Introduction
Alzheimer's disease (AD) is the most common form of
neurological disorder, presenting symptoms of irreversible and
progressive memory loss as well as cognitive decline as a result of
the death of brain cells (1). In
2017, a total of 5.5 million people suffered from AD in the USA, of
which 5.3 million were aged ≥65 years old (2). However, not all brain regions
associated with AD are simultaneously affected to the same extent
during disease progression (3,4).
Therefore, it is of critical importance to determine the different
underlying mechanisms affecting different brain regions during
AD.
As high-throughput technologies have advanced,
multi-level omics data are subsequently becoming widely available
(5). Due to the increasing
production of large-scale functional genomic datasets, single
biomarkers for AD are now being determined on a genome-wide scale
(6,7). Cellular genes demonstrating a
correlation in expression changes are likely to function as a gene
pair that perform similar functions (8,9).
However, gene pairs can be present in biological networks that
interact with numerous molecular regulators, such as micro (mi)RNAs
and long non-coding (lnc)RNAs, which serve important roles in
regulating transcription, post-transcriptional modification and
translation as well as non-coding (nc)RNAs in humans (10–12).
LncRNAs have numerous structural features that are similar to
messenger (m)RNAs; for example, lncRNAs may recognize complementary
sequences of mRNAs, and thus regulate mRNA processing (13). Whereas miRNAs negatively regulate
gene expression via the targeting of mRNAs (14). However, interactions between miRNAs
and mRNAs are not unidirectional, and mRNAs and lncRNAs (15) can function as competing endogenous
(ce)RNAs for binding with miRNAs (16). For example, the cell division cycle
42 AD associated gene and the ribonuclease P RNA component H1
lncRNA represent a compensatory mechanism in the early stage of AD
pathogenesis (17). Furthermore,
modularity has been considered to represent an important indicator
for the identification of key molecules present in multi-level
ncRNA-mediated gene regulatory networks in order to further the
understanding of AD in different brain regions.
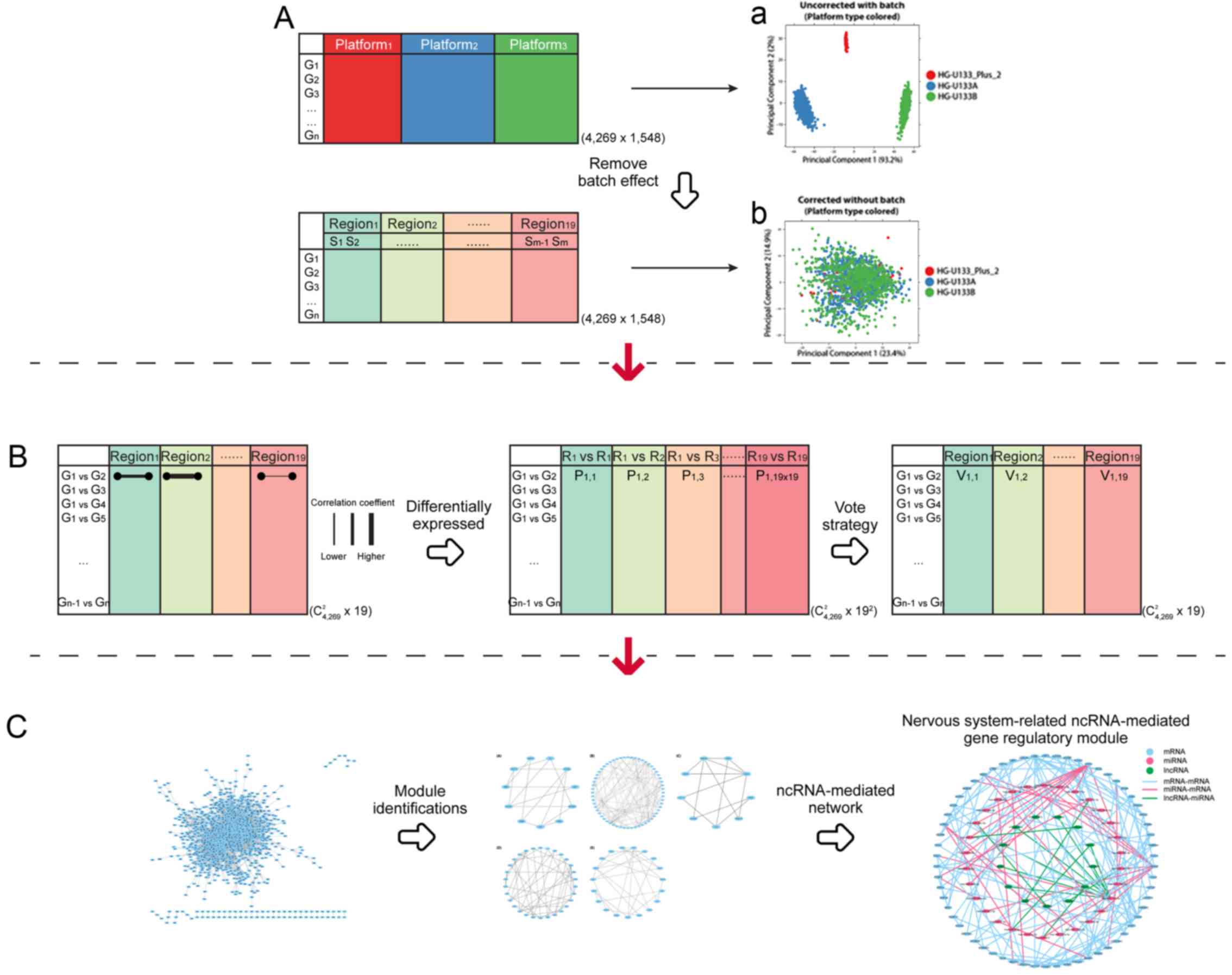
In the present study, based on 1,548 samples from a
cohort of 90 patients with AD spanning 19 brain regions from Gene
Expression Omnibus (GEO) database (database no. GSE84422) (3), a gene pair-based method was
established for the classification of 19 brain regions into
different groups according to exhibited symptoms associated with AD
(Fig. 1). Principal component
analysis (PCA) revealed that there were marked differences in the
effect of AD on the amygdala, caudate nucleus, nucleus accumbens,
precentral gyrus, putamen and temporal pole brain regions as well
as a cluster of a further 13 brain regions disparately affected by
AD. Five functional modules were identified to distinguish these
regional groupings based on the most strongly associated gene-pairs
revealed by PCA. Functional annotation of genes in two of the
modules suggested a strong correlation with pathways associated
with the nervous system, including cholinergic synapses, circadian
entrainment and dopaminergic synapses. Notably, following
integration of the two modules with interactions between miRNAs,
lncRNAs and mRNAs, as revealed by RNA Association Interaction
Database (RAID) v.2.0 analysis (18), one module demonstrated a close
association with miRNAs, which, on the contrary, interacted with
numerous lncRNAs associated with AD, such as metastasis associated
lung adenocarcinoma transcript 1 (MALAT1) and non-protein
coding taurine upregulated 1 (TUG1), which suggested that
mRNAs and lncRNAs may represent ceRNAs for binding with miRNAs.
These results suggested that this classification-represented
ncRNA-mediated gene regulatory module established by the
gene-pair-based method used in the present study may further the
understanding of the mechanisms underlying the pathogenesis of
AD.
Materials and methods
Gene expression and RNA-RNA
interactions
Gene expression data of 1,548 samples obtained from
a cohort of 90 patients with AD spanning 19 brain regions generated
by HG-U133A, HG-U133B and HG-U133_Plus_2 platforms were downloaded
from the GEO database (database no. GSE84422) (3). The 19 brain regions included the
amygdala, anterior cingulate, caudate nucleus, dorsolateral
prefrontal cortex, frontal pole, hippocampus, inferior frontal
gyrus, inferior temporal gyrus, middle temporal gyrus, nucleus
accumbens, occipital visual cortex, parahippocampal gyrus,
posterior cingulate cortex, precentral gyrus, prefrontal cortex,
putamen, superior parietal lobule, superior temporal gyrus and
temporal pole. Robust multi-array average-normalized microarray
data were downloaded (19).
Replicate genes were combined by taking the mean of their
expression values. In order to minimize variance induced by
technical variation, batch effects were removed using the
R/Bioconductor package sva v 3.24.4 (20), which is based on surrogate variable
analysis (21).
RNA-RNA interactions in humans, including 66
lncRNA-mRNA interactions, 85 lncRNA-miRNA interactions and 8,368
miRNA-mRNA interactions, were retrieved from RAID v.2.0 with a
confidence score of ≥0.8 (18).
Classification of brain regions using
a gene pair-based method
Gene pairs exhibiting correlated expression changes
are likely to collectively perform a common function rather than
such a function being performed by a single gene. Therefore, gene
pairs were predicted to be more strongly associated with AD
compared with genes functioning alone. A gene pair-based method was
established to classify brain regions into different regional
groups according to how each region is affected by AD.
Firstly, a C(2,N) × M correlation coefficient matrix
was calculated followed by a Spearman's rank correlation
coefficient, rnm, calculation for each pair of N genes
in each of the M regions (Fig.
1B).
rnm=ρgXm,gYm=cov(gXm,gYm)σgXmσgYm
n and m refers to the row and column in the matrix,
respectively; gXm and gYm refers to the
sample rank of gene X and gene Y in the region, respectively; ρ
represents the rank-based converted Pearson correlation
coefficient; cov and σ represent the covariance and the standard
deviations of the rank variables, respectively; and C(2,N) refers
to the 2-combinations of an N-element set.
Subsequently, the difference between each of the two
brain regions for each gene pair was determined via Spearman's rank
correlation coefficients and the sample size for each region was
determined using the R/Bioconductor package cocor v 1.1–3
(22) using a C(2,N) ×
M2 matrix calculation. Each gene pair was scored using a
sum-normalized vote strategy, which is the votes divided by the
sum, to determine the differential expression of correlated gene
pairs across regions.
Finally, PCA was used to calculate the contribution
score for each differentially expressed gene pair. Principal
component 1 (PC1) and principal component 2 (PC2) were used to
classify brain regions into different groups.
ncRNA-mediated gene regulatory module
identifications
A network was constructed using gene pairs that were
revealed to exhibit the top 1% contribution rate in classified
brain regions following PC1 and PC2 analyses. MCODE v 1.4.2
(23), a Cytoscape (24) plugin, was used to determine the
functional modules that represented highly interconnected clusters
of a network. Identified modules were integrated with 8,519 RNA-RNA
interactions using RAID v 2.0 (18) with a confidence score of ≥0.8 in
order to reveal a ncRNA-mediated gene regulatory module (Fig. 1C).
Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analysis
The enrichment of KEGG (25–27)
pathways was performed using Database for Annotation, Visualization
and Integrated Discovery (DAVID) v 6.8 (28). Significantly enriched pathways were
defined as P<0.05.
Heatmap analysis
Heatmap, representing data values using a
color-coding system, was generated using the R/Bioconductor package
gplots v 3.0.1 (29).
Results
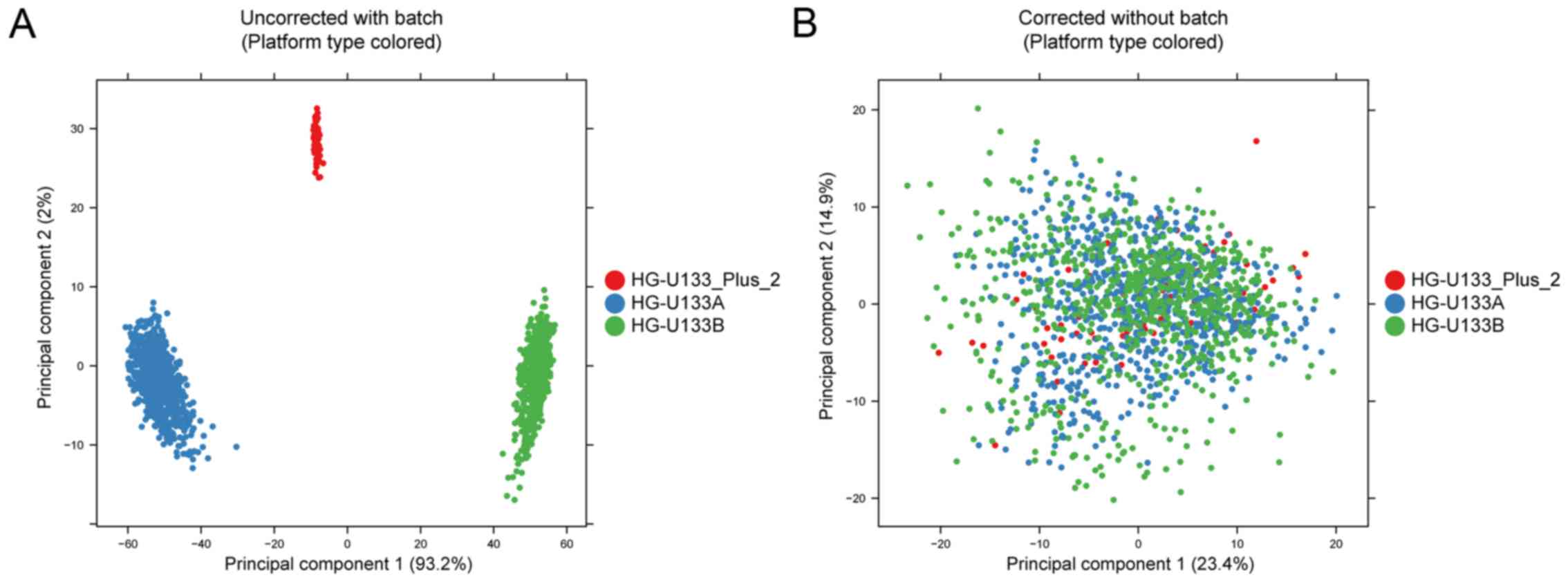
Batch effects induced by technical
variance are removed
Following the collection of gene expression data
from HG-U133A, HG-U133B, and HG-U133_Plus_2 platforms, it was
revealed that there were marked differences in the top 500 genes
based on the variance of gene expression between the three
platforms in all samples; however, no marked differences were
demonstrated between different brain regions (Fig. 2A). Following the removal of the
batch effect by the R/Bioconductor package sva v 3.24.4
(20), a satisfactory profile was
obtained when samples did not exhibit any marked differences based
on their generated platform (Fig.
2B).
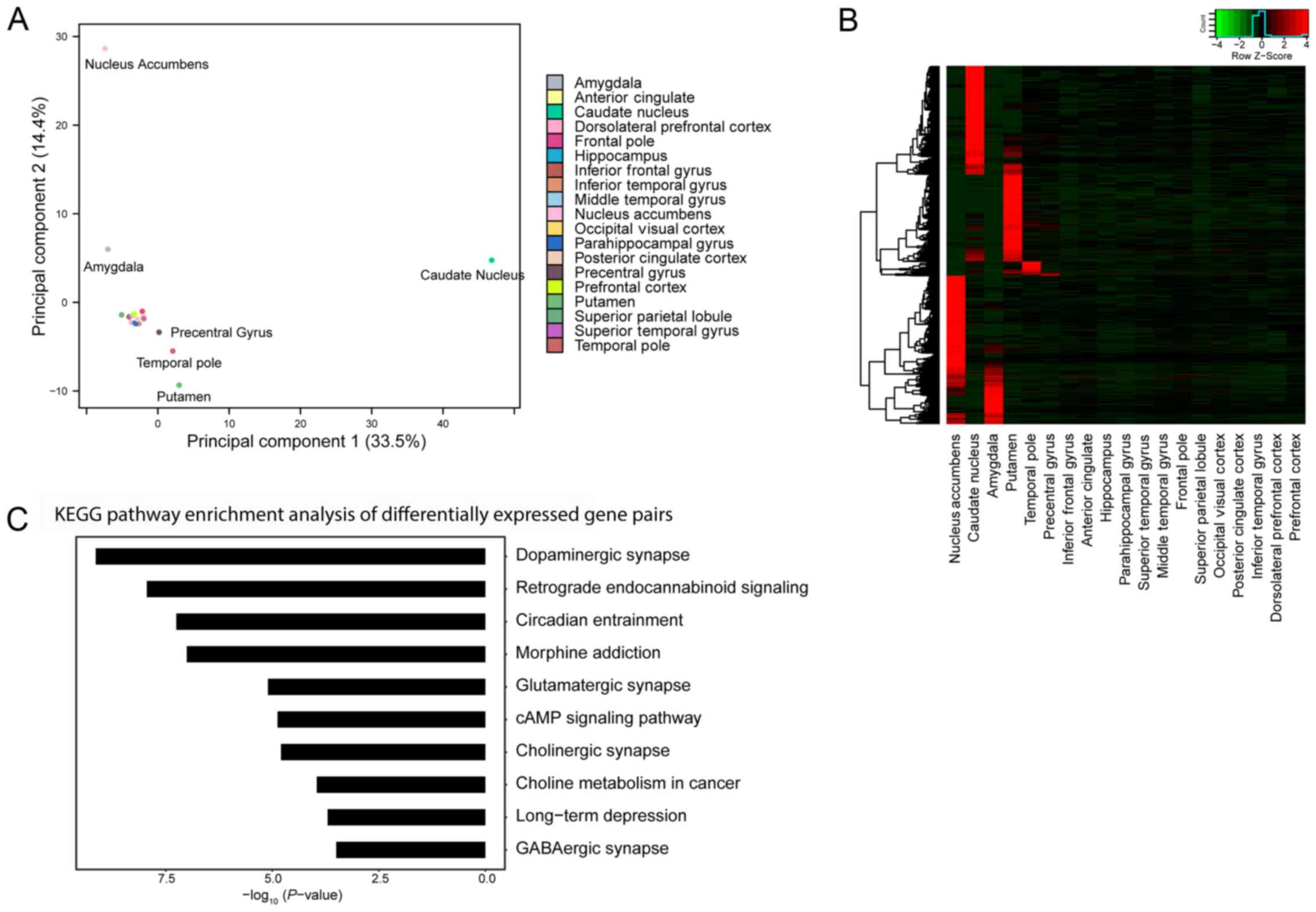
Gene pair-based method is used to
classify 19 brain regions
Based on the established gene pair-based method,
seven region groups were identified, including six out of 19 brain
regions (31.6%) exhibiting different patterns of expressed gene
pairs from each other, and an additional 13 clustered regions
exhibiting different patterns from the six regions as presented in
Fig. 3A, which suggested that
despite the 19 brain regions being situated physically adjacent to
one another, numerous regions, particularly the amygdala, caudate
nucleus, nucleus accumbens, precentral gyrus, putamen and temporal
pole regions, exhibited different expression levels of gene pairs.
In order to investigate the mechanisms underlying the differential
expression of gene-pairs in the identified brain regions, 4,731
gene-pairs (including 1,308 genes) that represented the top 1%
contribution in PC1 and PC2 (Fig.
3B) were used to classify the different brain regions.
Annotation enrichment analysis of these 1,308 genes using DAVID
(28) demonstrated that 63 KEGG
(25–27) pathways were significantly annotated
(P<0.05), the top 10 pathways of which are presented in Fig. 3C. The top five pathways, including
dopaminergic synapse, retrograde endocannabinoid signaling,
circadian entrainment, morphine addiction and glutamatergic
synapse, demonstrated a strong association between the 1,308 genes
and pathways associated with the nervous system. For example, the
majority of genes enriched in the dopaminergic synapse pathway were
regulated by the same genes (for example protein kinase A) or
regulated each other, highlighting the importance of these enriched
genes in this pathway.
The ncRNA-mediated gene regulatory
module is identified
To further analyze the functions and cellular
processes the 4,731 gene pairs are involved in, these were used to
construct an interaction network involving 1,308 genes and 4,731
interactions. Using a Cytoscape plugin MCODE (23,24),
5 highly interconnected modules were identified (Fig. 4A-E). Enrichment analysis of
functional Module 2 and Module 3 demonstrated a strong correlation
between pathways associated with the nervous system, including
cholinergic synapses (P=0.04), circadian entrainment (P=0.01) and
dopaminergic synapse (P=0.02). Notably, Module 2 was revealed to
exhibit numerous interactions with miRNAs, which were revealed via
integration with the ncRNA-mediated network to interact with a
number of lncRNAs to form a ncRNA-mediated gene regulatory module,
involving 105 molecules and 144 interactions (Fig. 4F). For example, surplus
neurotrophic receptor tyrosine kinase (NTRK3) and peptidyl
arginine deiminase 1 (PADI1) genes were demonstrated to
interact with surplus miRNAs, including hsa-miR-9-5p,
hsa-miR-125a-5p, hsa-miR-125b-5p and hsa-miR-145-5p;
which were also revealed to interact with surplus lncRNAs,
including long intergenic non-protein coding RNA, regulator of
reprogramming; MALAT1, POU class 5 homeobox (POU5F)1B,
POU5F1 pseudogene (P)3, POU5F1P4, POU5F1P5, TUG1 and urothelial
cancer associated 1 (UCA1). This suggested that these genes
and lncRNAs may act as competing endogenous (ce)RNAs, by competing
for binding of miRNAs. Many of the predicted ceRNAs have been
previously reported as genes and lncRNAs associated with AD,
including NTRK3, PADI1, MALAT1, POU5F1B, POU5F1P4, TUG1 and
UCA1 (30–35). Therefore, the results suggested
that lncRNAs, including POU5F1P3 and POU5F1P5, may
perform similar functions related to the nervous system like
POU5F1B and POU5F1P4, which have previously been
demonstrated to be associated with AD. In conclusion, this
indicated that the ncRNA-mediated gene regulatory module may aid
the understanding of the mechanisms underlying AD, as well as the
development of novel therapeutic treatment strategies for patients
with AD.
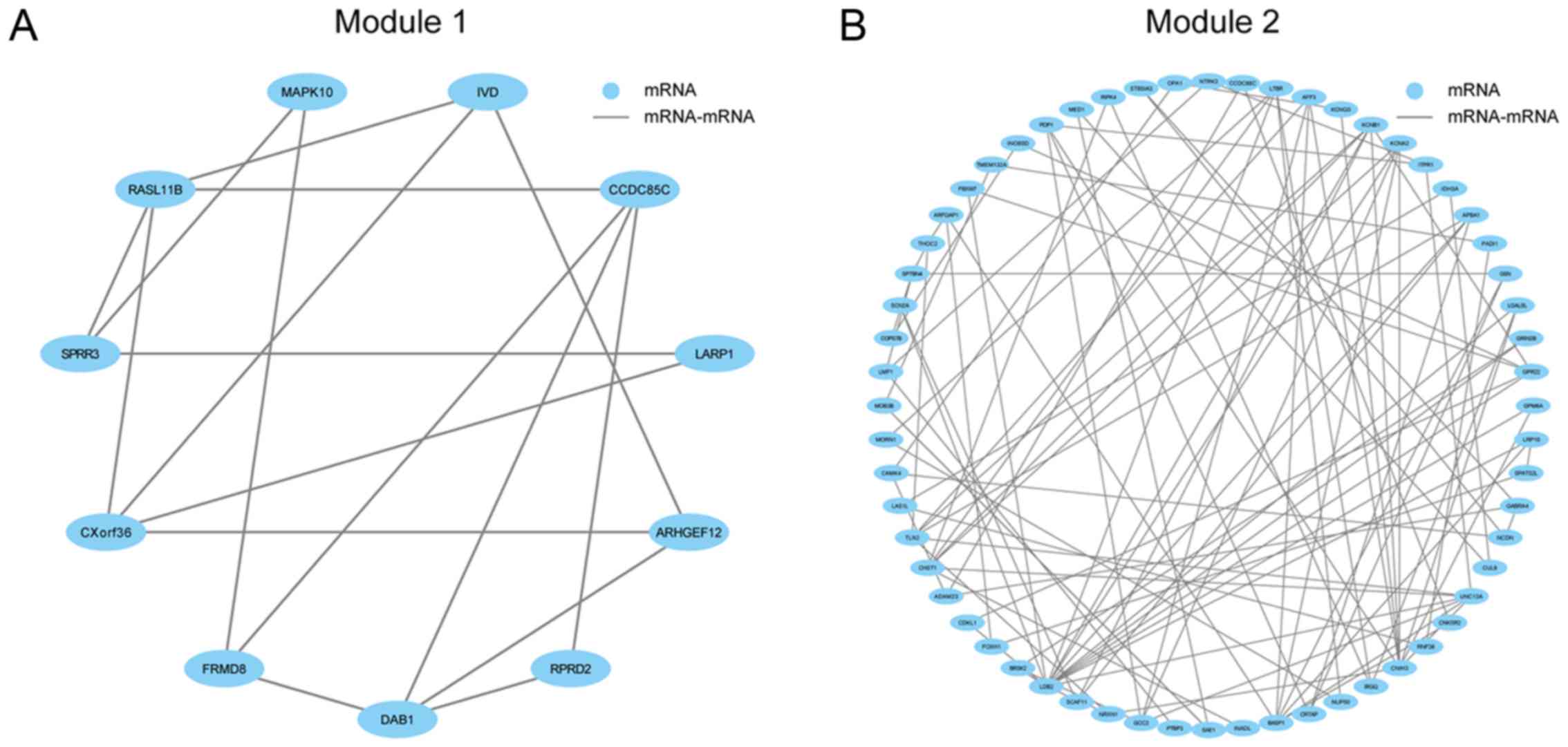 | Figure 4.(A) Module 1, (B) Module 2, (C)
Module 3, (D) Module 4 and (E) Module 5 are five highly
interconnected modules detected by the network constructed using
the gene pair-based method. The nodes and the edges indicated the
mRNAs and the interactions between them. (F) A ncRNA-mediated gene
regulatory module associated with the nervous system was
established. The nodes in blue, red and green represent mRNAs,
miRNAs and lncRNAs, respectively. The edges in blue, red and green
represent interactions between mRNAs, miRNAs and mRNAs, and lncRNAs
and miRNAs, respectively. mRNA, messenger RNA; miRNA, microRNA;
ncRNA, non-conding RNA; lncRNA, long non-coding RNA. |
Discussion
AD represents the most common neurological disorder
worldwide, and has been previously reported to differentially
affect various brain regions (3,4). The
aim of the present study was to identify five functional modules of
brain regions associated with AD in 90 patients, which may be
utilized to classify 19 brain regions into seven groups, including
marked disparate groupings of six single regions and a cluster of
another 13 regions using a gene pair-based method. Following this,
five modules were used to further analyze associations with ncRNAs
using the ncRNA-mediated network. Finally, genes involved in one of
the ncRNA-mediated gene regulatory modules identified by the
present study were revealed to be significantly enriched in
pathways associated with the nervous system, such as the
cholinergic synapse pathway. In addition, in agreement with
previous studies that revealed that NTRK3 and PADI1
genes, as well as MALAT1 and TUG1 lncRNAs in this
module were associated with AD (30–35),
it can be inferred that POU5F1P3 and POU5F1P5 lncRNAs
compete for binding a pool of miRNAs with NTRK3 and
PADI1 genes, which provides novel insight into the
mechanisms underlying AD.
Unlike traditional biomarker identification that
identifies single molecules, the gene pair-based method established
in the present study, which integrated the ncRNA-mediated gene
regulatory network, may better represent the complex mechanisms
underlying AD, as it includes associations between lncRNAs, miRNAs
and mRNAs. For example, in the ncRNA-mediated gene regulatory
module identified in the present study, hepatocellular carcinoma
upregulated lncRNA and forkhead box O3B pseudogene lncRNAs were
revealed to compete for binding with surplus miRNAs, such as
hsa-miR-182-5p and hsa-miR-107. Furthermore, opa
interacting protein 5-antisense RNA 1 and cyclin dependent kinase 4
pseudogene 1 lncRNAs as well as the inositol 1,4,5-trisphosphate
receptor type 1 gene were revealed to compete for binding with
miRNAs, such as hsa-miR-424-5p and hsa-miR-34c-5p.
Therefore, such ceRNAs and miRNAs may represent novel candidate
biomarkers for AD.
In conclusion, the present study established a gene
pair-based method incorporated with ncRNA-mediated gene regulatory
networks to identify module biomarkers associated with different
brain regions, including lncRNAs, miRNAs and mRNAs. The results of
the present study suggested that in order to comprehensively
develop biomarker prediction methods and therapeutic strategies for
the treatment of patients with AD, the interactions among cellular
molecules must be determined.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LY and SW conceived and designed the experiments. LY
and SQ analyzed the data. LY and SW wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AD
|
Alzheimer's disease
|
|
lncRNA
|
long non-coding RNA
|
|
miRNA
|
microRNA
|
|
ncRNA
|
non-coding RNA
|
|
PCA
|
principal component analysis
|
|
ceRNA
|
competing endogenous RNA
|
References
|
1
|
Kumar A, Singh A and Ekavali: A review on
Alzheimer's disease pathophysiology and its management: An update.
Pharmacol Rep. 67:195–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maiese K: Late onset Alzheimer's disease:
Novel clinical prospects for the future. Curr Neurovasc Res.
14:892017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang M, Roussos P, McKenzie A, Zhou X,
Kajiwara Y, Brennand KJ, De Luca GC, Crary JF, Casaccia P, Buxbaum
JD, et al: Integrative network analysis of nineteen brain regions
identifies molecular signatures and networks underlying selective
regional vulnerability to Alzheimer's disease. Genome Med.
8:1042016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X, Michaelis ML and Michaelis EK:
Functional genomics of brain aging and Alzheimer's disease: Focus
on selective neuronal vulnerability. Curr Genomics. 11:618–633.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Collins FS, Morgan M and Patrinos A: The
Human Genome Project: Lessons from large-scale biology. Science.
300:286–290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Constantinides VC, Paraskevas GP,
Emmanouilidou E, Petropoulou O, Bougea A, Vekrellis K, Evdokimidis
I, Stamboulis E and Kapaki E: CSF biomarkers β-amyloid, tau
proteins and a-synuclein in the differential diagnosis of
Parkinson-plus syndromes. J Neurol Sci. 382:91–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin PP, Chen WL, Yuan F, Sheng L, Wu YJ,
Zhang WW, Li GQ, Xu HR and Li XN: An UHPLC-MS/MS method for
simultaneous quantification of human amyloid beta peptides Aβ1-38,
Aβ1-40 and Aβ1-42 in cerebrospinal fluid using micro-elution solid
phase extraction. J Chromatogr B Analyt Technol Biomed Life Sci.
1070:82–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chopra P, Lee J, Kang J and Lee S:
Improving cancer classification accuracy using gene pairs. PLoS
One. 5:e143052010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jin N, Wu H, Miao Z, Huang Y, Hu Y, Bi X,
Wu D, Qian K, Wang L, Wang C, et al: Network-based
survival-associated module biomarker and its crosstalk with cell
death genes in ovarian cancer. Sci Rep. 5:115662015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Knoll M, Lodish HF and Sun L: Long
non-coding RNAs as regulators of the endocrine system. Nat Rev
Endocrinol. 11:151–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
ENCODE Project Consortium, . Birney E,
Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH,
Weng Z, Snyder M, Dermitzakis ET, et al: Identification and
analysis of functional elements in 1% of the human genome by the
ENCODE pilot project. Nature. 447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bertone P, Stolc V, Royce TE, Rozowsky JS,
Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et
al: Global identification of human transcribed sequences with
genome tiling arrays. Science. 306:2242–2246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sheinerman KS, Toledo JB, Tsivinsky VG,
Irwin D, Grossman M, Weintraub D, Hurtig HI, Chen-Plotkin A, Wolk
DA, McCluskey LF, et al: Circulating brain-enriched microRNAs as
novel biomarkers for detection and differentiation of
neurodegenerative diseases. Alzheimers Res Ther. 9:892017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cesana M, Cacchiarelli D, Legnini I,
Santini T, Sthandier O, Chinappi M, Tramontano A and Bozzoni I: A
long noncoding RNA controls muscle differentiation by functioning
as a competing endogenous RNA. Cell. 147:358–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tay Y, Kats L, Salmena L, Weiss D, Tan SM,
Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al:
Coding-independent regulation of the tumor suppressor PTEN by
competing endogenous mRNAs. Cell. 147:344–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cai Y, Sun Z, Jia H, Luo H, Ye X, Wu Q,
Xiong Y, Zhang W and Wan J: Rpph1 upregulates CDC42 expression and
promotes hippocampal neuron dendritic spine formation by competing
with miR-330-5p. Front Mol Neurosci. 10:272017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li
Y, Liu L, Hou P, Cui T, Tan P, et al: RAID v2.0: An updated
resource of RNA-associated interactions across organisms. Nucleic
Acids Res. 45:(D1). D115–D118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leek JT, Johnson WE, Parker HS, Jaffe AE
and Storey JD: The sva package for removing batch effects and other
unwanted variation in high-throughput experiments. Bioinformatics.
28:882–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leek JT and Storey JD: Capturing
heterogeneity in gene expression studies by surrogate variable
analysis. PLoS Genet. 3:1724–1735. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diedenhofen B and Musch J: Cocor: A
comprehensive solution for the statistical comparison of
correlations. PLoS One. 10:e01219452015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M, Sato Y, Kawashima M, Furumichi
M and Tanabe M: KEGG as a reference resource for gene and protein
annotation. Nucleic Acids Res. 44:(D1). D457–D462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Warnes G, Bolker B, Bonebakker L,
Gentleman R, Liaw Andy WH, Lumley T, Maechler M, Magnusson A,
Moeller S, Schwartz M and Venables B: gplots: Various R programming
tools for plotting data. R package. version 3.0.3. 2016.
|
|
30
|
Braskie MN, Kohannim O, Jahanshad N,
Chiang MC, Barysheva M, Toga AW, Ringman JM, Montgomery GW, McMahon
KL, de Zubicaray GI, et al: Relation between variants in the
neurotrophin receptor gene, NTRK3, and white matter integrity in
healthy young adults. NeuroImage. 82:146–153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chavanas S, Adoue V, Méchin MC, Ying S,
Dong S, Duplan H, Charveron M, Takahara H, Serre G and Simon M:
Long-range enhancer associated with chromatin looping allows AP-1
regulation of the peptidylarginine deiminase 3 gene in
differentiated keratinocyte. PLoS One. 3:e34082008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yao J, Wang XQ, Li YJ, Shan K, Yang H,
Wang YN, Yao MD, Liu C, Li XM, Shen Y, et al: Long non-coding RNA
MALAT1 regulates retinal neurodegeneration through CREB signaling.
EMBO Mol Med. 8:346–362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Costa V, Esposito R, Aprile M and
Ciccodicola A: Non-coding RNA and pseudogenes in neurodegenerative
diseases: ‘The (un)Usual suspects’. Front Genet. 3:2312012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson R: Long non-coding RNAs in
Huntington's disease neurodegeneration. Neurobiol Dis. 46:245–254.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng J, Yi D, Liu Y, Wang M, Zhu Y and
Shi H: Long nonding RNA UCA1 regulates neural stem cell
differentiation by controlling miR-1/Hes1 expression. Am J Transl
Res. 9:3696–3704. 2017.PubMed/NCBI
|