Introduction
Heart failure is thought to be comparable to an
engine that has run out of fuel, due to the key involvement of
mitochondrial dysfunction (1).
Therefore, improving the cellular energy supply may provide a
promising therapeutic strategy for individuals with heart failure.
Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α
(PGC-1α), a major regulator of energy metabolism, is abundantly
expressed in the mitochondria and initiates multiple responses,
including adaptive thermogenesis (2), fatty acid oxidation (FAO) (3) and mitochondrial biogenesis (4). Thus, PGC-1α may be a candidate target
for the treatment of heart disease, as fatty acid metabolism
supplies more adenosine 5′-triphosphate (ATP) than carbohydrate
metabolism (5). Furthermore, as
FAO is decreased when heart failure or cardiac hypertrophy emerges,
strategies promoting the re-utilization of fatty acids by the heart
appear to be a promising therapeutic approach for the treatment of
heart failure (6).
PGC-1α inactivation impairs cardiac output in
isolated working hearts or cardiac responses in transgenic mice
challenged with physiological stress, and ultimately contributes to
increased mortality in response to stress (7–9). By
contrast, PGC-1α overexpression enhances mitochondrial biogenesis
by increasing oxygen consumption and ATP production (3,10).
However, constitutive PGC-1α overexpression leads to abnormal
mitochondrial genesis and cardiomyopathy (11). This outcome is abolished by
decreasing PGC-1α expression to basal levels (11). Although PGC-1α overexpression
results in cardiac function deterioration in a mouse model of
transverse aortic constriction (TAC) (9), Pereira et al (12) demonstrated that the use of a
transgene to maintain PGC-1α expression in TAC mice preserves
angiogenic function rather than contractile and mitochondrial
functions. However, investigation in cardiomyocytes indicated that
the expression of full-length PGC-1α (FL-PGC-1α) is downregulated
in the presence of pro-hypertrophic factors such as high-glucose
and angiotensin (Ang) II, and may be involved in autophagy and
anti-inflammatory processes (13,14).
Despite the beneficial effects reported by certain studies, further
study is required to determine whether increasing PGC-1α expression
under pathological conditions has a favorable impact (15,16).
In addition, various subtypes of PGC-1α have not been fully
investigated; therefore, the present study aimed to explore the
function of N-terminal truncated PGC-1α (NT-PGC-1α) in
cardiomyocytes.
NT-PGC-1α is an alternative splice variant of
full-length PGC-1α that contains the first 270 amino acids of the
full-length protein (17).
NT-PGC-1α has a longer half-life than PGC-1α and is predominantly
located in the cytoplasm (18,19).
NT-PGC-1α is sensitive to oxidative stress, and is downregulated in
the brains of patients with diabetes and Alzheimer's disease
(20). However, the function of
NT-PGC-1α in cardiomyocytes remains unclear. In the present study,
NT-PGC-1α was suggested as a metabolic regulator in neonatal rat
cardiomyocytes (NRCMs). It was demonstrated that NT-PGC-1α
activated fatty acid metabolism-associated downstream molecules,
prevented a decrease in mitochondrial membrane potential and ATP
concentration, reduced reactive oxygen species (ROS) generation,
decreased lipid droplet accumulation and increased oxygen
consumption. This suggested that NT-PGC-1α may be beneficial in
mitochondrial impairment prevention and may be involved in
mitochondrial fatty acid metabolism. Thus, NT-PGC-1α represents a
potential therapeutic target in heart failure.
Materials and methods
Experimental animals
Neonatal Sprague-Dawley rats (1–3 days old) were
obtained from the Laboratory Animal Center of Southern Medical
University (Guangzhou, China) and sacrificed immediately. The
present study was approved by the Southern Medical University
review board and the animal protocols used complied with the Guide
for the Care and Use of Laboratory Animals (21).
Isolation and culture of NRCMs. Neonatal
Sprague-Dawley rats were sacrificed via 2% isoflurane inhalation
and subsequent cervical dislocation. Hearts were removed, dissected
and enzymatically digested with 0.2% pancreatin overnight at 4°C.
Cells were subsequently isolated by magnetic stirring with
collagenase II (1 mg/ml) in a sterile glass vial. Following 90 min
of differential adhesion at 37°C, isolated cells were plated in a
culture dish in the presence of 0.1 mM 5-bromo-2′-deoxyuridine
(BrdU; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) to inhibit
fibroblast proliferation with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) in Dulbecco's modified
Eagle's medium (DMEM) medium (Gibco; Thermo Fisher Scientific,
Inc.). Following 48 h at 37°C, spontaneously contracting NRCMs were
treated with 10 µM phenylephrine (PE; Selleck Chemicals, Shanghai,
China) for 48 h at 37°C, 1 µM Ang II (Abcam, Cambridge, MA, USA)
for 24 h, JC-1 (Beyotime Institute of Biotechnology, Haimen, China)
for 20 min or
4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoic
acid (BODIPY™ FL C12) lipid probe (Thermo Fisher
Scientific, Inc.) for 30 min in DMEM containing penicillin and
streptomycin (100:1) (Gibco; Thermo Fisher Scientific, Inc.)
without FBS. PE and Ang II are pro-hypertrophic factors that were
applied to induce cardiac hypertrophy. JC-1 is a probe used to
detect the mitochondrial membrane potential and FL-C12 is used to
measure the lipid accumulation in cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The protocols used for RT-qPCR were in accordance
with the manufacturer's guidelines. Briefly, total RNA from NRCMs
was extracted using RNAiso Plus (Takara Biotechnology Co., Ltd.,
Dalian, China). Total RNA (1 µg/reaction) was reverse transcribed
to cDNA with reverse transcriptase and random hexamer primers
(PrimeScript™RT reagent kit with gDNA Eraser, Takara Biotechnology
Co., Ltd.). RT was conducted at 37°C for 15 min, 85°C for 5 sec and
stop at 4°C. qPCR was conducted at 95°C 15 sec for 1 cycle, 95°C 5
sec and 60°C for 5 sec under 40 cycles, using synthesized cDNA in a
10 µl reaction volume (SYBR Green PCR kit; Takara Biotechnology
Co., Ltd.) with a LightCycler 480 system (Roche Diagnostics, Basel,
Switzerland). Gene expression levels were normalized to β-actin and
quantified using the 2−ΔΔCq method (22). The primers used are listed in
Table I (Sangon Biotech Co., Ltd.,
Shanghai, China).
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| β-actin |
TGGACAGTGAGGCAAGGATAG |
TACTGCCCTGGCTCCTAGCA |
| Acadm |
GTCGCCCCAGACTACGATAA |
GCCAAGACCACCACAACTCT |
| Acadvl |
TGGACAAAGGAAAGGAACTCA |
ACTCAGACCACTGCCAATCC |
| PDK4 |
ACCGTCGTCTTGGGAAAAG |
CGTTGGAGCAGTGGAGTATG |
| CPT1B |
AAGAACACGAGCCAACAAGC |
TACCATACCCAGTGCCATCA |
| CPT-2 |
CTGTCCACCAGCACTCTGAA |
GCAGCCTATCCAGTCATCGT |
| SOD2 |
CTGGCTTGGCTTCAATAAGG |
CGTGCTCCCACACATCAAT |
| SOD3 |
TCTGCAACCTGCTACTGGTG |
AGTGCGTGTCGCCTATCTTC |
| PPAR-α |
GACAAGGCCTCAGGATACCA |
TCTTGCAGCTTCGATCACAC |
| NT-PGC-1α |
ACCACAAACGATGACCCTCC |
CTGCGGTTGTGTATGGGACT |
| FL-PGC-1α |
GGCACGCAGTCCTATTCATT |
CATCCTTTGGGGTCTTTGAG |
Recombinant adenovirus infection
Recombinant adenoviruses including mCherry-NT-PGC-1α
and NT-PGC-1α (virus titer: 9×109 cfu/ml) were purchased
from Obio Technology Corp., Ltd. (Shanghai, China). Overexpression
of NT-PGC-1α in NRCMs was achieved by infection with the
recombinant adenovirus at a multiplicity of infection (MOI) of 100
(cells were at a density of 1.5×106 in 60-mm plate).
Following 4 h of infection at 37°C, an equal volume of fresh DMEM
with 10% FBS was added to the culture. The cells were incubated for
a further 24 h at 37°C to allow the virus to achieve maximum
effect. The corresponding vehicle control were adenoviruses without
NT-PGC-1α.
Immunofluorescence
NRCMs were washed with PBS and fixed with 4%
paraformaldehyde for 15 min at 4°C. Following a further wash, cells
were blocked with a 1% goat serum albumin (Beyotime Institute of
Biotechnology, Haimen, China) for 1 h and permeabilized in PBS with
0.1% Triton X-100 for 5 min at room temperature.
Anti-PGC-1α-N-terminal antibody (1:100; cat. no. ab191838; Abcam)
was applied overnight at 4°C. Fluorescein isothiocyanate-conjugated
goat anti-rabbit immunoglobulin G (IgG) secondary antibody (1:200;
cat. no. sc-2012; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
was subsequently added for 2 h at room temperature. For the
visualization of nuclei, cells fixed at 4°C were incubated with
DAPI for 10 min at room temperature. In addition, live cells were
infected with mCherry-NT-PGC-1α or mCherry adenovirus and incubated
with Hoechst 33258 for 10 min. Cells were observed under a confocal
microscope (Olympus FV10i' magnifications, ×10 or 60).
Western blot analysis
Protein was extracted from cultured NRCMs using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors (1:100;
Sigma-Aldrich; Merck KGaA) and quantified with a bicinchoninic acid
(BCA) protein assay. Total nuclear and cytoplasmic protein was
separated using a Nuclear and Cytoplasmic Extraction Reagent kit
(Thermo Fisher Scientific. Inc.). Equal amounts of cell lysates (30
µg/lane) were determined via a Bicinchoninic Acid protein assay
(Thermo Fisher Scientific, Inc.), were separated on a 10–12%
SDS-PAGE gel, electro-transferred to 0.22 µm polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA) and blocked
using 10% bovine serum albumin (BSA) for 1 h at room temperature.
Membranes were incubated with the following primary antibodies at
4°C for 14–16 h: Anti-PGC-1α-N-terminal (1:1,000; cat. no.
ab191838), anti-ANP (1:500; cat. no. ab180649), anti-histone H3
(1:1,000; cat. no. ab1791), anti-acyl-coenzyme A
dehydrogenase-medium chain (Acadm; 1:10,000; cat. no. ab92461),
anti-PPAR-α (1:1,000; cat. no. ab8934; all Abcam), anti-superoxide
dismutase 2 (SOD2; 1:1,000; cat. no. sc30080; Santa Cruz
Biotechnology, Inc.) and anti-β-actin (cat. no. bs0061R; 1:1,000;
BIOSS, Beijing, China). Membranes were subsequently incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG secondary
antibody (cat. no. sc-2012; 1:5,000; Santa Cruz Biotechnology,
Inc.) for 1 h at room temperature. Immunoreactive bands were
detected with Pierce enhanced chemiluminescence substrate (Pierce;
Thermo Fisher Scientific, Inc.) and the GeneGnome imaging system
(Syngene, Frederick, MD, USA). Bands were quantified using ImageJ
software v 1.49 (National Institutes of Health, Bethesda, MD,
USA).
Detection of intracellular ATP
concentration
An Enhanced ATP Assay kit (Beyotime Institute of
Biotechnology) was obtained to measure cellular ATP levels
according to the manufacturer's instructions. This kit utilizes a
firefly luciferase-based method of detection. Briefly, NRCMs were
lysed according to the aforementioned kit at 4°C and centrifuged at
12,000 × g for 5 min. Protein concentration in the supernatant was
detected using a BCA protein assay (Thermo Fisher Scientific, Inc.)
and the supernatants were mixed with ATP detection working buffer
in a white 96-well plate. Luminescence was measured in relative
light units using a Multiskan Spectrum SpectraMax M3 (Molecular
Devices, LLC, Sunnyvale, CA, USA). ATP levels were expressed as
nmol/mg protein.
Measurement of mitochondrial membrane potential
(MMP). MMP was detected using a JC-1 fluorescent probe (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. When the MMP increases, JC-1 is aggregated in the
matrix of the mitochondria and releases red fluorescence; when the
MMP decreases, JC-1 forms monomers and emits green fluorescence.
Thus, the ratio of red/green fluorescence reflects the level of
relative mitochondrial depolarization. Briefly, cells
(10,000/confocal dish) were incubated with JC-1 for 30 min at 37°C
and fluorescence images were obtained using an Olympus FV10i
confocal microscope (magnification, ×10) (Olympus Corporation,
Tokyo, Japan). The intensity of the red (excitation wavelength, 490
nm; emission wavelength, 530 nm) and green (excitation wavelength,
525 nm; emission wavelength, 590 nm) fluorescence in NRCMs cultured
in a 96-well plate was determined using a SpectraMax M3 microplate
reader. Cells treated with 50 mM carbonyl cyanide
m-chlorophenylhydrazone at 37°C exhibited a depolarized MMP and
were used as a positive control.
Measurement of intracellular ROS
levels
A Reactive Oxygen Species Assay kit (Beyotime
Institute of Biotechnology) was used to determine ROS levels based
on the oxidation of 2′,7′-dichlorofluorescin diacetate (DCFH-DA).
The experimental procedures were performed strictly according to
the manufacturer's guidelines. Following isolation, NRCMs
(10,000/confocal dish) were seeded in culture dishes that were
observable under a confocal microscope to obtain fluorescence
images and fluorescence intensities. Briefly, following treatment
with PE and infection with adenovirus expressing either NT-PGC-1α
(Adv-NT-PGC-1α) or a control vector, cells were harvested and
seeded in a 96-well plate at a density of 1×105
cells/well. Following incubation with DCFH-DA for 20 min at 37°C,
the plate was read at excitation and emission wavelengths of 488
and 525 nm, respectively, using a SpectraMax M3 microplate reader
to measure fluorescence intensity. Additionally, NRCMs were
visualized with a Olympus FV10i confocal microscope
(magnifications, ×10 and ×60; Olympus Corporation).
Preparation of palmitate (PA)-bovine
serum albumin (BSA) and oleic acid (OA)-BSA conjugates
PA and OA were conjugated to fat-free BSA (FA-BSA)
as previously described by Choi et al (20). Sodium PA and OA were obtained from
Sigma-Aldrich (Merck KGaA). FA-BSA was purchased from Roche
Diagnostics. PA and OA were added to 30% BSA at 70°C in a water
bath for 30 min. Conjugated molecules were aliquoted and stored at
−20°C.
Lipid droplet (LD) staining and
detection
NRCMs were incubated with low glucose DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 1 g/l glucose and 100 µM
OA-BSA conjugate for 24 h at 37°C and subsequently fixed with 4%
paraformaldehyde for 30 min at 4°C. Following washing with PBS,
cells were stained with 1 µM BODIPY™ FL C12 lipid probe (Thermo
Fisher Scientific, Inc.) and DAPI (Beyotime Institute of
Biotechnology) for 30 and 5 min at 37°C, respectively. Cells were
subsequently washed with PBS for 5 min three times. Images of fixed
cells were captured with an Olympus FV10i confocal laser scanning
microscope (magnifications, ×10 or ×60; Olympus Corporation).
BODIPY™ FL C12 was observed as green fluorescence and used to
visualize LDs. The number of LDs in each cell was subsequently
calculated by Image-Pro Plus 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA) for statistical analysis.
Extracellular oxygen consumption
An Extracellular O2 Consumption Assay kit
was purchased from Abcam. The procedure was performed strictly
according to the manufacturer's protocol. Briefly, NRCMs were
seeded in a black-walled 96-well plate with a clear, flat bottom at
a density of 1×105 cells/well. Following infection with
an Adv-NT-PGC-1α adenovirus or empty virus, cells were treated with
10 µM PE or 500 nM MK886 (Abcam) for 48 or 6 h at 37°C,
respectively. The medium was subsequently replaced with 150 µl
low-glucose DMEM and 50 µM PA-BSA, containing 10 µl Extracellular
Oxygen Consumption reagent; control cells were treated with 10 µl
of the low glucose DMEM medium. Following this, pre-warmed (37°C)
high-sensitivity mineral oil was rapidly added to limit back
diffusion of ambient oxygen, and the plate was read in a pre-warmed
(37°C) SpectraMax M3 microplate reader (excitation, 380 nm;
emission, 650 nm). In this assay, the oxygen consumption reagent
reflects the oxygen levels in the surrounding medium. As the test
material respires, oxygen is depleted in the surrounding
environment, which is measured as an increase in the
phosphorescence signal.
Statistical analysis
All experiments were performed in triplicate. Data
were analyzed by SPSS 20.0 software (IBM Corp., Armonk, NY, USA).
Quantitative data were presented as the mean ± standard error of
the mean. A normal distribution test was performed to determine
whether parametric or non-parametric tests should be applied.
Comparisons of parameters between two experimental groups were
performed using a two-tailed t-test, whereas comparisons among
three or more groups were performed using two-way analysis of
variance, followed by the Least Significant Difference test or
Dunnett's T3 test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
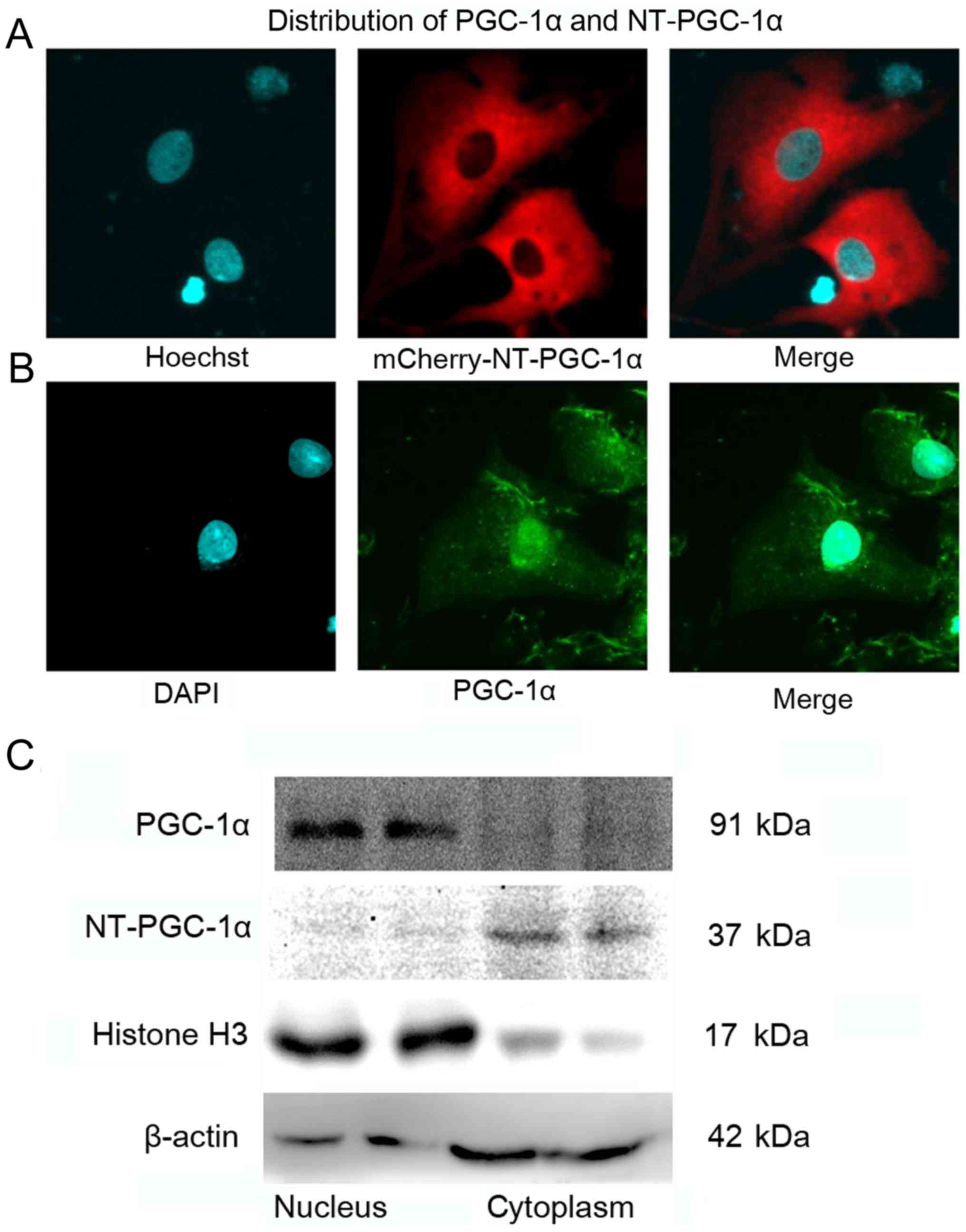
NT-PGC-1α is predominantly expressed
in the cytoplasm of NRCMs
NRCMs were infected with mCherry-Adv-NT-PGC-1α for
24 h, stained with Hoechst 33258 for 10 min and observed by
confocal microscopy to examine the subcellular localization of
NT-PGC-1α. Immunofluorescence staining of NRCMs was performed to
detect total PGC-1α, including FL-PGC-1α and NT-PGC-1α. NT-PGC-1α
was primarily located in the cytoplasm, with a small amount
distributed in the nucleus (Fig.
1A), whereas endogenous PGC-1α was predominantly located in the
nucleus (Fig. 1B). In addition,
western blot analysis of cytoplasmic and nuclear protein extracts
from mouse heart tissues demonstrated that the distribution of
NT-PGC-1α was markedly different from that of PGC-1α (Fig. 1C).
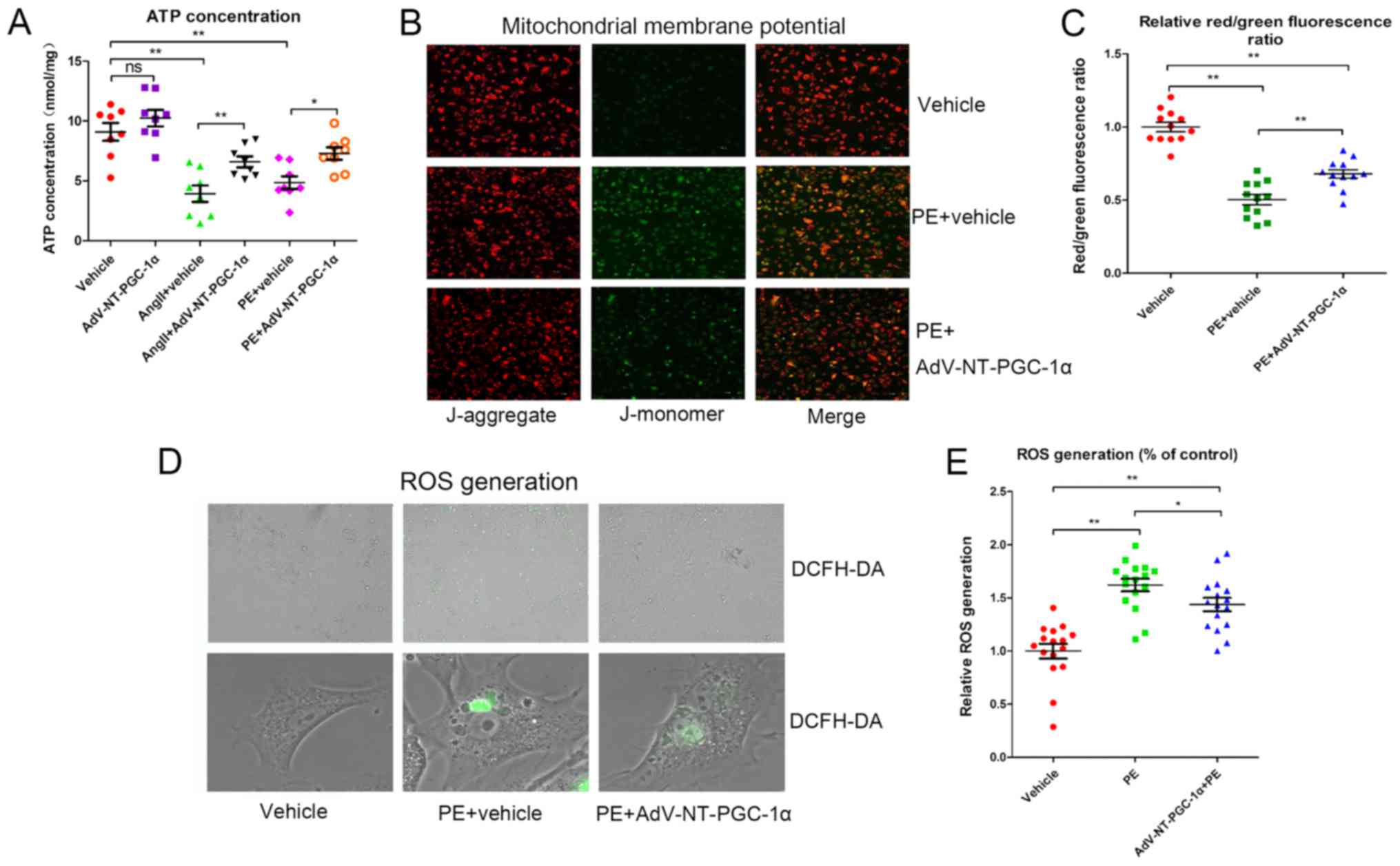
NT-PGC-1α overexpression attenuates
reductions in MMP, ATP and ROS generation
Cells were infected with Adv-NT-PGC-1α or control
adenovirus and subsequently exposed to 10 µM PE or 1 µM Ang II for
48 h. Intracellular ATP concentrations were measured using a
microplate reader. Cells incubated with PE (4.851±0.535 nmol/mg;
n=8) or Ang II (3.931±0.685 nmol/mg; n=8) had significantly reduced
ATP levels compared with cells infected with the control adenovirus
alone (9.100±0.745 nmol/mg; P<0.01; Fig. 2A). This impairment in ATP
generation was significantly alleviated by NT-PGC-1α overexpression
induced by Adv-NT-PGC-1α infection, when compared with PE treatment
(4.851±0.535 vs. 7.288±0.519 nmol/mg; n=7–8; P<0.05) or Ang II
treatment (3.931±0.685 vs. 6.588±0.453 nmol/mg; n=8; P<0.01) in
the control adenovirus groups (Fig.
2A).
MMP and ROS generation were assessed to further
investigate the effect of NT-PGC-1α on mitochondrial function.
Following infection with Adv-NT-PGC-1α and incubation of NRCMs with
PE, cells were treated with either JC-1 or DCFH-DA. The relative
MMP levels, indicated by the red/green fluorescence ratio, were
significantly decreased in the PE+vehicle group (0.504±0.035; n=12)
when compared with that of the vehicle control (1.000±0.033; n=12;
P<0.01). AdV-NT-PGC-1α infection in PE-treated cells
significantly reversed the effect of PE treatment on MMP levels
(0.679±0.029 vs. 0.504±0.035; n=12; P<0.01; Fig. 2B and C). Additionally, PE resulted
in increased ROS levels compared with the corresponding empty virus
control (1.619±0.059 vs. 1.000±0.069; n=16; P<0.01), whereas
NT-PGC-1α overexpression inhibited the ROS generation induced by PE
(1.437±0.063 vs. 1.619±0.059; n=16; P<0.05; Fig. 2D and E). These results indicated
that NT-PGC-1α may have improved mitochondrial function.
NT-PGC-1α increases fatty acid
metabolism-associated gene expression
RT-qPCR demonstrated that Adv-NT-PGC-1a infection
effectively increased NT-PGC-1a expression when compared with the
empty vehicle control (P<0.01; Fig.
3A). NT-PGC-1α overexpression significantly enhanced the
transcription of fatty acid metabolism-associated genes (Fig. 3B), including carnitine
palmitoyltransferase 1b (CPT1b; 1.000±0.208 vs. 8.603±2.822; n=8;
P<0.05), carnitine palmitoyltransferase 2 (CPT-2; 1.000±0.225
vs. 2.846±0.542; n=8; P<0.01), acyl-coenzyme A
dehydrogenase-very long chain (Acadvl; 1.000±0.337 vs. 7.027±2.144,
n=8; P<0.05), Acadm (1.000±0.150 vs. 2.712±0.607; n=8;
P<0.05), pyruvate dehydrogenase kinase 4 (PDK4; 1.000±0.228 vs.
7.717±2.072; n=8; P<0.01), PPAR-α (1.000±0.306 vs. 6.309±1.518;
n=6; P<0.05) and superoxide dismutase 3 (SOD3; 1.000±0.147 vs.
5.565±1.972; n=8; P<0.05). However, no significant differences
were detected in the levels of SOD2 (1.000±0.123 vs. 3.925±1.414;
n=8; P=0.058) and FL-PGC-1α (1.000±0.134 vs. 2.402±0.857, n=8).
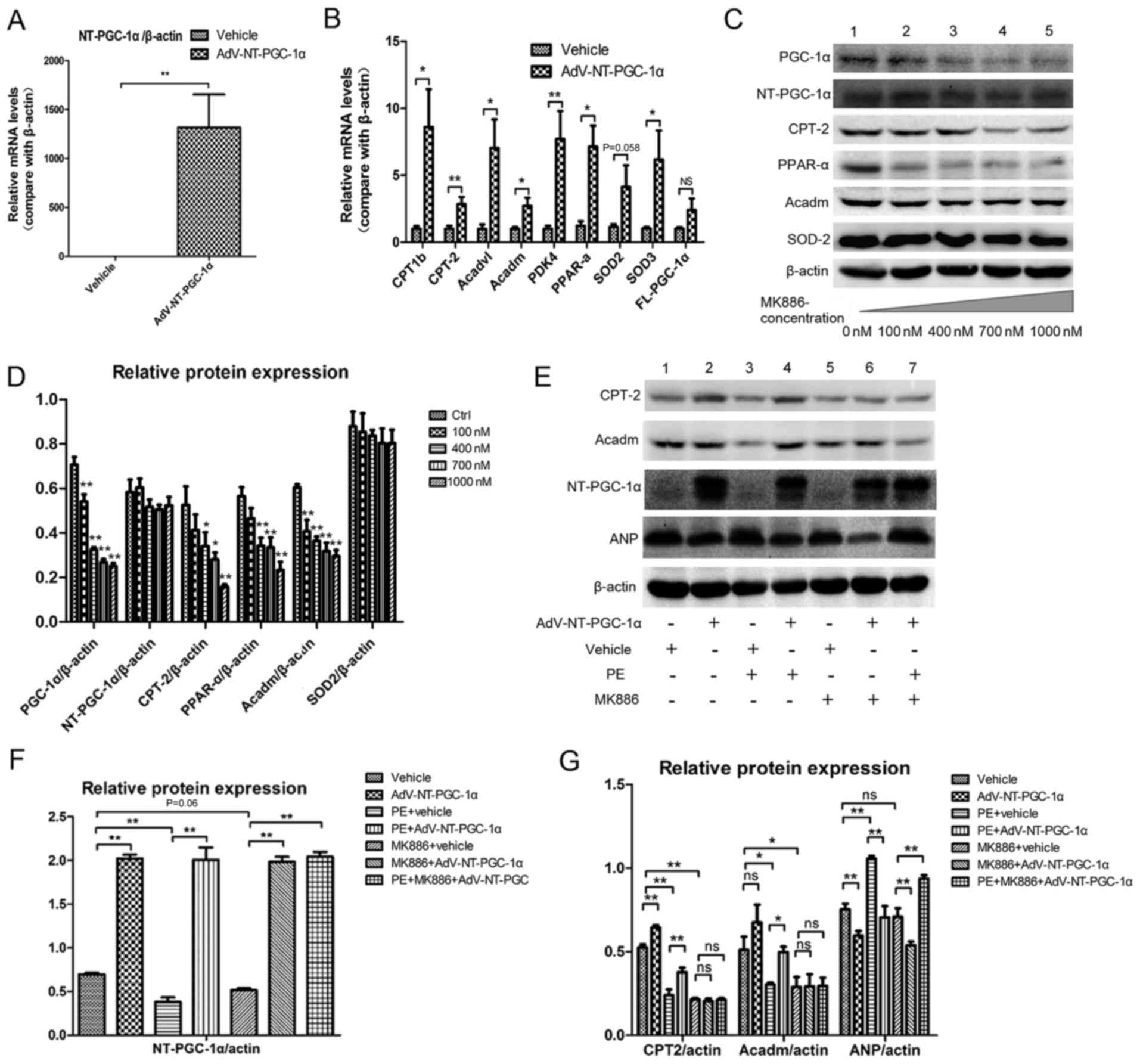 | Figure 3.NT-PGC-1α overexpression increases
fatty acid metabolism-associated gene expression. (A) NT-PGC-1α
overexpression in NRCMs induced by adenoviral infection (n=6–8 per
group). (B) Effect of NT-PGC-1α overexpression on the expression of
downstream target genes, including enzymes involved in fatty acid
metabolism and anti-oxidant enzymes (n=6–8 per group). (C)
Alterations in the expression of PGC-1a, NT-PGC-1α, CPT-2, PPAR-α,
Acadm and SOD2 in response to treatment with MK886, a PPAR-α
inhibitor. (D) Quantification of western blot results by
densitometry (n=3–5 per group). (E-G) Effect of NT-PGC-1α
overexpression, PE and MK886 (500 nM) on fatty acid
metabolism-associated protein expression (n=5 per group).
*P<0.05 and **P<0.01, as indicated or compared with the
control. AdV, adenovirus; NT, N-terminal truncated; PGC-1α,
peroxisome proliferator-activated receptor-γ coactivator-1α; CPT-2,
carnitine palmitoyltransferase 2; PPAR-α, peroxisome
proliferator-activated receptor α; Acadm, medium-chain specific
acyl-coenzyme A dehydrogenase, mitochondrial; SOD2, superoxide
dismutase 2; PE, phenylephrine; ns, not significant. |
Cells were treated with MK886, a fatty acid
metabolism inhibitor, to investigate the potential pathways by
which NT-PGC-1α overexpression may have altered the expression of
these genes. MK886 is a PPAR-α antagonist and was demonstrated to
inhibit the expression of PPAR-α and its downstream proteins CPT-2
and Acadm, as well as PGC-1α, in a dose-dependent manner (Fig. 3C and D). The western blot results
indicated that the expression levels of CPT-2 (0.505±0.019 vs.
0.239±0.034 for β-actin; n=5; P<0.01) were significantly
decreased in cells exposed to PE, and Acadm (0.511±0.078 vs.
0.303±0.012 for β-actin; n=5, P<0.01) also decreased when
exposed to PE (Fig. 3E and G;
Lanes 1 and 3), and these effects were reversed by NT-PGC-1α
overexpression (CPT-2, 0.377±0.026; P<0.01; Acadm, 0.498±0.032;
P<0.05; Fig. 3E and G; Lane 4).
In addition, NT-PGC-1α overexpression also reversed the PE-induced
increase detected in atrial natriuretic peptide (ANP) levels
(P<0.01; Fig. 3E and G; Lane
4). Additionally, the expression levels of NT-PGC-1α in different
groups were evaluated to ensure the success of Adv-NT-PGC-1a
infection (Fig. 3F). These effects
of NT-PGC-α overexpression on fatty acid metabolism were attenuated
by MK886 (Fig. 3E and G; between
lanes 1 and 2; lanes 5 and 6).
NT-PGC-1α overexpression decreases LD
accumulation in high lipid medium
NRCMs overexpressing NT-PGC-1α were incubated with
OA-BSA for 24 h and subsequently treated with MK886 for 6 h to
investigate the mechanism by which NT-PGC-1α regulated fatty acid
metabolism. Notably, the number of LDs (Fig. 4) was significantly increased in
cells incubated with OA-BSA compared with vehicle control cells
(111.10±9.86 vs. 1.73±0.48; n=30 and 27, respectively; P<0.01;
Fig. 4A, B and E). NT-PGC-1α
overexpression significantly decreased the number of LDs compared
with the OA-BSA+vehicle group (61.90±8.69 vs. 111.10±9.86; n=50 and
30, respectively; P<0.01; Fig. 4B,
C and E). As expected, the effect of NT-PGC-1α was markedly
reduced in cells exposed to MK886 (Fig. 4C, D and E; P<0.01). Taken
together, these results demonstrated that NT-PGC-1α overexpression
decreased lipid accumulation in NRCMs, indicating a role for
NT-PGC-1α in fatty acid metabolism.
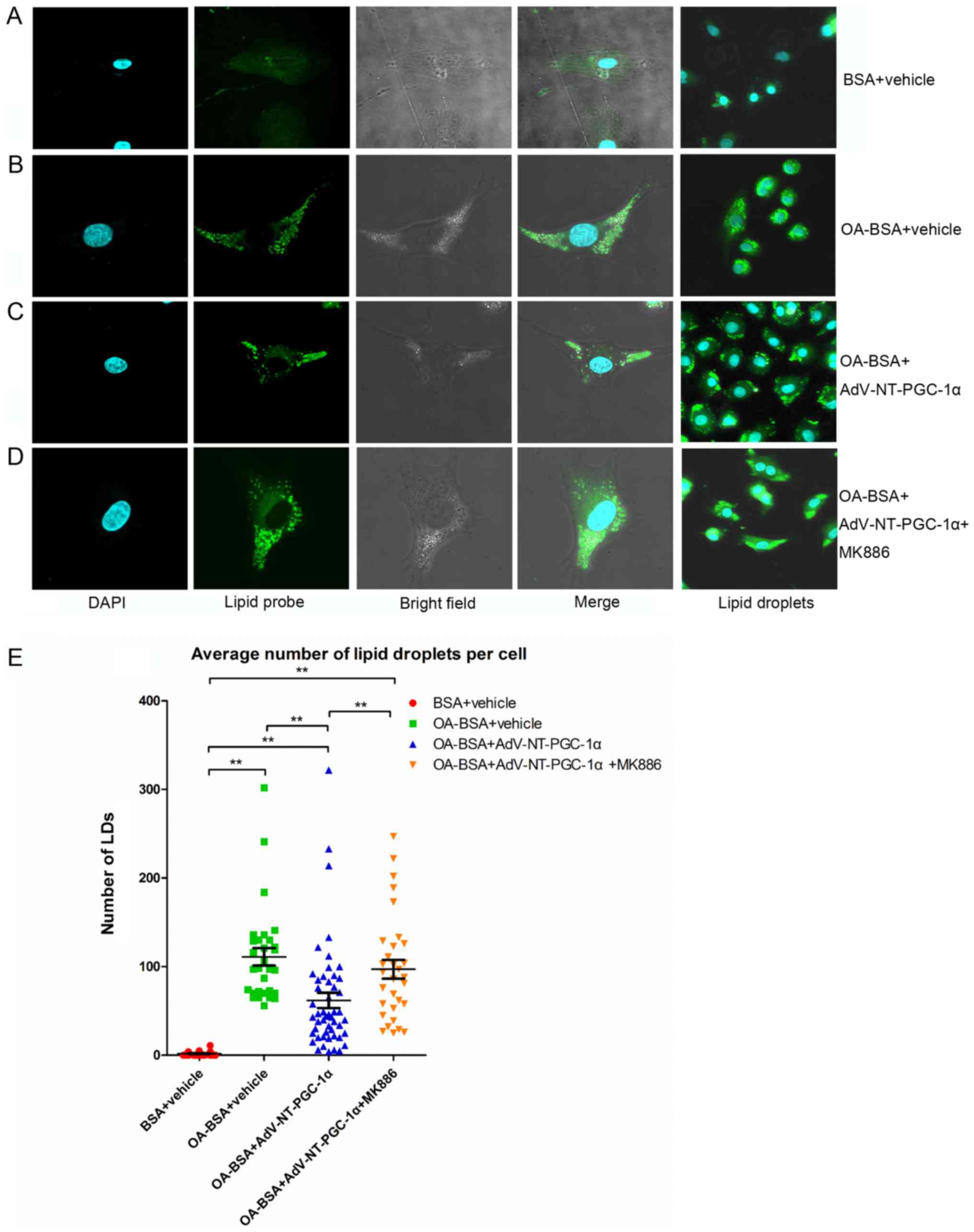 | Figure 4.NT-PGC-1α overexpression reduces LD
accumulation in NRCMs. Representative images of LD staining of
NRCMs cultured in high lipid medium from (A) the BSA+vehicle group,
(B) OA-BSA+vehicle group, (C) OA-BSA+AdV-NT-PGC-1α group and (D)
OA-BSA+AdV-NT-PGC-1α+MK886 group. (E) NT-PGC-1α overexpression
reduced LD accumulation in NRCMs. The number of LDs in each group
was counted. **P<0.01, as indicated. BSA+vehicle, n=27;
OA-BSA+vehicle, n=30; OA-BSA+AdV-NT-PGC-1α, n=50 and
OA-BSA+AdV-NT-PGC-1α+MK886, n=31. AdV, adenovirus; NT-PGC-1α,
N-terminal truncated peroxisome proliferator-activated receptor-γ
coactivator-1α; LDs, lipid droplets; NRCMs, neonatal rat
cardiomyocytes; OA, oleic acid; BSA, bovine serum albumin. |
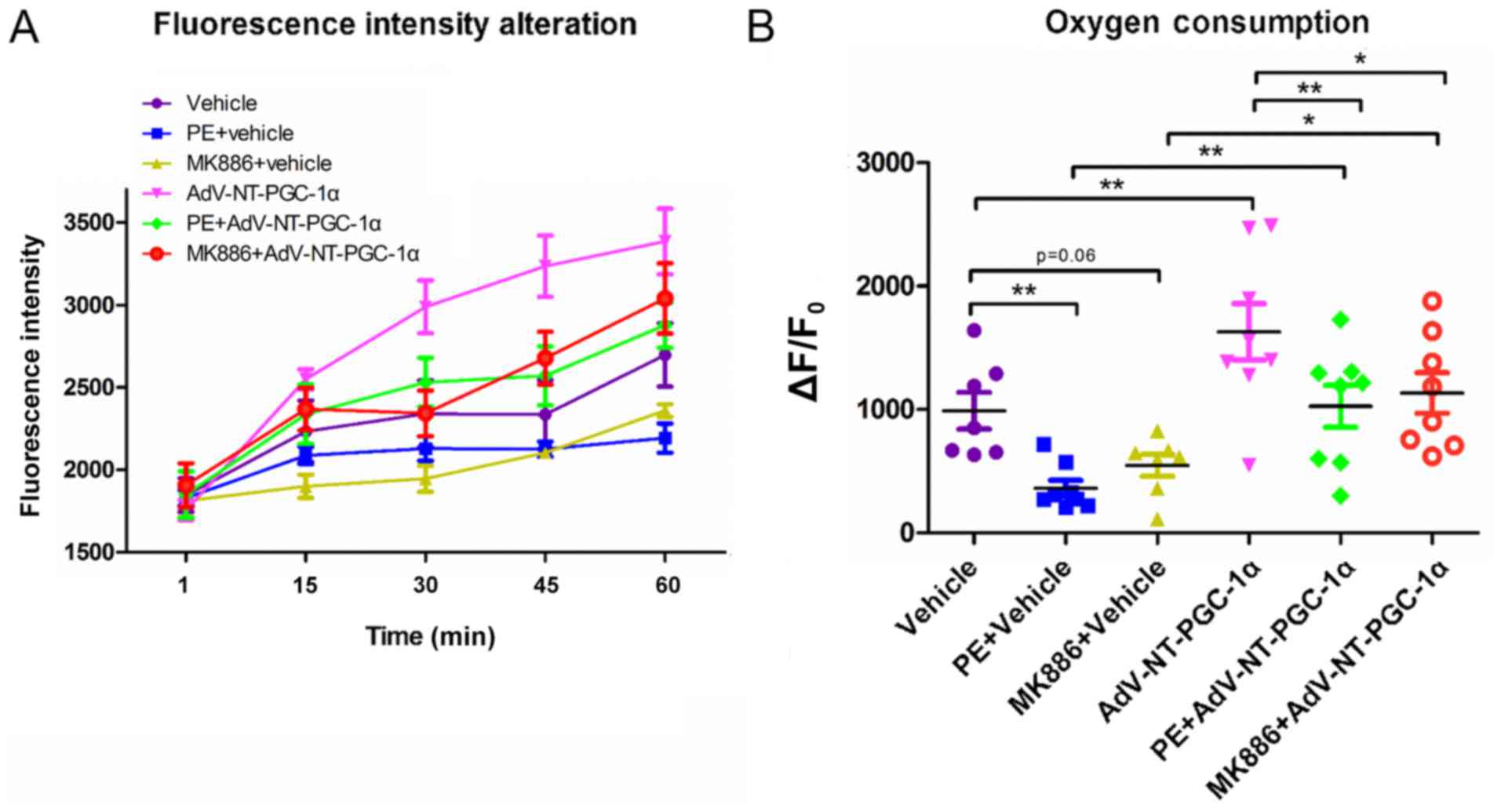
NT-PGC-1α overexpression increases the
oxygen consumption in high lipid medium
Based on results obtained by LD staining and western
blot analyses of fatty acid metabolism-associated enzymes, the
present study investigated the oxygen consumption of NRCMs in high
lipid medium (50 µM PA). NT-PGC-1α-overexpressing NRCMs were
stimulated with PE or MK886 and extracellular oxygen consumption
rates were analyzed. As indicated in Fig. 5A, oxygen consumption steadily
increased in control empty virus cells during the monitoring
period, whereas cells infected with Adv-NT-PGC-1α exhibited a
significant increase in oxygen consumption. In addition,
NT-PGC-1α-overexpressing NRCMs exposed to 10 µM PE or 500 nM MK886
displayed an increasing trend in oxygen consumption, that was
slower compared with NT-PGC-1α-overexpressing NRCMs that had not
been exposed to PE or MK886. Furthermore, PE or MK886 exposure
reduced oxygen consumption in cells lacking NT-PGC-1α
overexpression, with only a marginal increase observed over time
(Fig. 5A). Further comparison of
oxygen consumption among the different groups at the end of the
observational period revealed significant differences in
consumption between the vehicle (989.88±147.89) and PE+vehicle
(362.00±64.54.05; n=7–8; P<0.01) and Adv-NT-PGC-1α
(1,629.82±228.42; n=8; P<0.01) groups. In addition, consumption
was significantly different between the PE+vehicle
(362.00±64.54.05) and PE+Adv-NT-PGC-1α (1,026.82±170.29; n=8;
P<0.01) groups, as well as between the MK886+vehicle
(546.84±89.08) and MK886+Adv-NT-PGC-1α (1,133.90±164.19; n=7–8;
P<0.05) groups (Fig. 5B).
Discussion
In the present study, the role of NT-PGC-1α in NRCMs
was examined. It was demonstrated that NT-PGC-1α influenced MMP and
fatty acid metabolism in cardiomyocytes. NT-PGC-1α is primarily
located in the cytoplasm, unlike FL-PGC-1α, and has been reported
to regulate fatty acid metabolism in adipose tissue in the absence
of FL-PGC-1α (18,23). Consistent with these results, the
present study examined the cellular distribution of FL-PGC-1α and
NT-PGC-1α by immunofluorescence and western blotting to determine
that NT-PGC-1α was primarily expressed in the cytoplasm and may
perform similar roles in fatty acid metabolism in NRCMs. Previous
studies on the effects of NT-PGC-1α have primarily been examined in
adipocytes or skeletal muscle cells (24,25);
however, limited reports have investigated the expression of
NT-PGC-1α in human heart tissue. Thus, the present study further
explored the effects of NT-PGC-1α in cardiomyocytes.
ATP and ROS production, as well as the MMP were
detected to examine the function of NT-PGC-1α in NRCMs. NT-PGC-1α
overexpression alleviated PE- or Ang II-induced reductions in ATP
production and PE-induced ROS generation. These effects may have
contributed to its role in mitochondrial function improvement, as
NT-PGC-1α also reversed PE-induced MMP impairment. PE and Ang II
are G-protein-coupled receptor agonists, which have potent
pro-hypertrophic effects in the heart, leading to increases in ANP
and brain natriuretic peptide, which are indicators of heart
failure or heart impairment (26).
Furthermore, they are well-known stimulants for establishing
cardiac hypertrophy and heart failure experimental models (27), despite also exerting critical
effects in blood pressure maintenance. Additionally, PE may inhibit
the expression of PPAR-α (28),
which is associated with energy metabolic dysfunction in heart
disease. Therefore, it was postulated that NT-PGC-1α overexpression
may have ameliorated the impairments in mitochondrial function
induced by PE and Ang II in the present study.
Although NT-PGC-1α has a structure similar to
FL-PGC-1α, FL-PGC-1α has anti-oxidative effects and is versatile in
transcription (17,29). Thus, NT-PGC-1α may have a similar
function to PGC-1α and exert its effects by associating with
transcription factors via its nuclear domain in the nucleus, or may
be directly associated with mitochondria via protein
interactions.
To the best of our knowledge, the role of NT-PGC-1α
in NRCM fatty acid metabolism has not been previously investigated.
However, in a recent study focused on obesity, NT-PGC-1α was
reported to have a profound impact on FAO in mice (23). In addition, Zhang et al
(17) observed that NT-PGC-1α
overexpression upregulates the expression of its downstream target
CPT-2 (17). Furthermore,
crosstalk between NT-PGC-1α and phosphatase and tensin
homolog-induced putative kinase 1 (PINK1) is involved in FAO
(30), and PINK1 silencing impairs
PGC-1α-associated lipid metabolism (20). Thus, NT-PGC-1α is important in
fatty acid metabolism and may ameliorate the metabolic deficits
observed in patients with cardiac hypertrophy and heart
failure.
In support of this hypothesis, the present study
demonstrated that NT-PGC-1α overexpression increased the mRNA
expression of PPAR-α-associated genes and alleviated PE-induced
reductions in CPT-2 and Acadm expression, enzymes that are involved
in transferring fatty acids to mitochondria and initiating the
mitochondrial fatty acid β-oxidation pathway. In the present study,
NT-PGC-1α-overexpressing NRCMs were treated with MK886, a
lipoxygenase inhibitor that antagonizes PPAR-α activity (31). As expected, MK886 reduced the
effect of NT-PGC-1α on the expression of fatty acid
metabolism-associated proteins. Thus, NT-PGC-1α may have exerted
metabolic effects, as well as influencing the PPAR-α signaling
pathway activity to an extent.
Furthermore, the effects of NT-PGC-1α overexpression
on free fatty acid (FFA) metabolism were examined. LDs accumulated
in NRCMs when cells were exposed to OA-BSA, whereas NT-PGC-1α
overexpression reduced the number of LDs that accumulated in cells.
In addition, the LD number increased in NRCMs infected with
Adv-NT-PGC-1α and treated with MK886. Strategies that enhance FAO
increase the levels of FFA and β-oxidation (32); however, as FFA content in the
culture medium was at a constant level, the fatty acid was mostly
from exogenous sources rather than lipid mobilization in the
present study. Based on these results, NT-PGC-1α may alleviate the
lipotoxicity of FFA by enhancing its metabolism, rather than via
lipid mobilization.
To further investigate the function of NT-PGC-1α in
fatty acid metabolism, the present study measured oxygen
consumption in NT-PGC-1α-overexpressing NRCMs exposed to PE and/or
MK886 in a high lipid medium. NT-PGC-1α overexpression resulted in
increased oxygen consumption, consistent with the findings of
protein expression levels. Therefore, NT-PGC-1α may function by
shuttling into the nucleus and activating the associated signaling
pathway, or it may originate and function in the nucleus. It is
also possible that it directly participates in crosstalk with
mitochondria in the cytoplasm. If NT-PGC-1α functions in the
nucleus, then forced NT-PGC-1α nuclear translocation would likely
enhance its function; a cyclic adenosine monophosphate (cAMP)
analog was reported to activate PGC-1α and increase the nuclear
localization of NT-PGC-1α (17),
and the function of cAMP in myocardial contractility is well
established (33). However,
additional studies are required to verify whether nuclear NT-PGC-1α
acts as an effector molecule in response to cAMP stimulation.
Recent research demonstrated that NT-PGC-1α directly interacts with
mitochondria (34); therefore, it
may exert effects in the nucleus and the cytoplasm. Combined with
the findings of these previous reports, the present study validated
that NT-PGC-1α has several roles in fatty acid metabolism in
various cell types, and may have broad applications in the
treatment of obesity, heart failure and atherosclerosis.
There are certain limitations and drawbacks to the
present study. First, endogenous NT-PGC-1α was not detected by
immunofluorescence; FL-PGC-1α contains all of the antigen epitopes
in NT-PGC-1α, whereas NT-PGC-1α lacks the C-terminal epitopes of
FL-PGC-1α (35). Thus, a
N-terminal PGC-1α antibody would detect NT-PGC-1α and FL-PGC-1α,
while a C-terminal PGC-1α antibody would only detect FL-PGC-1α.
Therefore, evaluation of NT-PGC-1α expression in NRCMs without
transfection failed due to antibody limitations, preventing the
immunofluorescent detection of NT-PGC-1α. Therefore, the most
reliable way to accurately detect endogenous NT-PGC-1α at present
may be to separate cytoplasmic and nuclear proteins. Secondly, FAO
was not examined directly, and the results of western blotting, LD
accumulation and oxygen consumption only provide indirect evidence.
Furthermore, application of small interfering RNA rather than an
inhibitor to interfere with PPAR-α expression may provide more
conclusive data to support the proposed pathway of NT-PGC-1α. In
addition, the roles of NT-PGC-1α in the cytoplasm and its effects
in animal heart tissue require further exploration.
In conclusion, the present study demonstrated that
NT-PGC-1α alleviated PE-induced mitochondrial function impairment
and influenced fatty acid metabolism in NRCMs, likely through the
PPAR-α-associated signaling pathway. Thus, NT-PGC-1α may provide a
promising target for the treatment of cardiac hypertrophy and heart
failure.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270320).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DX, HR and QZe made substantial contributions to the
design of the present study experiments. QZh and WL conducted cell
cultures. ZL, JH and WC performed experiments and analyzed data.
ZL, JH and QZh produced the figures. ZL, JH and WL prepared the
manuscript. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Southern
Medical University review board and the animal protocols used
complied with the Guide for the Care and Use of Laboratory Animals
(21).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neubauer S: The failing heart-an engine
out of fuel. N Engl J Med. 356:1140–1151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kong X, Banks A, Liu T, Kazak L, Rao RR,
Cohen P, Wang X, Yu S, Lo JC, Tseng YH, et al: IRF4 is a key
thermogenic transcriptional partner of PGC-1alpha. Cell. 158:69–83.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang TY, Zheng D, Houmard JA, Brault JJ,
Hickner RC and Cortright RN: Overexpression of PGC-1alpha increases
peroxisomal and mitochondrial fatty acid oxidation in human primary
myotubes. Am J Physiol Endocrinol Metab. 331–2016. 2017.
|
|
4
|
Wu Z, Puigserver P, Andersson U, Zhang C,
Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC and
Spiegelman BM: Mechanisms controlling mitochondrial biogenesis and
respiration through the thermogenic coactivator PGC-1. Cell.
98:115–124. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Depre C, Vanoverschelde JL and Taegtmeyer
H: Glucose for the heart. Circulation. 99:578–588. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arumugam S, Sreedhar R, Thandavarayan RA,
Karuppagounder V and Watanabe K: Targeting fatty acid metabolism in
heart failure: Is it a suitable therapeutic approach? Drug Discov
Today. 21:1003–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin J, Wu PH, Tarr PT, Lindenberg KS,
St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM,
et al: Defects in adaptive energy metabolism with CNS-linked
hyperactivity in PGC-1alpha null mice. Cell. 119:121–135. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leone TC, Lehman JJ, Finck BN, Schaeffer
PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N,
Bernal-Mizrachi C, et al: PGC-1alpha deficiency causes multi-system
energy metabolic derangements: Muscle dysfunction, abnormal weight
control and hepatic steatosis. PLoS Biol. 3:e1012005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Karamanlidis G, Garcia-Menendez L, Kolwicz
SJ, Lee CF and Tian R: Promoting PGC-1alpha-driven mitochondrial
biogenesis is detrimental in pressure-overloaded mouse hearts. Am J
Physiol Heart Circ Physiol. 307:H1307–H1316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
LeBleu VS, O'Connell JT, Gonzalez HK,
Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A,
Domingos CL, Rocha RM, et al: PGC-1alpha mediates mitochondrial
biogenesis and oxidative phosphorylation in cancer cells to promote
metastasis. Nat Cell Biol. 16:992–1003, 1–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Russell LK, Mansfield CM, Lehman JJ,
Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML,
McDonald JA and Kelly DP: Cardiac-specific induction of the
transcriptional coactivator peroxisome proliferator-activated
receptor gamma coactivator-1alpha promotes mitochondrial biogenesis
and reversible cardiomyopathy in a developmental stage-dependent
manner. Circ Res. 94:525–533. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pereira RO, Wende AR, Crum A, Hunter D,
Olsen CD, Rawlings T, Riehle C, Ward WF and Abel ED: Maintaining
PGC-1alpha expression following pressure overload-induced cardiac
hypertrophy preserves angiogenesis but not contractile or
mitochondrial function. FASEB J. 28:3691–3702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie J, Cui K, Hao H, Zhang Y, Lin H, Chen
Z, Huang X, Cao S, Liao W, Bin J, et al: Acute hyperglycemia
suppresses left ventricular diastolic function and inhibits
autophagic flux in mice under prohypertrophic stimulation.
Cardiovasc Diabetol. 15:1362016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Li J, Hou Z, Yu Y and Yu B: KLF5
overexpression attenuates cardiomyocyte inflammation induced by
oxygen-glucose deprivation/reperfusion through the
PPARgamma/PGC-1alpha/TNF-alpha signaling pathway. Biomed
Pharmacother. 84:940–946. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gundewar S, Calvert JW, Jha S,
Toedt-Pingel I, Ji SY, Nunez D, Ramachandran A, Anaya-Cisneros M,
Tian R and Lefer DJ: Activation of AMP-activated protein kinase by
metformin improves left ventricular function and survival in heart
failure. Circ Res. 104:403–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patten IS and Arany Z: PGC-1 coactivators
in the cardiovascular system. Trends Endocrinol Metab. 23:90–97.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Huypens P, Adamson AW, Chang JS,
Henagan TM, Boudreau A, Lenard NR, Burk D, Klein J, Perwitz N, et
al: Alternative mRNA splicing produces a novel biologically active
short isoform of PGC-1alpha. J Biol Chem. 284:32813–32826. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trausch-Azar J, Leone TC, Kelly DP and
Schwartz AL: Ubiquitin proteasome-dependent degradation of the
transcriptional coactivator PGC-1{alpha} via the N-terminal
pathway. J Biol Chem. 285:40192–40200. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang JS, Huypens P, Zhang Y, Black C,
Kralli A and Gettys TW: Regulation of NT-PGC-1alpha subcellular
localization and function by protein kinase A-dependent modulation
of nuclear export by CRM1. J Biol Chem. 285:18039–18050. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi J, Ravipati A, Nimmagadda V, Schubert
M, Castellani RJ and Russell JW: Potential roles of PINK1 for
increased PGC-1alpha-mediated mitochondrial fatty acid oxidation
and their associations with Alzheimer disease and diabetes.
Mitochondrion. 18:41–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guide for the Care and Use of Laboratory
Animals (8th Edition). 2011.https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdfPubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim J, Fernand VE, Henagan TM, Shin J,
Huypens P, Newman S, Gettys TW and Chang JS: Regulation of brown
and white adipocyte transcriptome by the transcriptional
coactivator NT-PGC-1alpha. PLoS One. 11:e1599902016.
|
|
24
|
Jun HJ, Joshi Y, Patil Y, Noland RC and
Chang JS: NT-PGC-1alpha activation attenuates high-fat diet-induced
obesity by enhancing brown fat thermogenesis and adipose tissue
oxidative metabolism. Diabetes. 63:3615–3625. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Popov DV, Bachinin AV, Lysenko EA, Miller
TF and Vinogradova OL: Exercise-induced expression of peroxisome
proliferator-activated receptor γ coactivator-1α isoforms in
skeletal muscle of endurance-trained males. J Physiol Sci.
64:317–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Keys JR, Zhou RH, Harris DM, Druckman CA
and Eckhart AD: Vascular smooth muscle overexpression of G
protein-coupled receptor kinase 5 elevates blood pressure, which
segregates with sex and is dependent on Gi-mediated signaling.
Circulation. 112:1145–1153. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schlegel P, Reinkober J, Meinhardt E,
Tscheschner H, Gao E, Schumacher SM, Yuan A, Backs J, Most P,
Wieland T, et al: G protein-coupled receptor kinase 2 promotes
cardiac hypertrophy. PLoS One. 12:e1821102017. View Article : Google Scholar
|
|
28
|
Huang Q, Huang J, Zeng Z, Luo J, Liu P,
Chen S, Liu B, Pan X, Zang L and Zhou S: Effects of
ERK1/2/PPARalpha/SCAD signal pathways on cardiomyocyte hypertrophy
induced by insulin-like growth factor 1 and phenylephrine. Life
Sci. 124:41–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geng T, Li P, Yin X and Yan Z: PGC-1alpha
promotes nitric oxide antioxidant defenses and inhibits FOXO
signaling against cardiac cachexia in mice. Am J Pathol.
178:1738–1748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Choi J, Batchu VV, Schubert M, Castellani
RJ and Russell JW: A novel PGC-1alpha isoform in brain localizes to
mitochondria and associates with PINK1 and VDAC. Biochem Biophys
Res Commun. 435:671–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kehrer JP, Biswal SS, La E, Thuillier P,
Datta K, Fischer SM and Vanden HJ: Inhibition of
peroxisome-proliferator-activated receptor (PPAR)alpha by MK886.
Biochem J. 356:899–906. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pascual F and Coleman RA: Fuel
availability and fate in cardiac metabolism: A tale of two
substrates. Biochim Biophys Acta. 1861:1425–1433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Beca S, Ahmad F, Shen W, Liu J, Makary S,
Polidovitch N, Sun J, Hockman S, Chung YW, Movsesian M, et al:
Phosphodiesterase type 3A regulates basal myocardial contractility
through interacting with sarcoplasmic reticulum calcium ATPase type
2a signaling complexes in mouse heart. Circ Res. 112:289–297. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang JS and Ha K: An unexpected role for
the transcriptional coactivator isoform NT-PGC-1alpha in the
regulation of mitochondrial respiration in brown adipocytes. J Biol
Chem. 292:9958–9966. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Villena JA: New insights into PGC-1
coactivators: Redefining their role in the regulation of
mitochondrial function and beyond. FEBS J. 282:647–672. 2015.
View Article : Google Scholar : PubMed/NCBI
|