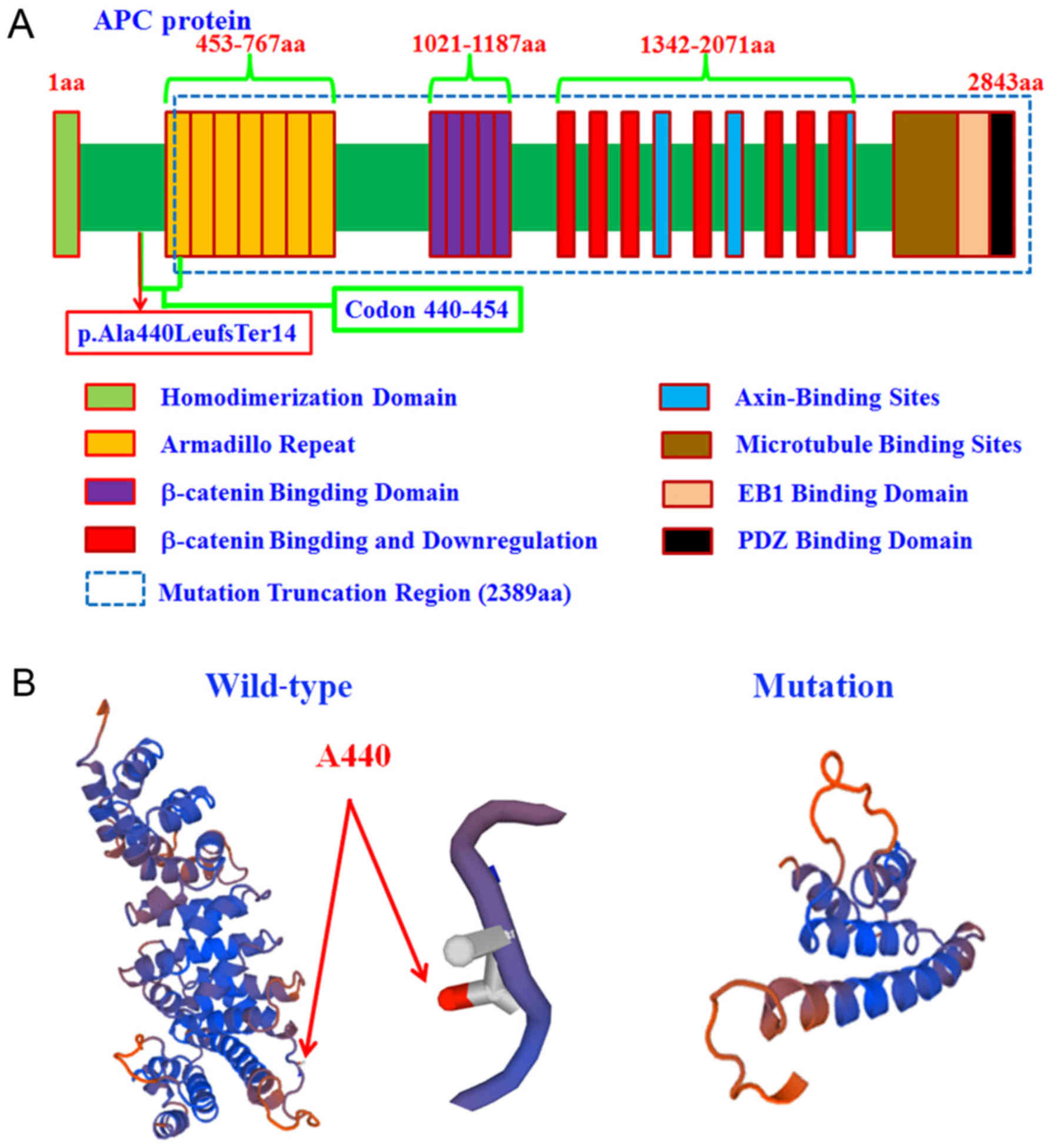
downstream of APC protein (Fig.
4). To further study the pathogenicity of this mutation,
protein tertiary structures of wild type and p.Ala440LeufsTer14
mutation of APC were predicted using the SWISS-MODEL. According to
the tetramer, the mutated APC protein lost C-terminal helices
(2,389 amino acids downstream), which was an important region to
bind with β-catenin, microtubule and other elements (Fig. 4).
Brief literature review of the APC
mutations in Han Chinese
All the mutations that were hitherto reported were
listed in Table II. We have
retrieved 20 individual studies (including the current study). In
total, there were 79 Han Chinese FAP families included, and 54
different APC mutations reported. Among the mutations, 59.3%
(32/54) were frameshift mutations, 24.1% (13/54) were nonsense
mutations, 3.7% (2/54) were missense mutations, 7.4% (4/54) were
large deletions and 5.5% (3/54) were intronic variants.
| Table II.Reported APC mutations causing
familial adenomatous polyposis in the Han Chinese population. |
Table II.
Reported APC mutations causing
familial adenomatous polyposis in the Han Chinese population.
| Author, year | Mutation in
APC gene (Han Chinese) | Codon | Mutation types | Occurrence | (Refs.) |
|---|
| Liu et al,
2005 |
c.366delG(p.Phe123LeufsX2) | 123 | Frameshift | 1 | (20) |
| Liu et al,
2005 |
c.387insT(p.Glu129AspfsX10) | 129 | Frameshift | 1 | (20) |
| Liu et al,
2005 |
c.541insA(p.Gln181ThrfsX12) | 181 | Frameshift | 1 | (20) |
| Liu et al,
2005 |
c.637C>T(p.Arg213X) | 213 | Nonsense | 1 | (20) |
| Pang et al,
2001 | c.646C>T
(p.Arg216X) | 216 | Nonsense | 1 | (21) |
| Cai et al,
2008 |
c.694C>T(p.Arg232X) | 232 | Nonsense | 1 | (22) |
| Cai et al,
2008 |
c.763_764insA(p.His255GlnfsX2) | 255 | Frameshift | 1 | (22) |
| Zhang et al,
2016 | c.794_795insG
(p.Val266SerfsX11) | 266 | Frameshift | 1 | (23) |
| Liu et al,
2005; |
c.847C>T(p.Arg283X) | 283 | Nonsense | 2 | (20,21) |
| Pang et al,
2001 |
| Liu et al,
2005 |
c.1213C>T(p.Arg405X) | 405 | Nonsense | 1 | (20) |
| Pang et al,
2018 | c.1317delA
(p.Ala440LeufsX14) | 440 | Frameshift | Novel | The present
study |
| Sheng et al,
2010 | c.1327G>T
(p.Glu443X) | 443 | Nonsense | 1 | (24) |
| Liu et al,
2005 |
c.1495C>T(p.Arg499X) | 499 | Nonsense | 1 | (20) |
| Jang et al,
2010 | c.1548G>C
(p.Lys516Asn) | 516 | Missense | 1 | (25) |
| Jang et al,
2010 | c.1766T>A
(p.Leu589X) | 589 | Nonsense | 1 | (25) |
| Song et al,
2013 | c.1828_1829insG
(p.Asp610GlyfsX23) | 610 | Frameshift | 1 | (26) |
| Chen et al,
2011 | c.1999 C>T (p.
Gln667X) | 667 | Nonsense | 1 | (27) |
| Jang et al,
2010 | c.2016_2047del
(p.Ser673LeufsX10) | 673 | Frameshift | 1 | (25) |
| Cai et al,
2008 | c.2031_2034delCAGT
(p.Ser678MetfsX39) | 678 | Frameshift | 1 | (22) |
| Zhang et al,
2016 | c.2142_2143insG
(p.His715AlafsX19) | 715 | Frameshift | 1 | (23) |
| Cai et al,
2008 |
c.2240C>G(p.Ser747X) | 747 | Nonsense | 1 | (22) |
| Sheng et al,
2010 | c.2336_2337insT
(p.Leu779PhefX9) | 779 | Frameshift | 1 | (24) |
| Jang et al,
2010 | c.2510C>G
(p.Ser837X) | 837 | Nonsense | 1 | (25) |
| Liu et al,
2005 | c.2547_2550delTAGA
(p.Asp849GlufsX11) | 849 | Frameshift | 1 | (20) |
| Li et al,
2017 | c.2971G>T
(p.Glu991X) | 991 | Nonsense | 1 | (28) |
| Pang et al,
2001 | c.3067_3068insA
(p.Thr1023AsnfsX6) | 1,023 | Frameshift | 1 | (21) |
| Cai et al,
2008 | c.3182_3183delAA
(p.Lys1061ThrfsX3)a | 1,061 | Frameshift | 1 | (22) |
| Liu et al,
2005; |
c.3183_3187delACAAA | 1,061 | Frameshift | 5 | (20–24) |
| Pang et al,
2001; |
(p.Lys1061LysfsX2)a |
|
|
|
|
| Cai et al,
2008; |
|
|
|
|
|
| Sheng et al,
2010 |
|
|
|
|
|
| Wang et al,
2008; |
c3184_3187delCAAA | 1,061 | Frameshift | 3 | (29–31) |
| Chen et al,
2015; |
(p.Gln1061AspfsX59) |
|
|
|
|
| Chen et al,
2015 |
|
|
|
|
|
| Jang et al,
2010 | c.3180_3184del
(p.Gln1062X) | 1,062 | Nonsense | 1 | 25 |
| Cai et al,
2008; |
c.3202_3205delTCAA | 1,068 | Frameshift | 3 | (22,24) |
| Sheng et al,
2010 |
(p.Ser1068GlyfsX57) |
|
|
|
|
| Zhang et al,
2017 | c.3418delC
(p.Pro1140Leufx25) | 1,140 | Frameshift | 1 | (32) |
| Cai et al,
2008 |
c.3576delA(p.Lys1192AsnfsX73) | 1,192 | Frameshift | 1 | (22) |
| Zhu et al,
2012 |
c.3587C>A(p.Ser1196X) | 1,196 | Frameshift | 1 | (33) |
| Liu et al,
2005 | c.3667delC
(p.Ser1223LeufsX42) | 1,223 | Frameshift | 1 | (20) |
| Chen et al,
2015 | c.3921_3924delAAAA
(p.Ile1307MetfsX13)a | 1,307 | Frameshift | 1 | (31) |
| Sheng et al,
2010 |
c.3923_3929delAAGAAAA
(p.Lys1308ArgfsX11)a | 1,308 | Frameshift | 1 | (24) |
| Chen et al,
2015 |
c.3926_3929delAAAAa | 1,309 | Frameshift | 1 | (30) |
| Liao et al,
2014 | c.3922_3925 del
AAAG (p. Glu1309Argfs ×11) | 1,309 | Frameshift | 1 | (34) |
| Wang et al,
2008; |
c.3926_3930delAAAAG | 1,309 | Frameshift | 6 | (29–31,34) |
| Chen et al,
2015; |
(p.Glu1309AspfsX4) |
|
|
|
|
| Chen et al,
2015; |
|
|
|
|
|
| Liao et al,
2014 |
|
|
|
|
|
| Liu et al,
2005; |
c.3927_3931delAAAGA | 1,309 | Frameshift | 10 | (20,22,24,35-37) |
| Cai et al,
2008; |
(p.Glu1309AspfsX4) |
|
|
|
|
| Sheng et al,
2010; |
|
|
|
|
|
| Zhang et al,
2016; |
|
|
|
|
|
| Pan et al,
2014; |
|
|
|
|
|
| Gan et al,
1994 |
|
|
|
|
|
| Chen et al,
2015 | c4127_4126delAT
(p.Tyr1376LysfsX9) | 1,376 | Frameshift | 1 | (31) |
| Sheng et al,
2010 |
c.4179_4180GAdelinsT
(p.Asp1394LeufsX21) | 1,394 | Frameshift | 1 | (24) |
| Cai et al,
2008 | c.4209delC
(p.Ser1404ProfsX11) | 1,404 | Frameshift | 1 | (22) |
| Liu et al,
2005 | c.4391_4394delAGAG
(p.Glu1464ValfsX8) | 1,464 | Frameshift | 1 | (20) |
| Wang et al,
2008; | c.5432C>T (p.
Ser1811Leu)a | 1,811 | Missense | 3 | (29–31) |
| Chen et al,
2015; |
|
|
|
|
|
| Chen et al,
2015 |
|
|
|
|
|
| Liu et al,
2005 | c.5947_5950delGAAA
(p.Glu1983MetfsX60) | 1,983 | Frameshift | 1 | (20) |
| Jiang et al,
2015 | c.1936-2148
del | / | Large deletion | 1 | (3) |
| Sheng et al,
2010 | 10A (Alternative
exon) and Exon11 | / | Large deletion | 1 | (24) |
| Sheng et al,
2010 | Exon15 start | / | Large deletion | 1 | (24) |
| Zhang et al,
2016 | c.423_8532del | / | Large deletion | 1 | (38) |
| Sheng et al,
2010 | c.532-2A>T | / | Intronic | 1 | (24) |
| Sheng et al,
2010 | c.657+1G>A | / | Intronic | 1 | (24) |
| Zhang et al,
2016 | c.1312+1G>A | / | Intronic | 1 | (23) |
There were three main hotspot mutation sites in the
APC gene: i) c.3180-c.3187/codon 1061–1062, which was in the
β-catenin binding domain of APC, accounted for 12.7% (10/79) of the
occurrence; ii) c.3921-c.3931/codon 1307–1309, which was in the
mutation cluster region (MCR), accounted for 25.3% (20/79) of the
occurrence; and iii) c.5432C>T (p.Ser1811Leu), which was in the
β-catenin binding and down-regulation domain, accounted for 3.8%
(3/79) of the occurrence. In total, these hotspot mutations
accounted for 41.8% of the overall occurrence.
Discussion
Familial adenomatous polyposis (FAP) refers to an
autosomal dominant inherited condition, which is characterized by
the early onset of numerous adenomatous colorectal polyps.
Approximately 50% of FAP patients develop adenomas at the age of
15, and it increases to 95% by the age of 35 years old (10). In many FAP patients, extra colonic
manifestations are observed, including gastric and duodenal polyps,
congenital hypertrophy of the retinal pigment epithelium (CHRPE),
sebaceous cysts, and tumors in the upper gastrointestinal tract,
thyroid gland and brain (5,6). In
this pedigree, a novel frameshift mutation c.1317delA
(p.Ala440LeufsTer14) in the APC gene was identified. The proband
has the heterozygous mutation, so theoretically he inherited the
mutant strand from his father (who was also a patient) and the
wild-type strand from his mother (who was healthy). But
unfortunately we are not able to validate the phenotypes of the
proband's father and mother, because both of them passed away.
However, we noticed that the novel mutation was correlated to extra
colonic manifestations featuring by duodenal polyposis and
sebaceous cysts. In addition, FAP patients also have a high
lifetime risk of colorectal cancer (CRC), which is one of the most
common forms of cancer in the world (10,11).
In this family, patient III:7 (proband) and III:15 have been
already diagnosed with CRC at the age of 42 and 43 respectively.
Considering FAP-related CRC takes many years to develop, and the
overall mean age of onset for CRC is 40 years old (3), unfortunately it's possible that other
patients in this family may still have a high risk of CRC as time
goes by. As a result, early diagnosis and routine medical
monitoring are remarkably crucial for the FAP patients.
Genetic diagnosis mainly focuses on the APC gene in
chromosome 5q22.2, as mutations in this gene almost account for all
cases of FAP (12). The APC gene
encodes a tumor suppressor protein, which is responsible for
ubiquitination and degradation of β-catenin and functions as an
antagonist of the Wnt signaling pathway (13,14).
β-catenin is an important factor in cell cycle entry,
proliferation, differentiation, migration, apoptosis, and
progression. Abnormal accumulation of β-catenin and aberrant
activation of Wnt/β-catenin pathway leads to progression of several
major human cancers (15,16). In addition, APC also involves in
other processes including cell migration and adhesion,
transcriptional activation, microtubule stabilization and
apoptosis. Inactivation of APC can lead to defective chromosome
segregation and aberrant mitosis (17–19).
In the present study, we detected a novel frame shift mutation
c.1317delA (p.Ala440LeufsTer14) in the exon 10 of the APC gene in a
large Chinese FAP pedigree. This mutation could result in a
truncation of 2,389 amino acids downstream of APC protein. Protein
tertiary structure analysis revealed that the mutated APC protein
lost C-terminal helices, which was an important region to bind with
β-catenin, microtubule and other elements.
Hitherto, more than 4,600 APC mutations have been
reported according to Leiden Open Variation Database (LOVD;
databases.lovd.nl/shared/genes/APC). However, these mutations are
related to numerous diseases, mainly gastric cancer. And they are
not confined to a certain population. Given that FAP-related CRC
only accounts for approximately 1% of overall CRC (2) and ethnical difference might exist
among different populations, we conducted a brief literature review
and focused on the APC mutation spectrum only in the Han Chinese
FAP population. According to our findings, among the 4,600 reported
mutations, only 54 were related to Han Chinese FAP. Truncating
mutations, including frameshift mutations (59.3%), nonsense
mutations (24.1%) and large deletions (7.4%), account for an
overall percentage of 90.8%. While non-truncating mutations,
including missense mutations (3.7%) and intronic mutations (5.5%),
account for the rest 9.2%. These findings support that direct
sequencing of APC gene could still be the gold standard and the
most accurate method in genetic diagnosis of FAP. In fact, there
are several other alternative methods for gene testing, such as
protein truncation test (PTT), multiplex ligation-dependent probe
amplification (MLPA), southern blot, and real-time quantitative PCR
(RT-PCR). But the major disadvantage of these methods is the
inability to detect the non-truncating mutations, which account for
9.2% in the Han Chinese population. As a result, we still recommend
that FAP testing should be performed using direct sequencing of the
whole APC gene to detect the small-scale mutations along with MLPA
to identify large insertion and deletions. If no mutation is
detected, then testing for large gene rearrangements should be
conducted.
To sum up, we reported that a novel heterozygous
frame shift mutation c.1317delA (p.Ala440LeufsTer14) in the APC
gene was responsible for FAP in a large Chinese pedigree. And we
revealed a genotype-phenotype correlation between this novel
mutation and extra colonic manifestations featuring by duodenal
polyposis and sebaceous cysts. In the meantime, we conducted a
literature review, piling up all the reported APC mutations causing
FAP in the Han Chinese population. In summary, these data enhance
understanding of the mutation spectrum of the APC gene causing FAP
in Han Chinese and inform the development of effective guidelines
in its genetic diagnosis and clinical management.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of China (grant nos. 81371048 and
81670853), The Department of Science and Technology of Sichuan
Province, China (grant no. 2015HH0031) and The Department of
Sichuan Provincial Health (grant no. 130163).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding authors on reasonable
request.
Authors' contributions
BG and ZY designed the study. MP, XiH, XuH, NH, PL,
LL, JF and KW recruited the participants and collected the clinical
information. ZY and JY performed the sequencing analysis. YL and MP
conducted the literature review and analyzed the data. ZY wrote the
initial draft, and BG corrected the English spelling and grammar.
All authors critically revised, reviewed and approved the
publication of this manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Boards of Sichuan Academy of Medical Sciences and Sichuan
Provincial People's Hospital (Sichuan, China). Written informed
consents were obtained from the family prior to the study.
Consent for publication
Written informed consents were obtained from the
family prior to the study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Half E, Bercovich D and Rozen P: Familial
adenomatous polyposis. Orphanet J Rare Dis. 4:222009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galiatsatos P and Foulkes WD: Familial
adenomatous polyposis. Am J Gastroenterol. 101:385–398. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang SS, Li JJ, Li Y, He LJ, Wang QJ,
Weng DS, Pan K, Liu Q, Zhao JJ, Pan QZ, et al: A novel pathogenic
germline mutation in the adenomatous polyposis coli gene in a
Chinese family with familial adenomatous coli. Oncotarget.
6:27267–27274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sung JJ, Lau JY, Goh KL and Leung WK: Asia
Pacific Working Group on Colorectal Cancer: Increasing incidence of
colorectal cancer in Asia: Implications for screening. Lancet
Oncol. 6:871–876. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang YH, Lim SB, Kim MJ, Chung HJ, Yoo HW,
Byeon JS, Myung SJ, Lee W, Chun S and Min WK: Three novel mutations
of the APC gene in Korean patients with familial adenomatous
polyposis. Cancer Genet Cytogenet. 200:34–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burt RW, DiSario JA and Cannon-Albright L:
Genetics of colon cancer: Impact of inheritance on colon cancer
risk. Annu Rev Med. 46:371–379. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stenson PD, Ball EV, Mort M, Phillips AD,
Shiel JA, Thomas NS, Abeysinghe S, Krawczak M and Cooper DN: Human
gene mutation database (HGMD): 2003 update. Hum Mutat. 21:577–581.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fearnhead NS, Britton MP and Bodmer WF:
The ABC of APC. Hum Mol Genet. 10:721–733. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Korinek V, Barker N, Morin PJ, van Wichen
D, de Weger R, Kinzler KW, Vogelstein B and Clevers H: Constitutive
transcriptional activation by a beta-catenin-Tcf complex in APC-/-
colon carcinoma. Science. 275:1784–1787. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leoz ML, Carballal S, Moreira L, Ocaña T
and Balaguer F: The genetic basis of familial adenomatous polyposis
and its implications for clinical practice and risk management.
Appl Clin Genet. 8:95–107. 2015.PubMed/NCBI
|
|
11
|
Ibrahim A, Barnes DR, Dunlop J, Barrowdale
D, Antoniou AC and Berg JN: Attenuated familial adenomatous
polyposis manifests as autosomal dominant late-onset colorectal
cancer. Eur J Hum Genet. 22:1330–1333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aihara H, Kumar N and Thompson CC:
Diagnosis, surveillance, and treatment strategies for familial
adenomatous polyposis: Rationale and update. Eur J Gastroenterol
Hepatol. 26:255–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van de Wetering M, Sancho E, Verweij C, de
Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D,
Haramis AP, et al: The beta-catenin/TCF-4 complex imposes a crypt
progenitor phenotype on colorectal cancer cells. Cell. 111:241–250.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sieber OM, Tomlinson IP and Lamlum H: The
adenomatous polyposis coli (APC) tumour suppressor-genetics,
function and disease. Mol MedToday. 6:462–469. 2000.
|
|
15
|
Moon RT, Kohn AD, De Ferrari GV and Kaykas
A: WNT and beta-catenin signalling: Diseases and therapies. Nat Rev
Genet. 5:691–701. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang Y, Simoneau AR, Liao WX, Yi G, Hope
C, Liu F, Li S, Xie J, Holcombe RF, Jurnak FA, et al: WIF1, a Wnt
pathway inhibitor, regulates SKP2 and c-myc expression leading to
G1 arrest and growth inhibition of human invasive urinary bladder
cancer cells. Mol Cancer Ther. 8:458–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sulekova Z, Reina-Sanchez J and Ballhausen
WG: Multiple APC messenger RNA isoforms encoding exon 15 short open
reading frames are expressed in the context of a novel exon
10A-derived sequence. Int J Cancer. 63:435–441. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zumbrunn J, Kinoshita K, Hyman AA and
Nathke IS: Binding of the adenomatous polyposis coli protein to
microtubules increases microtubule stability and is regulated by
GSK3 beta phosphorylation. Curr Biol. 11:44–49. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adamcikova Z, Wachsmannova L, Hainova K,
Stevurkova V, Holec V, Ciernikova S, Cierna Z, Janega P, Babal P,
Mego M, et al: Study of the APC gene function in the mouse
APC+/APC1638N model. Neuro Endocrinol Lett. 33:26–33.
2012.PubMed/NCBI
|
|
20
|
Liu XR, Shan XN, Friedl W, Uhlhaas S,
Propping P and Wang YP: Analysis of germline mutations in the APC
gene in familial adenomatous polyposis patients. Zhonghua Yi Xue Yi
Chuan Xue Za Zhi. 22:261–264. 2005.PubMed/NCBI
|
|
21
|
Pang CP, Fan DS, Keung JW, Baum L, Tang
NL, Lau JW and Lam DS: Congenital hypertrophy of the retinal
pigment epithelium and APC mutations in Chinese with familial
adenomatous polyposis. Ophthalmologica. 215:408–411. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai SR, Zhang SZ and Zheng S: Detection of
adenomatous polyposis coli gene mutations in 31 familial
adenomatous polyposis families by using denaturing high performance
liquid chromatography. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
25:164–167. 2008.(In Chinese). PubMed/NCBI
|
|
23
|
Zhang S, Qin H, Lv W, Luo S, Wang J, Fu C,
Ma R, Shen Y, Chen S and Wu L: Novel and reported APC germline
mutations in Chinese patients with familial adenomatous polyposis.
Gene. 577:187–192. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sheng JQ, Cui WJ, Fu L, Jin P, Han Y, Li
SJ, Fan RY, Li AQ, Zhang MZ and Li SR: APC gene mutations in
Chinese familial adenomatous polyposis patients. World J
Gastroenterol. 16:1522–1526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jang YH, Lim SB, Kim MJ, Chung HJ, Yoo HW,
Byeon JS, Myung SJ, Lee W, Chun S and Min WK: Three novel mutations
of APC gene in Chinese patients with familial adenomatous
polyposis. Cancer Genet Cytogenet. 200:34–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song G, Yuan Y, Zheng F and Yang N: Novel
insertion mutation p. Asp610GlyfsX23 in APC gene causes familial
adenomatous polyposis in Chinese families. Gene. 516:204–208. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen S, Zhou J, Zhang X, Zhou X, Zhu M,
Zhang Y, Ma G and Li J: Mutation analysis of the APC Gene in a
Chinese FAP Pedigree with unusual Phenotype. ISRN Gastroenterol.
2011:9091212011.PubMed/NCBI
|
|
28
|
Li H, Zhang L, Jiang Q, Shi Z and Tong H:
Identification a nonsense mutation of APC gene in Chinese patients
with familial adenomatous polyposis. Exp Ther Med. 13:1495–1499.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang TT, Chen SQ and Zhang XM: Germline
mutation of adenomatous polyposis coli gene in Chinese patients
with familial adenomatous polyposis. Zhonghua Yi Xue Yi Chuan Xue
Za Zhi. 25:199–202. 2008.PubMed/NCBI
|
|
30
|
Chen Q, Liu S, Feng J, Zhang X, Chen S, Ma
G, Zhu M, Zhang Y and Yu J: Analysis of C.3925_3929 deletional
mutations of APC gene in pedigrees with familial adenomatous
polyposis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 32:524–528.
2015.(In Chinese). PubMed/NCBI
|
|
31
|
Chen QW, Zhang XM, Zhou JN, Zhou X, Ma GJ,
Zhu M, Zhang YY, Yu J, Feng JF and Chen S: Analysis of small
fragment deletions of the APC gene in chinese patients with
familial adenomatous polyposis, a precancerous condition. Asian Pac
J Cancer Prev. 16:4915–4920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Z, Liang S, Wang D, Liang S, Li Y,
Wang B, Jiang T, Zhao G, Zhang X and Banerjee S: A novel pathogenic
single nucleotide germline deletion in APC gene in a four
generation Chinese family with familial adenomatous polyposis. Sci
Rep. 7:123572017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu Z, Huang J, Dong J, et al: Analysis of
APC mutation in five kindreds of familial adenomatous polyposis.
Med J West China. 24:1654–1657. 2012.
|
|
34
|
Liao DX, Li B, Du XM, Yu JH, Chang H, Wu
ZQ, Hao HJ, Wang YX, Han WD, Cheng SJ and Luo CH: Two Chinese
pedigrees for adenomatous polyposis coli: New mutations at codon
1309 and predisposition to phenotypic variations. Fam Cancer.
13:361–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Li Z, Huang X and Ye J: Clinical
and molecular characteristics of a child with familial adenomatous
polyposis. Zhonghua Er Ke Za Zhi. 54:205–208. 2016.PubMed/NCBI
|
|
36
|
Pan H, Gao HL, Wu QY, et al: Diagnosis of
APC gene mutation in a patient with familial adenomatous polyposis.
Mil Med J Southeast China. 16:566–168. 2014.
|
|
37
|
Gan YB, Zheng S and Cai XH: Detection of a
gene mutation in familial adenomatous polyposis families by
PCR-RFLP method. Zhonghua Yi Xue Za Zhi. 74:352–354, 390–391.
1994.(In Chinese). PubMed/NCBI
|
|
38
|
Zhang Z, Liang S, Huang H, Wang D, Zhang
X, Wu J, Chen H, Wang Y, Rong T, Zhou Y and Banerjee S: A novel
pathogenic large germline deletion in adenomatous polyposis coli
gene in a Chinese family with familial adenomatous polyposis.
Oncotarget. 7:50392–50400. 2016.PubMed/NCBI
|