Introduction
Bone homeostasis is tightly regulated by the balance
between osteoblasts (OBs), the bone-forming cells, and osteoclasts
(OCs), the bone-resorbing cells (1). Pathological conditions such as
osteoporosis, inflammation, and cancer break this balance in favor
of the augmentation of OC differentiation and activity, leading to
net bone loss (1). As a result,
patients with these diseases suffer from increased risk of bone
fracture. Therefore, uncovering potential therapeutic targets by
understanding OC differentiation and function are important
objectives to treat these diseases (2).
OC differentiation is regulated by macrophage
colony-stimulating factor (M-CSF) and receptor activator of nuclear
factor-κB ligand (RANKL) (3,4).
M-CSF is crucial for myeloid cell survival and differentiation to
pre-osteoclasts, and RANKL elicits pre-osteoclast fusion,
polykaryon maturation, and OC activation. Mechanistically, the
binding of RANKL to its cell surface receptor, RANK, generates the
activation of multiple downstream pathways such as the nuclear
factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs),
phosphatidylinositol 3-kinase/Akt (PI3K/Akt), and phospholipase C
gamma 2 (PLCγ2)-Ca2+-CREB signaling pathways (5–7),
which leads to subsequent nuclear translocation of nuclear factor
of activated T cells, cytoplasmic 1 (NFATc1), a master
transcription factor for osteoclastogenesis (8–10).
Cysteinyl leukotrienes (CysLTs) are potent lipid
mediators of several immune and inflammatory stimuli (11). Three different CysLTs
(LTC4, LTD4, and LTE4) arise from
a single intracellular synthetic event by successive enzymatic
conversions. Arachidonic acid is oxidized by 5-lipoxygenase (5-LO)
to generate the unstable precursor leukotriene A4
(LTA4), and subsequently, leukotriene C4
synthase (LTC4S) converts LTA4 to the parent
CysLT, LTC4. After energy-dependent export from the
cell, LTC4 is sequentially converted to LTD4,
and then to the final and most stable CysLT, LTE4. The
actions of CysLTs are mediated via at least two types of CysLT
receptors, designated as CysLT receptor 1 (CysLTR1) and 2
(CysLTR2). CysLTs-CysLTR1 signaling is considered a major
pharmacological target for the treatment of asthma and allergic
rhinitis (11,12). Several CysLTR1 antagonists such as
montelukast have been approved by the Food and Drug Agency (FDA)
and are in the market for the treatment of these diseases (12). Recently, expanded roles of this
signaling pathway in the pathogenesis of cardiovascular diseases,
cerebrovascular diseases, malignant tumors, fibrosis, and immune
host defense have been suggested (11,13),
thereby indicating that it could be involved in various
pathophysiological conditions as well as in inflammation in a
tissue-specific manner.
Although several studies reported the involvement of
leukotriene in OC differentiation and function (14), little is known about the effect of
CysLTR1 on OCs and bone loss. Thus, we explored the detailed
mechanism by which CysLTR1 regulates OC formation and its
protective effect on bone loss using CysLTR1 antagonist to
re-position the CysLTR1 as a promising drug target in bone-related
diseases in association with excessive osteoclastogenesis.
Materials and methods
Reagents
CysLTR1 antagonist (Montelukast) and leukotriene
C4 were purchased from Cayman Chemical Company, (Ann
Arbor, MI, USA). P2Y12 antagonist (MRS2395) and 2-(Methylthio)-ADP
were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Antibodies against phospho-PLCγ2, phospho-Akt,
phospho-ERK, ERK, and c-Fos were purchased fromCell Signaling
Technology, Inc., (Danvers, MA, USA). Antibody against NFATc1 was
purchased from Santa Cruz Biotechnology, Inc. Antibody against
phospho-CREB was purchased from EMD Millipore (Billerica, MA, USA).
Antibody against β-actin was purchased from Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany). All other reagents were from
Sigma-Aldrich; Merck KGaA.
Animals
Male ICR (4- to 6-week-old) mice were purchased from
Samtako Inc., (Osan, Korea). Female C57BL/6J (6- to 7-week-old)
mice were purchased from DBL Inc., (Umsung, Korea). All mice were
maintained in the animal facility of the Sookmyung Women's
University in a 12:12 h light-dark cycle at 21±2°C, and were
allowed food and water ad libitum. All experiments were
performed in accordance with institutional guidelines approved by
the Sookmyung Women's University Care and Use Committee.
OC differentiation
Mouse bone marrow cells were obtained from the long
bones of 4- to 6-week-old male mice. Bone marrow cells were
cultured in α-MEM (10% FBS), supplemented with M-CSF (10 ng/ml;
R&D Systems, Inc., Minneapolis, MN, USA) for 12 h to separate
adherent and non-adherent cells. The non-adherent cells were then
harvested and cultured with M-CSF (30 ng/ml). After 3 days of
culture, the floating cells were removed and the attached cells
used as bone marrow-derived macrophages (BMMs). BMMs
(1×104) were plated in 96-well plates and differentiated
into OCs by further treatment with M-CSF (30 ng/ml) and RANKL (100
ng/ml). Cells were then fixed and permeabilized with an equal
volume mixture of acetone and ethanol for 30 s and then treated
with tartrate resistant acid phosphatase (TRAP) staining solution
(0.01% naphthol AS-MX phosphate (Sigma-Aldrich; Merck KGaA) and
0.06% Fast Red Violet LB salt (Sigma-Aldrich; Merck KGaA) in 50 mM
sodium tartrate dehydrate and 45 mM sodium acetate (pH 5.0)).
TRAP-positive multinucleated cells (>3 nuclei/cell) were counted
as mature osteoclasts.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was purified with easy-BLUE (iNtRON
Biotechnology, Seoul, Korea), and cDNA was prepared from 5 µg of
RNA using Revert Aid™ First-Strand cDNA Synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
RT-qPCR reactions were performed in a total volume of 20 µl using
SYBR−Green PCR Master mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Thermocycling was performed using a 7500 real-time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with the
following conditions: initial hold at 95°C for 10 min, followed by
40 cycles of denaturation at 95°C for 15 sec, annealing at 58°C,
and extension at 60°C for 1 min. Specific primer sequences for
RT-qPCR were designed as follows: calcitonin receptor (CTR),
5′-tttcaagaaccttagctgccagag-3′ (forward), 5′-caa ggc acg gac aat
gtt gag aag-3′ (reverse); cathepsin K, 5′-ctt cca ata cgt gca gca
ga-3′ (forward), 5′-acg cac caa tat ctt gca cc-3′ (reverse);
Atp6v0d2, 5′-tca gat ctc ttc aag gct gtg ctg-3′ (forward), 5′-gtg
cca aat gag ttc aga gtg atg-3′ (reverse); DC-STAMP, 5′-tgg aag ttc
act tga aac tac gtg-3′ (forward), 5′-ctc ggt ttc ccg tca gcc tctc
tc-3′ (reverse); αv-integrin, 5′-cct cag aga ggg aga tgt tca cac-3′
(forward), 5′-aac tgc caa gat gat cac cca cac-3′ (reverse);
β3-integrin, 5′-gat gac atc gag cag gtg aaa gag-3′ (forward),
5′-ccg gtc atg aat ggt gat gag tag-3′ (reverse); GAPDH, 5′-tgc acc
acc aac tgc tta gc-3′ (forward), 5′-ggc atg gac tgt ggt cat gag-3′
(reverse). Data were analyzed using 7500 System Sequence Detection
Software v2.0 (Applied Biosystems; Thermo Fisher Scientific, Inc.).
An index mRNA level was assessed using a threshold cycle (Cq) value
and normalized against GAPDH expression. qPCR results were
calculated using the 2−ΔΔCq method (15).
Western blot analysis
Total cell lysates were separated by SDS-PAGE and
transferred onto Immobilon-P membranes (EMD Millipore). The
membranes were blocked with 5% non-fat-milk in PBS-T, and then
immunostained with anti-phospho-PLCγ2 (1:1,000), anti-phospho-Akt
(1:1,000), anti-phospho CREB (1:1,000), anti-phospho ERK (1:1,000),
anti-ERK (1:1,000), anti-c-Fos (1:1,000), anti-NFATc1 (1:200), or
anti-β-actin (1:4,000), followed by secondary horseradish
peroxidase-conjugated antibody (1:5,000). The membranes were
developed using an enhanced chemiluminescence detection kit (GE
Healthcare Life Sciences, Little Chalfont, UK).
Actin ring staining
Cells were washed twice with PBS and fixed with 10%
formalin for 5 min. Cells were permeabilized with EtOH/Acetone
(1:1) for 1 min at room temperature, the solvent removed, and the
cells dried. Actin rings were stained with rhodamine-conjugated
phalloidin (Molecular Probes; Thermo Fisher Scientific, Inc.)
overnight in PBS at 4°C in the dark. After washing the cells twice
with PBS, the number of actin rings was counted using a
fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Bone resorption assay
BMMs were differentiated on dentin slices with M-CSF
and RANKL (PeproTech, Inc., Rocky Hill, NJ, USA) for 3 days, and
treated with montelukast or MRS2395 for 4 days. The cells were
removed from the dentin slice by wiping the surface, and then the
slices were stained with toluidine blue (1 µg/ml; J.T. Baker
Chemical Co., Phillipsburg, NJ, USA). The number of pits formed by
bone resorption on the dentin slices was counted.
Retroviral gene transduction
To generate retroviral stocks, PMX-puro GFP
(GFP-vector) or PMSCV-GFP NFATc1 (NFATc1) was transfected into the
packaging cell line platinum-E (Plat-E). Viral supernatant was
collected from culture media at 48 h after transfection using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. For infection with
retroviruses, BMMs were incubated with the viral supernatant (4
ml/dish), polybrene (10 µg/ml, Santa Cruz Biotechnology, Inc.), and
M-CSF (30 ng/ml) for 2 days, and selected by puromycin (2 µg/ml,
Sigma-Aldrich) for an additional 48 h.
siRNA transfection
BMMs were plated on 48-well plates at a density of
3.5×104 cells/well with 30 ng/ml M-CSF. After 24 h,
cells were transfected with 40 nM mouse P2Y12 on-target siRNAs
(sc-151960; Santa Cruz Biotechnology, Inc.) using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The control contained 40 nM
non-targeting siRNA (Qiagen GmbH, Hilden, Germany). The
transfection was performed in 2.5 ml of serum free medium for 6 h;
the cells were then cultured for 3 days in complete medium
containing 30 ng/ml M-CSF and 100 ng/ml RANKL to form
osteoclasts.
Lipopolysaccharide (LPS)-induced
osteoclast formation in vivo
ICR mice (5-week-old) were i.p. injected with
vehicle (EtOH) or montelukast (5 mg/kg) every day. The day after
the first injection of montelukast, LPS (0.5 mg) or PBS was
directly injected on the calvarium. Six days after LPS injection,
mice were euthanized and the calvaria were extracted (n=5 per each
group). Calvaria were fixed in 4% paraformaldehyde for 24 h at 4°C,
and then stained for TRAP. Image analysis was performed by using
ImageJ software (v1.32; National Institutes of Health, Bethesda,
MD, USA) according to the manufacturer's protocol.
Ovariectomy (OVX)-induced bone loss in
vivo
Female mice (9-week-old, C57BL/6J) were used for
ovariectomy. After 5 days, vehicle (1% methylcellulose) or
montelukast (5 mg/kg) was orally administered daily for 12 weeks
(n=7 per each group). Femurs were extracted and stored in 70%
ethanol at 4°C until analysis. Two-dimensional measurement was
performed with a micro-CT scanner and associated analysis software
(Model 1172; Bruker microCT, Kontich, Belgium) at 9-µm voxel size.
Image acquisition was performed at 35 kV of energy and 220 mA of
intensity (590 ms of integration time). The threshold was set to
segment the bone from the background, and the same threshold
setting was used for all samples.
Hind limb unloading model
Eight-week-old male ICR mice were orally
administered vehicle (1% methylcellulose) or montelukast (5 mg/kg)
daily. After a day, mice were subjected to hind limb unloading for
14 days. Hind limb unloading was conducted by applying a tape to
the surface of the hind limb to set a metal clip (16). The other end of the clip was fixed
to an overhead bar. The height of the bar was adjusted to maintain
the mice at 30° head down tilt with the hind limbs elevated above
the floor of the cage. Loaded control mice were also housed under
the same conditions except for hind limb unloading for the same
duration (n=7 per each group). Femurs were extracted and stored in
70% ethanol at 4°C and analyzed by micro-CT as described above.
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments. The statistical
significance of differences between groups was determined using
Student's t-test and analysis of variance with a least significant
difference post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
CysLTR1 antagonist inhibits
RANKL-induced OC formation and bone resorption in vitro
RANKL is essential and sufficient for the
differentiation of OC precursors into mature OCs in the presence of
M-CSF (3,4). Thus, we examined the effects of
CysLTR1 antagonist on RANKL-induced OC formation from
bone-marrow-derived macrophages (BMMs). We used montelukast since
it is a commonly prescribed CysLTR1 antagonist and is generally
considered a safe drug with the occurrence of few adverse drug
reactions (17). When BMMs were
incubated with M-CSF and RANKL for 4 days, numerous TRAP-positive
multinucleated OCs were generated. Treatment of the same cultures
with montelukast completely suppressed OC formation (Fig. 1A). Consistently, the mRNA
expression levels of various OC markers such as calcitonin receptor
(CTR), cathepsin K, Atp6v0d2, DC-STAMP, αv-integrin, and
β3-integrin were increased in cultures treated with M-CSF and RANKL
for 4 days. This effect was dramatically suppressed by montelukast
treatment (Fig. 1B).
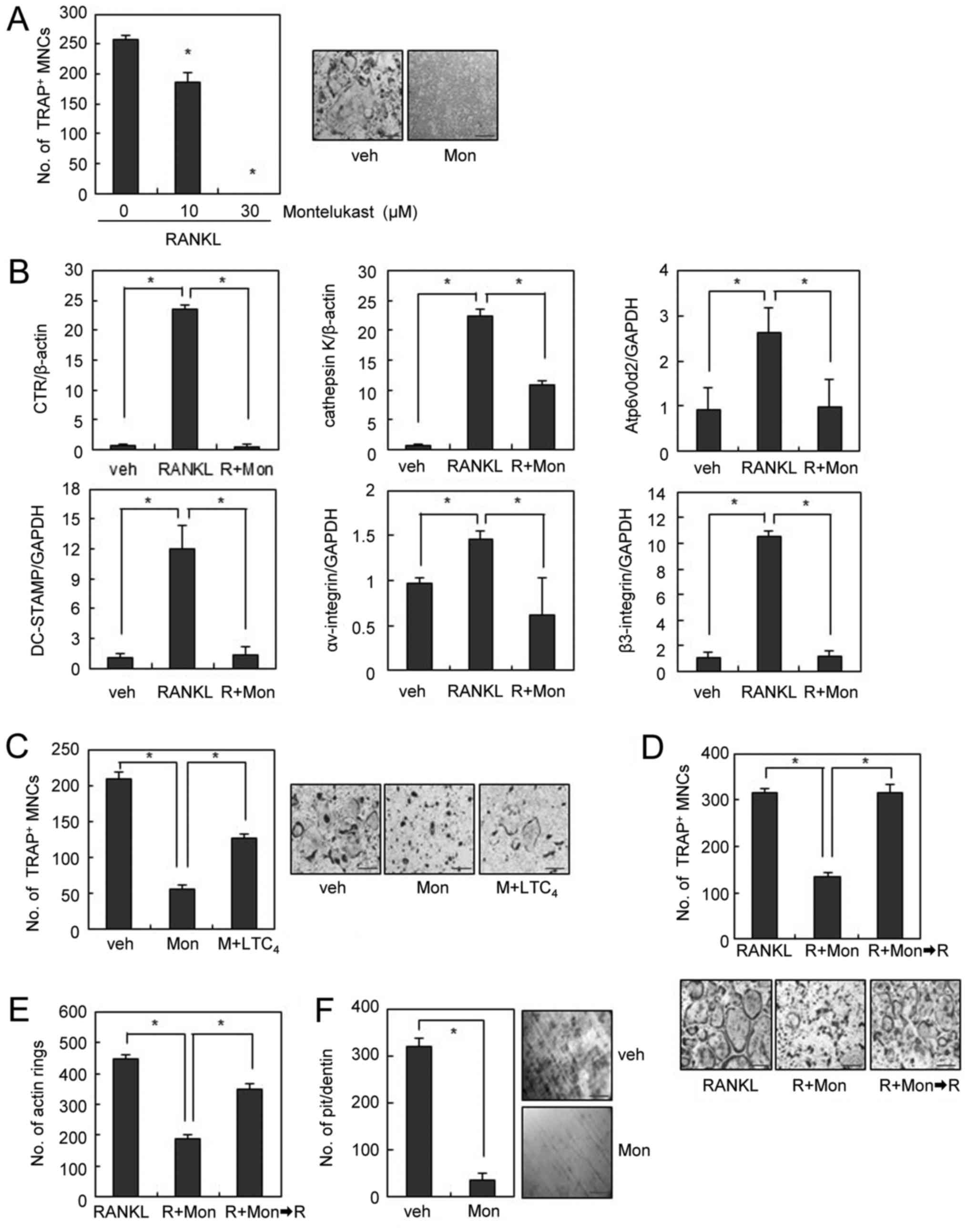 | Figure 1.Montelukast inhibits RANKL-induced OC
formation and bone resorption. (A) BMMs were cultured with M-CSF
(30 ng/ml) and RANKL (100 ng/ml) in the absence or presence of
montelukast for 4 days and stained for TRAP activity. TRAP-positive
cells that had >3 nuclei were counted as OCs. (B) mRNA
expression was determined by reverse transcription-quantitative
polymerase chain reaction. (C) BMMs were cultured with montelukast
(30 µM) in the absence or presence of leukotriene C4 (50
nM) during osteoclast differentiation and stained for TRAP
activity. (D and E) Mature OCs were treated with montelukast (30
µM). Following 24 h, montelukast was removed from the culture
medium and mature OCs were cultured for 24 h. (D) Actin rings or
(E) OCs were stained with rhodamine-conjugated phalloidin or TRAP
staining solution. (F) BMMs were differentiated on dentine slices
with M-CSF (30 ng/ml) and RANKL (100 ng/ml) for 3 days, and treated
with montelukast (30 µM) for 4 days. The resorbed pit numbers were
counted. Scale bars=200 µm. Data are expressed as mean ± standard
deviation from at least three independent experiments. *P<0.05,
as indicated. RANKL, receptor activator of nuclear factor-κB
ligand; OCs, osteoclasts; BMM, bone marrow-derived macrophages;
TRAP, tartrate resistant acid phosphatase; veh, vehicle; R, RANKL;
Mon, montelukast. |
Since montelukast is known to block the effects of
CysLTs by competitive binding to CysLTR1 (11), we next investigated whether
addition of CysLT rescues the inhibitory effect of montelukast on
OC formation. Suppression of osteoclastogenesis by montelukast was
partly rescued by addition of exogenous LTC4, suggesting
the partial involvement of CysLTR1 in the inhibition of OC
formation by montelukast (Fig.
1C).
We next investigated the effect of montelukast on
mature OCs. Mature OCs exhibit highly polarized morphological
features, which is an essential event to initiate bone resorption
(18). When mature OCs were
incubated with RANKL, a clear cytoplasm and a smooth periphery were
observed in polarized OCs. Conversely, the presence of montelukast
induced morphological changes such as a contracted cytoplasm and an
irregular cell periphery within 24 h (Fig. 1D). When we further observed the
morphology of OCs after removal of montelukast from the culture
medium, the number of mature OCs with a clear cytoplasm and a
smooth periphery increased along with the removal of montelukast
(Fig. 1D). Since actin rings are
believed to be one of the markers of polarized OCs (3), we examined whether the effect of
montelukast on actin ring formation was involved in the
morphological changes of OCs. F-actin staining showed that
montelukast efficiently disrupted the formation of actin rings in
mature OCs, which was recovered after removal of montelukast
(Fig. 1E). These data suggest that
montelukast reversibly induces the disruption of actin rings,
causing reversible morphological changes of mature OCs. To
investigate whether the effect of montelukast on the morphology of
OCs could be reflected in the osteoclastic activity, we performed
an in vitro resorption pit assay using dentine slices. Many
resorption pits were generated by RANKL-treated OCs (Fig. 1F). In contrast, montelukast
treatment strongly inhibited the formation of resorption pits by
RANKL-treated OCs (Fig. 1F).
Together, these results suggest that the CysLTR1 antagonist exerts
inhibitory effects on the morphology of OCs, which leads to reduced
bone resorption.
CysLTR1 antagonist regulates
osteoclastogenesis through NFATc1
The NFATc1 pathway plays a critical and fundamental
role in OC development and the lack of NFATc1 arrests
osteoclastogenesis (8). Thus, we
investigated the effect of montelukast on the NFATc1 pathway.
Montelukast abolished RANKL-induced NFATc1 expression in BMMs
(Fig. 2A). We next investigated
whether the effect of montelukast was rescued by the overexpression
of NFATc1. The NFATc1-transduced BMMs were cultured with M-CSF and
RANKL in the absence or presence of montelukast for 3 days. As
shown in Fig. 2B, suppression of
osteoclastogenesis by montelukast was efficiently overcome by the
forced expression of NFATc1, suggesting that the
anti-osteoclastogenic effect of montelukast is mainly due to a
reduction in NFATc1 expression.
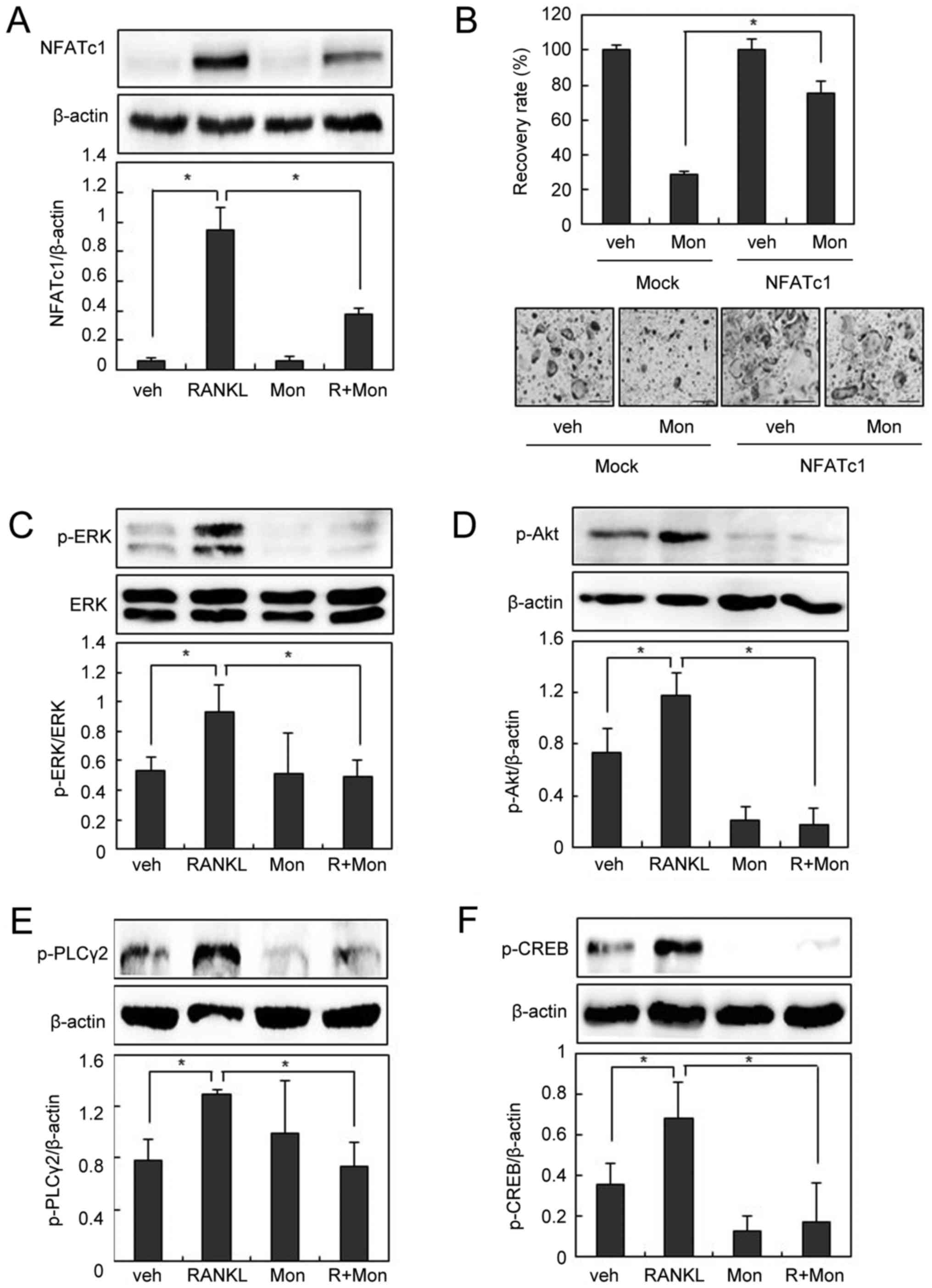 | Figure 2.Montelukast suppresses RANKL-induced
NFATc1 expression. (A) BMMs were pre-incubated in the absence or
presence of montelukast (30 µM) for 30 min and then cultured with
or without RANKL (200 ng/ml) for 24 h. Cell lysates were subjected
to western blotting analysis. (B) BMMs were infected through a
retrovirus packaging system. Infected BMMs were cultured with RANKL
(100 ng/ml) and macrophage colony-stimulating factor (30 ng/ml) in
the presence or absence of montelukast (30 µM) for 3 days.
TRAP-positive cells that had >3 nuclei were counted. The
recovery rate was defined as the percentage of OC formation in the
presence of montelukast. BMMs were pre-incubated in the absence or
presence of montelukast (30 µM) for 30 min, and then treated with
or without 200 ng/ml of RANKL for 15 min (p-ERK, p-Akt, and
p-PLCγ2) or 3 h (p-CREB). Cell lysates were then subjected to
western blot analysis for (C) p-ERK and ERK, (D) p-Akt, (E) p-
PLCγ2 and (F) p-CREB. Scale bars=200 µm. Data are expressed as
means standard deviation from at least three independent
experiments. *P<0.05, as indicated. RANKL, receptor activator of
nuclear factor-κB ligand; OCs, osteoclasts; BMM, bone
marrow-derived macrophages; TRAP, tartrate resistant acid
phosphatase; NFATc1, nuclear factor of activated T cells,
cytoplasmic 1; p-, phosphorylated; ERK, extracellular
signal-regulated kinase; Akt, protein kinase B; PLCγ2,
phospholipase Cγ2; CREB, cyclic adenosine monophosphate response
element-binding protein; veh, vehicle; R, RANKL; Mon,
montelukast. |
To identify the molecular mechanism underlying the
inhibitory effects of montelukast on osteoclastogenesis, we next
examined the effects of montelukast on early signaling pathways
such as ERK, Akt, and/or phospholipase Cγ2 (PLCγ2), which activate
the NFATc1 pathway (5–7).
As shown in Fig.
2C-E, phosphorylation of ERK, Akt, and PLCγ2 was observed 15
min after RANKL treatment, but suppressed by pretreatment with
montelukast. PLCγ2 phosphorylation increases intracellular calcium
levels, which in turn activates calcium-dependent transcriptional
factors such as cAMP response element-binding protein (CREB)
(7,19). Since CREB activation plays an
essential role in NFATc1 induction during osteoclastogenesis
(9,10), we further examined whether
montelukast inhibits CREB activation. RANKL stimulation led to CREB
phosphorylation, and montelukast interfered with this process
(Fig. 2F). Taken together, these
data suggest that montelukast regulates NFATc1 via ERK, Akt, and
PLCγ2 signaling pathways.
CysLTR1 antagonist regulates
osteoclastogenesis via CysLTR1 and P2Y12
Since the inhibition of osteoclastogenesis by
montelukast was only partially rescued by LTC4 (Fig. 1C), we speculated that the
inhibitory effect of montelukast on OC formation is likely to
involve mechanisms other than CysLTR1 blockage. In an effort to
identify an alternative receptor for montelukast, we found that the
addition of adenosine diphosphate (ADP), a P2Y12 agonist, also
rescued the blockade of OC formation by montelukast (Fig. 3A). These data suggest that the
inhibitory effect of montelukast on osteoclastogenesis was partly
due to its suppressive actions on P2Y12 receptor. To confirm the
regulatory role of P2Y12 on OC formation, we next investigated the
involvement P2Y12 receptor in OC differentiation. MRS2395, a P2Y12
antagonist, decreased the formation of TRAP-positive OCs in a
dose-dependent manner (Fig. 3B).
The suppression of osteoclastogenesis by MRS2395 was successfully
rescued by addition of exogenous ADP, suggesting the involvement of
P2Y12 in the inhibition of OC formation by MRS2395 (Fig. 3C). Similar inhibitory effects on
osteoclastogenesis were also obtained by using P2Y12-specific
siRNAs (Fig. 3D). Accordingly, the
RANKL-induced expression of various OC markers was suppressed by
MRS2395 treatment (Fig. 3E).
Furthermore, MRS2395 significantly inhibited the resorption pit
formation activity of OCs in dentine slices (Fig. 3F).
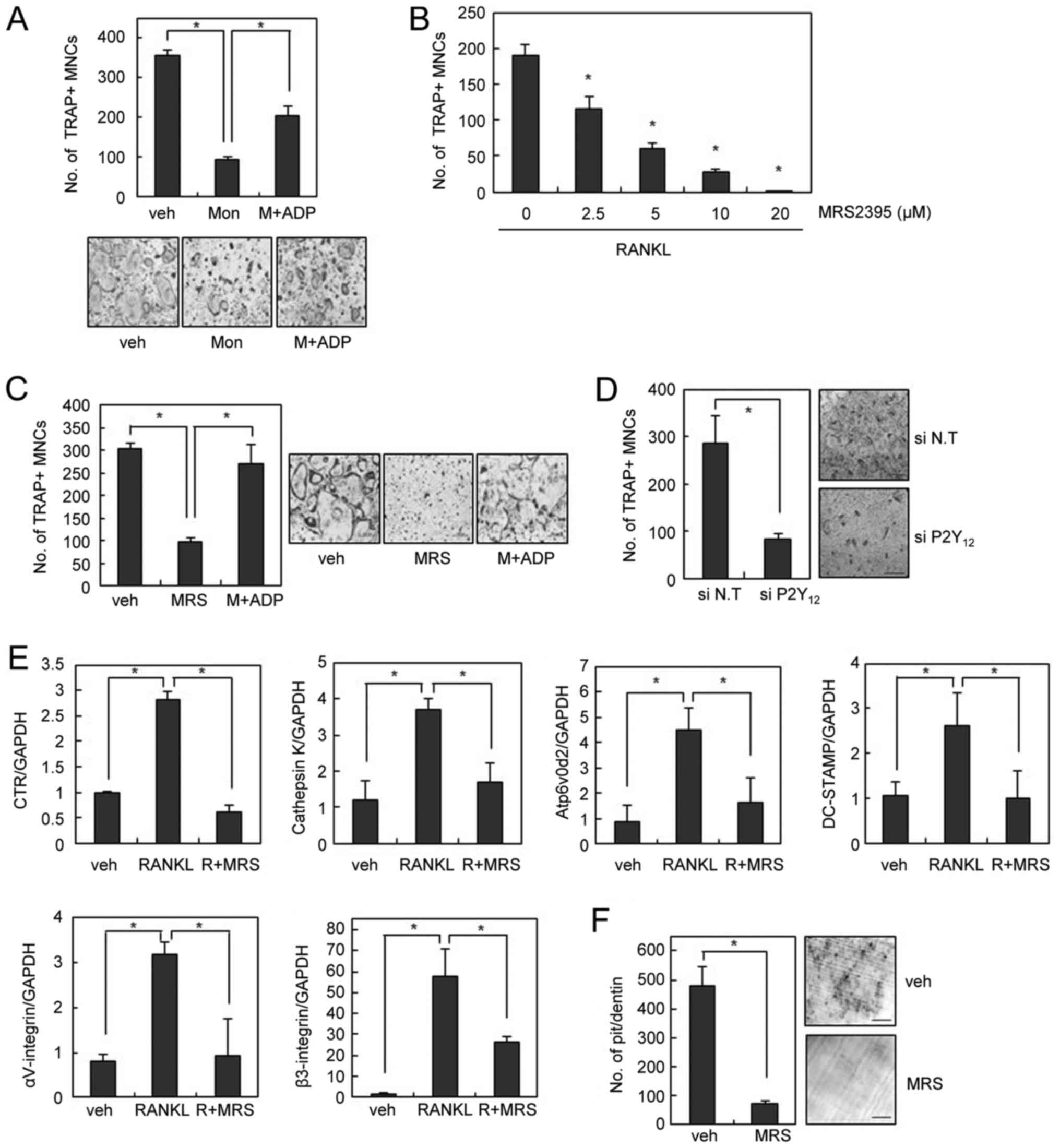 | Figure 3.P2Y12 blockade inhibits RANKL-induced
OC formation and bone resorption. (A) BMMs were cultured with
montelukast (30 µM) in the absence or presence of 2-MeS-ADP (10 µM)
during OC differentiation and stained for TRAP activity. (B) BMMs
were cultured with M-CSF (30 ng/ml) and RANKL (100 ng/ml) in the
absence or presence of MRS2395 for 4 days. (C) BMMs were cultured
with MRS2395 (20 µM) in the absence or presence of 2-MeS-ADP during
OC differentiation. (D) BMMs were transfected with 40 nM siRNA. The
siRNA-transfected BMMs were cultured with M-CSF (30 ng/ml) and
RANKL (100 ng/ml) for 3 days. (E) mRNA expression was determined by
reverse transcription-quantitative polymerase chain reaction. (F)
The resorbed pit numbers on dentine slices in the absence or
presence of MRS2395 (20 µM) were counted. Scale bars=200 µm. Data
are expressed as means ± standard deviation from at least three
independent experiments. *P<0.05, as indicated. RANKL, receptor
activator of nuclear factor-κB ligand; OCs, osteoclasts; BMM, bone
marrow-derived macrophages; TRAP, tartrate resistant acid
phosphatase; siRNA, small interfering RNA; veh, vehicle; R, RANKL;
Mon, montelukast; MRS, MRS2395; M-CSF, macrophage
colony-stimulating factor. |
We next examined the effect of P2Y12 blockade on
RANKL-induced NFATc1 expression. Pharmacological P2Y12 inhibition
by MRS2395 or down-regulation of P2Y12 by siRNA decreased
RANKL-induced NFATc1 expression (Fig.
4A and B). In addition, suppression of osteoclastogenesis by
MRS2395 was successfully overcome by the forced expression of
NFATc1 (Fig. 4C). MRS2395
abolished RANKL-induced activation of ERK and Akt, as montelukast
did (Fig. 4D and E). Taken
together, these data suggest that P2Y12 is involved in OC
differentiation, and the inhibitory effect of montelukast on OC
formation is partly due to P2Y12 blockade.
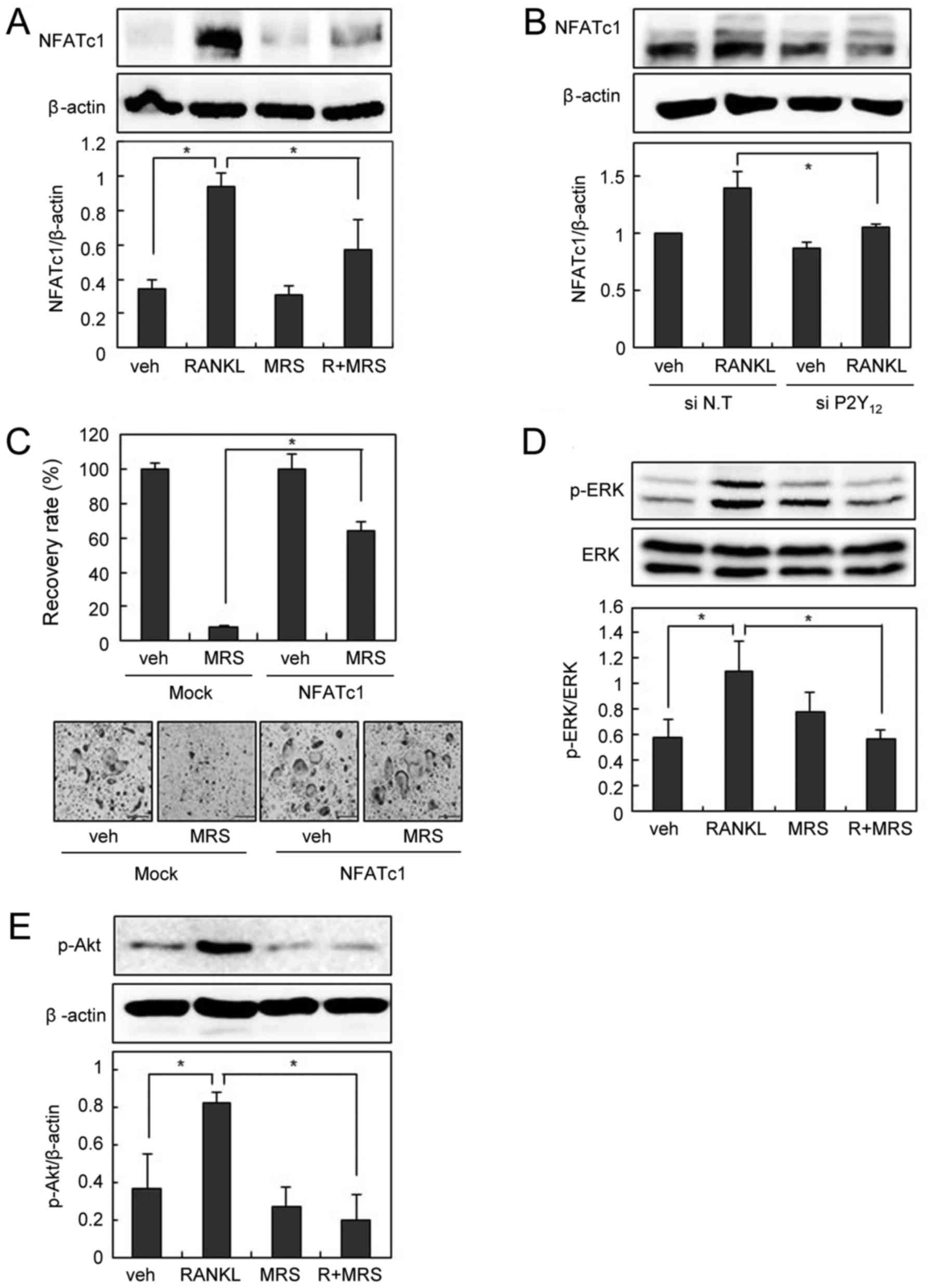 | Figure 4.P2Y12 blockade suppresses
RANKL-induced NFATc1 expression. (A) BMMs were pre-incubated in the
absence or presence of MRS2395 (20 µM) for 30 min and then cultured
with or without RANKL (200 ng/ml) for 24 h. Cell lysates were
subjected to western blotting analysis. (B) The siRNA-transfected
BMMs were cultured with RANKL (200 ng/ml) for 24 h. Cell lysates
were then subjected to western blotting analysis. (C)
Retrovirus-infected BMMs were cultured with RANKL (100 ng/ml) and
M-CSF (30 ng/ml) in the presence or absence of MRS2395 (20 µM) for
3 days. The recovery rate was defined as the percentage of
osteoclast formation in the presence of MRS2395. BMMs were
pre-incubated in the absence or presence of MRS2395 (20 µM) for 30
min, and then treated with or without 200 ng/ml of RANKL for 15
min. Cell lysates were then subjected to western blotting analysis
for (D) p-ERK and ERK, and (E) p-Akt. Scale bars=200 µm. Data are
expressed as means ± standard deviation from at least three
independent experiments. *P<0.05, as indicated. RANKL, receptor
activator of nuclear factor-κB ligand; OCs, osteoclasts; BMM, bone
marrow-derived macrophages; TRAP, tartrate resistant acid
phosphatase; NFATc1, nuclear factor of activated T cells,
cytoplasmic 1; p-, phosphorylated; ERK, extracellular
signal-regulated kinase; Akt, protein kinase B; siRNA, small
interfering RNA; veh, vehicle; R, RANKL; MRS, MRS2395. |
CysLTR1 antagonist prevented
ovariectomy- or unloading-induced bone loss in vivo
LPS is produced by gram-negative bacteria and
induces the secretion of TNF and other inflammatory cytokines,
leading to systemic inflammatory response. Injection of LPS in
animals can mimic OC-mediated bone destruction that occurs under
sepsis (20). To further
investigate the anti-osteoclastogenic effect of the CysLTR1
antagonist in vivo, we injected LPS into the supracalvarial
region of mice with or without montelukast. TRAP staining of whole
calvariae showed that LPS dramatically increased OC numbers
(Fig. 5A and B). In parallel with
its anti-osteoclastogenic effect in vitro, montelukast
notably reduced LPS-induced OC formation (Fig. 5A and B). These data suggest that
the CysLTR1 antagonist has an inhibitory effect on
inflammation-induced osteoclastogenesis in vivo.
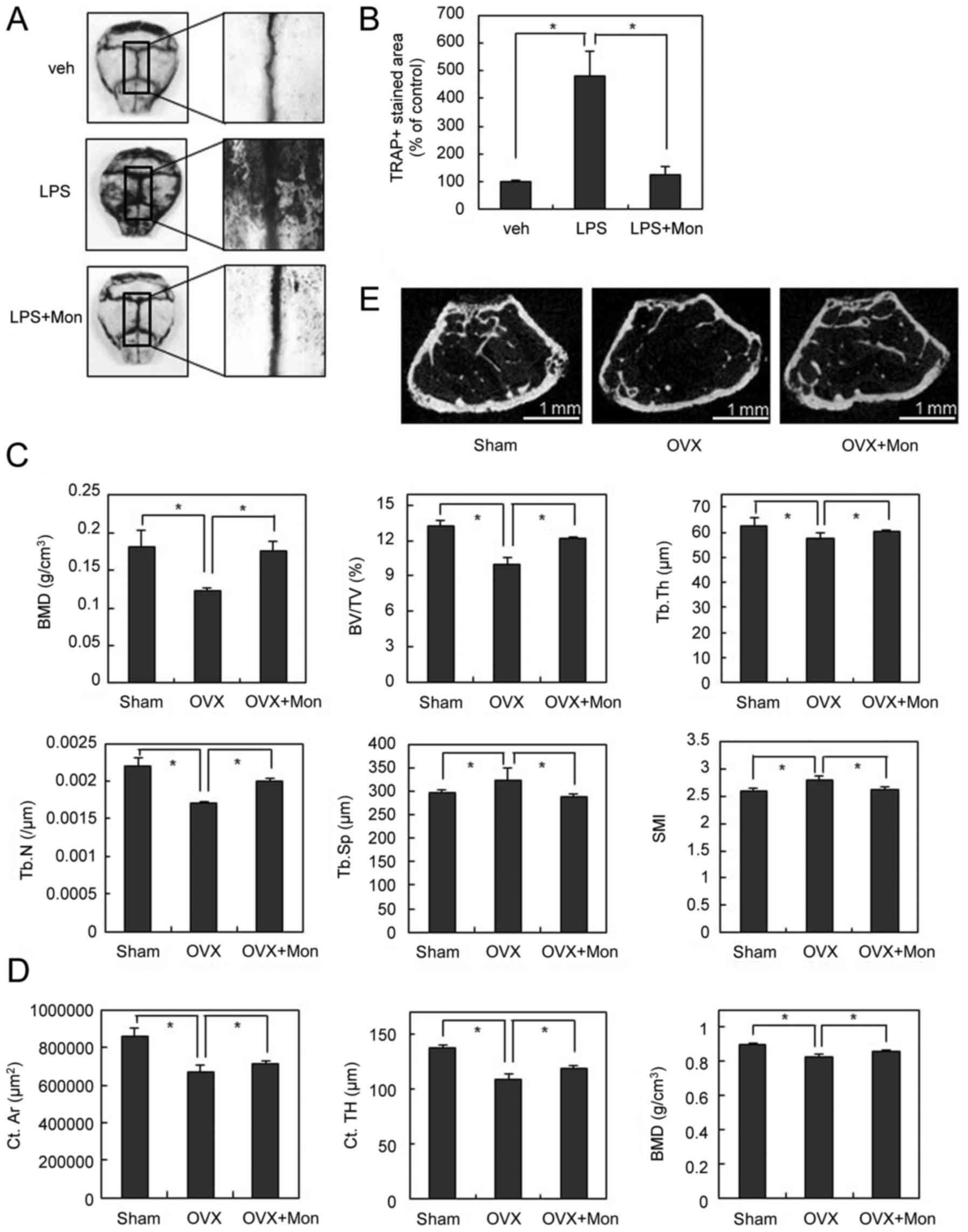 | Figure 5.Montelukast inhibits OC formation and
OVX-induced bone loss in vivo. (A) Calvariae of mice that
received vehicle, LPS or LPS plus montelukast (5 mg/kg) were
subjected to TRAP staining. (B) TRAP-positive stained areas in
calvariae were quantified by using ImageJ. (C) The femurs of
sham-operated, OVX and OVX plus montelukast mice were collected
following 12 weeks. Various bone parameters were analyzed by
micro-CT. (D) Two-dimensional micro-CT images of the distal
metaphysis of the femur. (E) Micro-CT analysis for cortical bone.
Scale bars=1 mm. Data are expressed as means ± standard deviation
from at least three independent experiments. *P<0.05, as
indicated. OCs, osteoclasts; TRAP, tartrate resistant acid
phosphatase; LPS, lipopolysaccharide; OVX, ovariectomy; CT,
computed tomography; BMD (g/cm3), bone mineral density;
BV/TV (%), bone volume per tissue volume; Tb.Th (µm), trabecular
thickness; Tb.N (/µm), trabecular number; Tb.Sp (µm), trabecular
separation; SMI, structural model index; Ct.Ar (µm2),
cortical area; Ct.TH (µm), cortical thickness; BMD
(g/cm3); veh, vehicle; Mon, montelukast. |
To further explore the therapeutic effects of
montelukast on pathological bone loss, we next used the
ovariectomized (OVX) or hind limb unloading mouse model to mimic
menopause- or disuse-induced osteoporosis. The µCT analysis
demonstrated that OVX significantly decreased the values of bone
mineral density (BMD), bone volume/tissue volume (BV/TV),
trabecular number (Tb.N), and trabecular thickness (Tb.Th), and
increased the values of trabecular separation (Tb.Sp) compared with
the sham operation. In contrast, in OVX mice, montelukast treatment
significantly inhibited the OVX-induced bone loss assessed by
measuring these parameters (Fig.
5C). In addition, the higher structure model index (SMI)
number, an indicator of increased fragility, in OVX mice was
significantly decreased by montelukast treatment (Fig. 5C). Similar results were
demonstrated by a two-dimensional visualization of the femoral area
(Fig. 5D). Furthermore, µCT
analysis for cortical bone confirmed that OVX significantly
decreased the values of cortical area (Ct.Ar), cortical thickness
(Ct.TH), and BMD compared with the sham operation (Fig. 5E). By treating montelukast,
OVX-induced cortical bone loss was suppressed (Fig. 5E). Montelukast also showed the
therapeutic effects in disuse osteoporosis. Two weeks after hind
limb unloading, the bone loss was less severe in
montelukast-treated mice than that in the vehicle-treated mice
(Fig. 6A). Montelukast-treated
mice showed higher BMD compared to vehicle-treated mice (Fig. 6B). In addition, unloading-induced
structural bone alterations, including decreases in BV/TV, Tb.Th,
and Tb.N and increases in Tb.Sp and SMI were abolished by
montelukast treatment (Fig. 6B).
Furthermore, µCT analysis for cortical bone confirmed that
unloading condition significantly decreased the values of cortical
area (Ct.Ar), cortical thickness (Ct.TH), and BMD compared with the
vehicle group mice. By treating montelukast, unloading-induced
cortical bone loss was suppressed (Fig. 6C). Taken together, these results
suggest that the CysLTR1 antagonist prevented OVX- or hind limb
unloading-induced bone destruction in vivo.
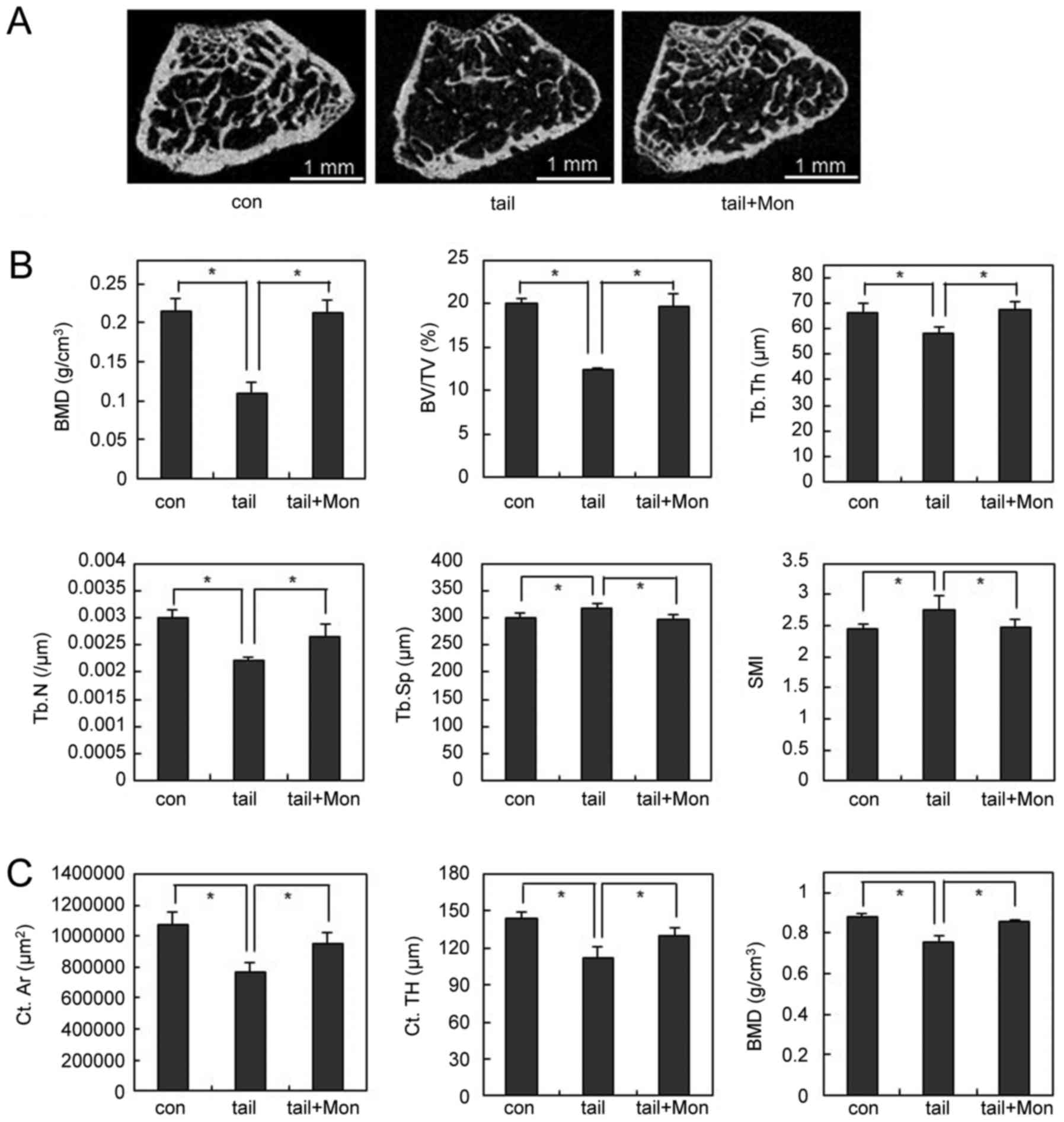 | Figure 6.Montelukast prevents
unloading-induced bone loss in vivo. The femurs of control,
unloading, and unloading plus montelukast mice were collected
following 2 weeks. (A) Two-dimensional micro-CT images of the
distal metaphysis of the femurs. Scale bars=1 mm. (B) Various bone
parameters of femurs were analyzed by micro-CT, including BMD
(g/cm3), BV/TV (%), Tb.Th (µm), Tb.N (/µm), Tb.Sp (µm)
and SMI. (C) Micro-CT analysis for cortical bone. Data are
expressed as means ± standard deviation from at least three
independent experiments. *P<0.05, as indicated. CT, computed
tomography; BMD (g/cm3), bone mineral density; BV/TV
(%), bone volume per tissue volume; Tb.Th (µm), trabecular
thickness; Tb.N (/µm), trabecular number; Tb.Sp (µm), trabecular
separation; SMI, structural model index; Ct.Ar (µm2),
cortical area; Ct.TH (µm), cortical thickness; BMD
(g/cm3); con, control; tail, hind limb unloading; Mon,
montelukast. |
Discussion
Osteoporosis is a bone disease with low bone mass
and bone fragility that increases the risk of bone fracture. It is
characterized as progressive and excessive bone resorption caused
by enhanced OC differentiation and/or activity (1,2).
RANKL is a critical factor for OC differentiation, and excessive
RANKL signaling results in enhanced OC formation and bone
resorption (3,4). As such, downregulation of RANKL
expression or its downstream signals may be a valuable approach to
the treatment of osteoporosis (2–4).
The CysLTs are a family of potent inflammatory lipid
mediators derived from arachidonic acid through the 5-LO pathway
(11). The CysLTR1 is a G-protein
coupled receptor (GPCRs) and its interaction with CysLTs plays a
central role in the pathophysiology of asthma and other
inflammatory diseases (11–13).
Several CysLTR1 antagonists have been developed to date and are
currently in clinical practice (13,21).
Montelukast, probably due to its once daily dosing schedule,
safety, and efficacy profile, is the most widely prescribed CysLTR1
antagonist worldwide. These agents are specifically used in the
treatment of allergic rhinitis, exercise- and aspirin-induced
asthma, and as add-on therapy for patients with asthma poorly
controlled by inhaled corticosteroid (ICS) monotherapy or ICS in
combination with long-acting β2-agonists (21,22).
In this study, we showed that montelukast potently
prevented RANKL-induced OC formation and bone loss in vivo.
Montelukast dramatically downregulated RANKL-induced expression of
NFATc1 in BMM culture. The forced expression of NFATc1 completely
restored the inhibition of osteoclastogenesis by montelukast,
suggesting that the inhibitory effect of montelukast is mediated at
least partially through NFATc1.
NFATc1 activation requires activation of the MAPKs,
Akt, and/or PLCγ2 signaling (5–7). Our
results further suggest that montelukast suppresses RANKL-induced
NFATc1 expression at least in part by inhibiting the ERK, Akt, and
PLCγ2 pathways in BMMs. Upon RANKL stimulation, immunoreceptor
tyrosine-based activation motif (ITAM)-bearing adaptors, Fc
receptor common γ subunit (FcRγ) and DNAX-activating protein 12
(DAP12), deliver co-stimulatory signals through activation of PLCγ.
Activated PLCγ generates inositol-1,4,5- triphosphate
(IP3), which mobilizes Ca2+ from the
endoplasmic reticulum stores through inositol triphosphate
receptors (IP3Rs) (23–26)
and, subsequently, generates Ca2+ oscillation. This
RANKL-induced Ca2+ oscillation activates CaMKIV,
followed by CREB activation (9),
which is critical for the activation of NFATc1 activation. Thus,
PLCγ2/Ca2 +/CREB/NFATc1 signaling is an important
pathway for OC differentiation and function in addition to MAPK
and/or Akt pathways. Given that the interaction of CysLTs with
CysLTR1 evokes Ca2 + oscillation (26,27),
montelukast seems to suppress OC formation partly by attenuating
the PLCγ2/Ca2 +/CREB/NFATc1 signaling pathway.
Recent evidence suggests that montelukast possesses
a range of secondary activities independent of CysLTR1, including
inhibition of 5-LO, histone acetyltransferase, cAMP
phosphodiesterase, and interference with purinergic P2Y12 receptors
(28). In particular, phylogenetic
analysis of the P2Y12 and CysLTR1 indicated that CysLTR1 is closely
related to P2Y12 (29).
Furthermore, a computer model predicted that CysLTs act as
surrogate ligands for the P2Y12 receptor (30), suggesting that CysLTR1 antagonists
interact functionally with P2Y12 receptor signaling pathways. In
this study, we demonstrated that the inhibitory effect of
montelukast on osteoclastogenesis was efficiently restored by
addition of P2Y12 agonist and by that of CysLT. Furthermore, P2Y12
antagonist inhibited osteoclast formation through signaling
pathways similar to those induced by montelukast. Thus, we suggest
that montelukast might exert its effect on OC formation by
antagonizing the CysLTR1 and P2Y12 signaling.
Drug repositioning is progressively getting
attention as a promising method for drug discovery. A repositioned
compound with proven bioavailability and known safety profile
presents many advantages such as an accelerated R&D process,
reduced development cost, and decreased failure rate due to safety
(31). Montelukast is an
FDA-approved anti-asthmatic drug used to treat asthma and allergic
rhinitis. Herein, we show that montelukast inhibits
osteoclastogenesis by suppressing the expression of NFATc1 via ERK,
Akt, and PLCγ2 pathways. Furthermore, montelukast prevented
ovariectomy- or disuse-induced bone loss. Taken together, our data
suggest that montelukast is a potential repositioned drug for
treating bone diseases associated with excessive bone
resorption.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Korean Health Technology R&D Project, Ministry of Health &
Welfare, Republic of Korea (grant no. HI14C24470000) and from the
National Research Foundation of Korea (NRF), funded by Ministry of
Science, ICT and Future Planning (MSIP; grant nos.
NRF-2014M1A3A3A02034917 and NRF-2016R1A2B4011636).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY participated in research design. JHK conducted
the experiments. JHK, HL, DSL and MY performed data analysis. MY
wrote the manuscript.
Ethics approval and consent to
participate
All experiments were performed in accordance with
institutional guidelines approved by the Sookmyung Women's
University Care and Use Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zaidi M: Skeletal remodeling in health and
disease. Nat Med. 13:791–801. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodan GA and Martin TJ: Therapeutic
approaches to bone diseases. Science. 289:1508–1514. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suda T, Takahashi N, Udagawa N, Jimi E,
Gillespie MT and Martin TJ: Modulation of osteoclast
differentiation and function by the new members of the tumor
necrosis factor receptor and ligand families. Endocr Rev.
20:345–357. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee ZH and Kim HH: Signal transduction by
receptor activator of nuclear factor kappa B in osteoclasts.
Biochem Biophys Res Commun. 305:211–214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon JB, Kim JH, Kim K, Youn BU, Ko A, Lee
SY and Kim N: Akt induces osteoclast differentiation through
regulating the GSK3β/NFATc1 signaling cascade. J Immunol.
188:163–169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Faccio R and Cremasco V: PLCgamma2: Where
bone and immune cells find their common ground. Ann N Y Acad Sci.
1192:124–130. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takayanagi H, Kim S, Koga T, Nishina H,
Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al:
Induction and activation of the transcription factor NFATc1 (NFAT2)
integrate RANKL signaling in terminal differentiation of
osteoclasts. Dev Cell. 3:889–901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sato K, Suematsu A, Nakashima T,
Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K,
Yamaguchi A, Takai T, et al: Regulation of osteoclast
differentiation and function by the CaMK-CREB pathway. Nat Med.
12:1410–1416. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Negishi-Koga T and Takayanagi H:
Ca2+-NFATc1 signaling is an essential axis of osteoclast
differentiation. Immunol Rev. 131:241–256. 2009. View Article : Google Scholar
|
|
11
|
Singh RK, Gupta S, Dastidar S and Ray A:
Cysteinyl leukotrienes and their receptors: Molecular and
functional characteristics. Pharmacology. 85:336–349. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nayak A: A review of montelukast in the
treatment of asthma and allergic rhinitis. Expert Opin
Pharmacother. 5:679–686. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Theron AJ, Steel HC, Tintinger GR, Gravett
CM, Anderson R and Feldman C: Cysteinyl leukotriene receptor-1
antagonists as modulators of innate immune cell function. J Immunol
Res. 2014:6089302014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hikiji H, Takato T, Shimizu T and Ishii S:
The roles of prostanoids, leukotrienes and platelet-activating
factor in bone metabolism and disease. Prog Lipid Res. 47:107–126.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–208. 2001.
View Article : Google Scholar
|
|
16
|
Ishijima M, Rittling SR, Yamashita T,
Tsuji K, Kurosawa H, Nifuji A, Denhardt DT and Noda M: Enhancement
of osteoclastic bone resorption and suppression of osteoblastic
bone formation in response to reduced mechanical stress do not
occur in the absence of osteopontin. J Exp Med. 193:399–404. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Virchow JC and Bachert C: Efficacy and
safety of montelukast in adults with asthma and allergic rhinitis.
Respir Med. 100:1952–1959. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wagner EF and Karsenty G: Genetic control
of skeletal development. Curr Opin Genet Dev. 11:527–532. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wada T, Nakashima T, Hiroshi N and
Penninger JM: RANKL-RANK signaling in osteoclastogenesis and bone
disease. Trends Mol Med. 12:17–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ha H, Lee JH, Kim HN, Kim HM, Kwak HB, Lee
S, Kim HH and Lee ZH: alpha-Lipoic acid inhibits inflammatory bone
resorption by suppressing prostaglandin E2 synthesis. J Immunol.
176:111–117. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riccioni G, Bucciarelli T, Mancini B, Di
Ilio C and D'Orazio N: Antileukotriene drugs: Clinical application,
effectiveness and safety. Curr Med Chem. 14:1966–1977. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Paggiaro P and Bacci E: Montelukast in
asthma: A review of its efficacy and place in therapy. Ther Adv
Chronic Dis. 2:47–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim N, Takami M, Rho J, Josien R and Choi
Y: A novel member of the leukocyte receptor complex regulates
osteoclast differentiation. J Exp Med. 195:201–209. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koga T, Inui M, Inoue K, Kim S, Suematsu
A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al:
Costimulatory signals mediated by the ITAM motif cooperate with
RANKL for bone homeostasis. Nature. 428:758–763. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ferron M, Boudiffa M, Arsenault M, Rached
M, Pata M, Giroux S, Elfassihi L, kisseleva M, Majerus PW, Rousseau
F and Vacher J: Inositol polyphosphate 4-phosphatase B as a
regulator of bone mass in mice and humans. Cell Metab. 14:466–477.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim H, Kim T, Jeong BC, Cho IT, Han D,
Takegahara N, Negishi-Koga T, Takayanagi H, Lee JH, Sul JY, et al:
Tmem64 modulates calcium signaling during RANKL-mediated osteoclast
differentiation. Cell Metab. 17:249–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Di Capite J, Shirley A, Nelson C, Bates G
and Parekh AB: Intercellular Ca2+ wave propagation involving
positive feedback between CRAC channels and cysteinyl leukotrienes.
FASEB J. 3:894–905. 2009. View Article : Google Scholar
|
|
28
|
Tintinger GR, Feldman C, Theron AJ and
Anderson R: Montelukast: More than a cysteinyl leukotriene receptor
antagonist? Sci World J. 10:2403–2413. 2010. View Article : Google Scholar
|
|
29
|
Fredriksson R, Lagerstrom MC, Lundin LG
and Schioth HB: The G-protein-coupled receptors in the human genome
form five main families. Phylogenetic analysis, paralogon groups
and fingerprints. Mol Pharmacol. 63:1256–1272. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nonaka Y, Hiramoto T and Fujita N:
Identification of endogenous surrogate ligands for human P2Y12
receptors by in silico and in vitro methods. Biochem Biophys Res
Commun. 337:281–288. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Finsterer J and Frank M: Repurposed drugs
in metabolic disorders. Curr Top Med Chem. 13:2386–2394. 2013.
View Article : Google Scholar : PubMed/NCBI
|














