Introduction
Islet amyloid polypeptide (IAPP), also known as
amylin, is a 37-amino-acid residue polypeptide (1), which is co-synthetized and secreted
with insulin by β cells (2,3).
Islet amyloid deposit, primarily derived from IAPP, is a
pathological feature of 90% of patients with type 2 diabetes
mellitus (T2DM) (4–7). Human-IAPP (hIAPP) has a propensity to
form toxic oligomers spontaneously (1,5).
Previous studies have demonstrated that aggregated hIAPP was toxic
to islets and β cells in vitro or in vivo (8,9).
Unlike hIAPP, rodent IAPP (rIAPP) that lacks β-sheet structure due
to the 20–29 region proline substitutions, is nonamyloidogenic and
nontoxic to β cells (10). The
mechanisms of hIAPP-mediated toxicity are not yet completely
elucidated. Therefore, further study of the underlying mechanisms
of hIAPP-induced cytotoxicity in order to prevent loss of β cell
mass is viewed as the clinical goal of treatment of T2DM.
As a result of imbalance between generation of
reactive oxygen species (ROS) and antioxidant system (11), overproduction of ROS leads to
oxidative stress. Previous studies have indicated that islet
amyloid deposition induces oxidative stress and is associated with
the decrease of β cell mass in patients with T2DM (4,12).
In vitro studies also demonstrated that hIAPP promotes
oxidative stress and that hIAPP-induced β cell death was alleviated
by antioxidants (13,14). Redox state can regulate autophagy
and ROS are generally accepted as inducers of autophagy (15). Autophagy is an evolutionarily
conserved cellular mechanism for degradation of cytoplasmic
components (16). Damaged
organelles and abnormal proteins are sequestrated by autophagosomes
(16) and subsequently transported
to lysosomes for degradation and recycling (16). Under oxidative stress conditions,
autophagy can degrade damaged mitochondria, which are important
sources of intracellular ROS (17). Autophagy also removes oxidized
proteins that are toxic to the cell (15). There is growing support for a
hypothesis that autophagy is essential to maintain the function and
mass of pancreatic β cells (18–20).
Activation of autophagy by rapamycin relieved palmitate-induced
damage to β cells (21). β cell
specific disruption of autophagy associated gene 7 in mice led to
reduced insulin secretion, glucose intolerance and loss of β cell
mass (20). Dysregulation of
autophagy also serves a pathogenic role in amyloidosis-associated
neurodegeneration, including Alzheimer's disease (22). However, in certain cases, the ROS
scavenger catalase is also degraded by autophagy, therefore
inhibition of autophagy decreases the accumulation of ROS and
rescued cells from death (23,24).
Therefore, the effect of autophagy on oxidative stress may be
altered under different pathological conditions.
The above evidence indicated that autophagy may be
involved in hIAPP-induced oxidative stress in β cells. The present
study was designed to verify this hypothesis, and the results
suggested that treatment with hIAPP promotes autophagic flux
through ROS-mediated adenosine 5′-phosphate-activated protein
kinase (AMPK) signaling pathway in INS-1 cells. Chemical activation
of autophagy through AMPK signaling significantly attenuated
hIAPP-induced INS-1 cell death and oxidative stress. Therefore,
pharmacological modulation of autophagy through the AMPK signaling
may offer an alternative therapeutic approach to prevent or slow β
cell failure in T2DM.
Materials and methods
Cell line and regents
INS-1 cell line was purchased from Cell Center of
Chinese Academy of Medical Sciences (Beijing, China). Compound C,
AMPK activator 5-aminoimidazole-4-carboxamide1-β-D-ribofuranoside
(AICAR), hIAPP, rIAPP, 3-methyladenine (3-MA), ammonium chloride
(NH4Cl) and MTT were obtained from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). RPMI-1640 medium and fetal bovine serum
(FBS) were from Hyclone (GE Healthcare Life Sciences, Logan, UT,
USA). Microtubule-associated protein 1 light chain 3 mouse
monoclonal antibody (LC3; cat. no. 2775; 1:1,000), phosphorylated
(p)-AMPKα rabbit monoclonal antibody (Thr172; cat. no. 4188;
1:1,000), AMPKα1 rabbit monoclonal antibody (cat. no. 5831;
1:1,000), antibodies were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA). P62 rabbit polycolonal antibody (cat. no.
AF5384; 1:1,000) and β-actin mouse monoclonal antibody (cat. no.
T0022; 1:3,000) were purchased from Affinity Biosciences
(Cincinnati, OH, USA). Secondary monoclonal antibodies, including
horseradish peroxidase (HRP)-labeled goat anti mouse immunoglobulin
G (IgG; H+L; cat. no. E030120-01; 1:5,000) and HRP-labeled goat
anti rabbit IgG (H+L; cat. no. E030110-01; 1:5,000) were purchased
from EarthOx, LLC (Millbrae, CA, USA). ROS and malondialdehyde
(MDA) content were measured with 2′,7′-dichlorofluorescin diacetate
(DCFHDA) assay kit and thiobarbituric acid (TBA) kit from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China).
Cell culture and intervention
INS-1 cells were maintained in RPMI-1640 medium
supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine,
50 mM mercaptoethanol, 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 g/ml
streptomycin and 100 U/ml penicillin at 37°C in a humidified
atmosphere containing 95% air and 5% CO2. For treatment,
hIAPP was dissolved in hexafluoroisopropanol (HFIP; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at room temperature (RT) for 8 h.
Subsequently, HFIP was removed by evaporation under N2
at RT for 1 h and stored at −80°C. Prior to use, hIAPP was freshly
dissolved in dimethylsulfoxide (DMSO) at 20 mg/ml and diluted to 10
µM by 20 mM PBS (pH 7.4). Cells were seeded in 96-well microplates
at a density of 1×104 cells/well for MTT assays. For
other assays, cells were seeded in 6-well plates at a density of
2×105/well. Cells were incubated with hIAPP (10 µM) or
rIAPP (10 µM) for 24 h to examine the effects on INS-1 cells. For
other treatments, cells were pretreated with 3-MA (0.5 nM),
N-acetyl-L-cysteine (NAC; 20 mM), AICAR (2 mM), compound C (10 µM),
NH4Cl (5 mM) for 2 h and subsequently incubated with or
without hIAPP (10 µM) for 24 h. The same volume of vehicle (DMSO
<0.1%) was used as negative control.
Transmission electron microscopy
(TEM)
INS-1 cells were fixed at 4°C in 2.5% glutaraldehyde
in PBS overnight. Following fixation in 1% osmium tetroxide at 4°C
for 30 min and dehydration in 50, 70, 90 and 100% ethanol in turn
at RT, each dehydration time was 15 min., Then cells were embedded
in epoxy resin at 60°C for 2 h. Subsequently, ultrathin section
were obtained (~50–60 nm) and stained with 3% uranyl acetate at RT
for 30 min. Images were captured by using HT7700 electron
microscope (Hitachi, Ltd., Tokyo, Japan). Each group randomly
selected 20 cells to count the number of intracellular
autophagosomes.
MTT assay
Cell viability was measured by MTT assay. INS-1
cells were plated in 96-well microplates and cultured at 37°C in a
humidified atmosphere for 48 h and treated as described above.
Following incubation, 20 µl/well MTT solution (5 mg/ml in PBS
buffer) was added and incubated for 4 h. Following removal of the
medium, 200 µl/well DMSO solution was added to dissolve the
formazan crystals. Absorbance of soluble dye was measured at a
wavelength of 570 nm with a microplate spectrophotometer (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
Measurement of ROS generation
Relative level of ROS in INS-1 cells was measured by
DCFHDA assay. Cells were seeded in 6-well plates for corresponding
treatment, as described above. Treated cells were washed with PBS
and incubated with DCFHDA (10 mM) in phenol red free medium (GE
Healthcare Life Sciences, Logan, UT, USA) at 37°C for 1 h. These
cells were washed and resuspended with PBS at density of
1×105 cells/ml. 2′,7′-dichlorofluorescein (DCF)
fluorescence was detected by an emission wavelength of 530 nm and
excitation wavelength of 502 nm. Unlabeled cells at the same cell
density in PBS were used as background control. The relative DCF
fluorescence intensities of the treated cells were expressed as
relative value to control. ROS in INS-1 cells were also directly
observed and photographed by fluorescence microscopy in 6-well
plates following incubation with DCFHDA for 1 h.
Determination of MDA
Following collection by centrifugation at RT for 5
min (250 × g), treated cells were washed with ice cold PBS and
lysed with 1% trition X-100 (Beijing Leagene Biotech Co., Ltd.,
Beijing, China) on ice for 20 min. Subsequently TBA method was used
to determine the levels of MDA. This assay is based on the
combination of MDA with TBA, which forms a red compound whose
absorbance can by determined at a wavelength of 532 nm. The MDA
content was expressed as nmol/mg protein.
Western blot analysis
Cells were harvested and washed with ice cold PBS,
then homogenized in radio immunoprecipitation assay buffer
(Solarbio Beijing Co., Ltd., Beijing, China). Lysates were mixed
and incubated on ice for 10 min. Cell debris was precipitated at
4°C for 10 min (16,099 × g). Protein concentrations were measured
by bicinchoninic acid protein assay. Proteins were separated by 10%
SDS-PAGE and electro-transferred to a polyvinylidene fluoride
membrane. Nonspecific binding was blocked by incubation in 5%
nonfat milk at RT for 2 h. Membranes were subsequently incubated
with the primary antibodies at 4°C overnight, washed 3 times with
TBST (0.05% Tween-20) and finally incubated with a HRP-conjugated
secondary antibody for 1 h. Protein bands were visualized using an
chemiluminescence (ECL) kit (EMD Millipore, Billerica, MA, USA).
Band intensities in the immunoblots were quantified by using image
J program, version 1.48 (National Institutes of Health, Bethesda,
MD, USA). P62 rabbit polycolonal antibody (cat. no. AF5384;
dilution; 1:1,000) and β-actin mouse monoclonal antibody (cat. no.
T0022; dilution, 1:3,000) were purchased from Affinity
Biosciences.
Statistical analysis
Results were presented as the mean ± standard
deviation. One-way analysis of variance with post hoc Tukey test
was used for multiple comparisons by SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
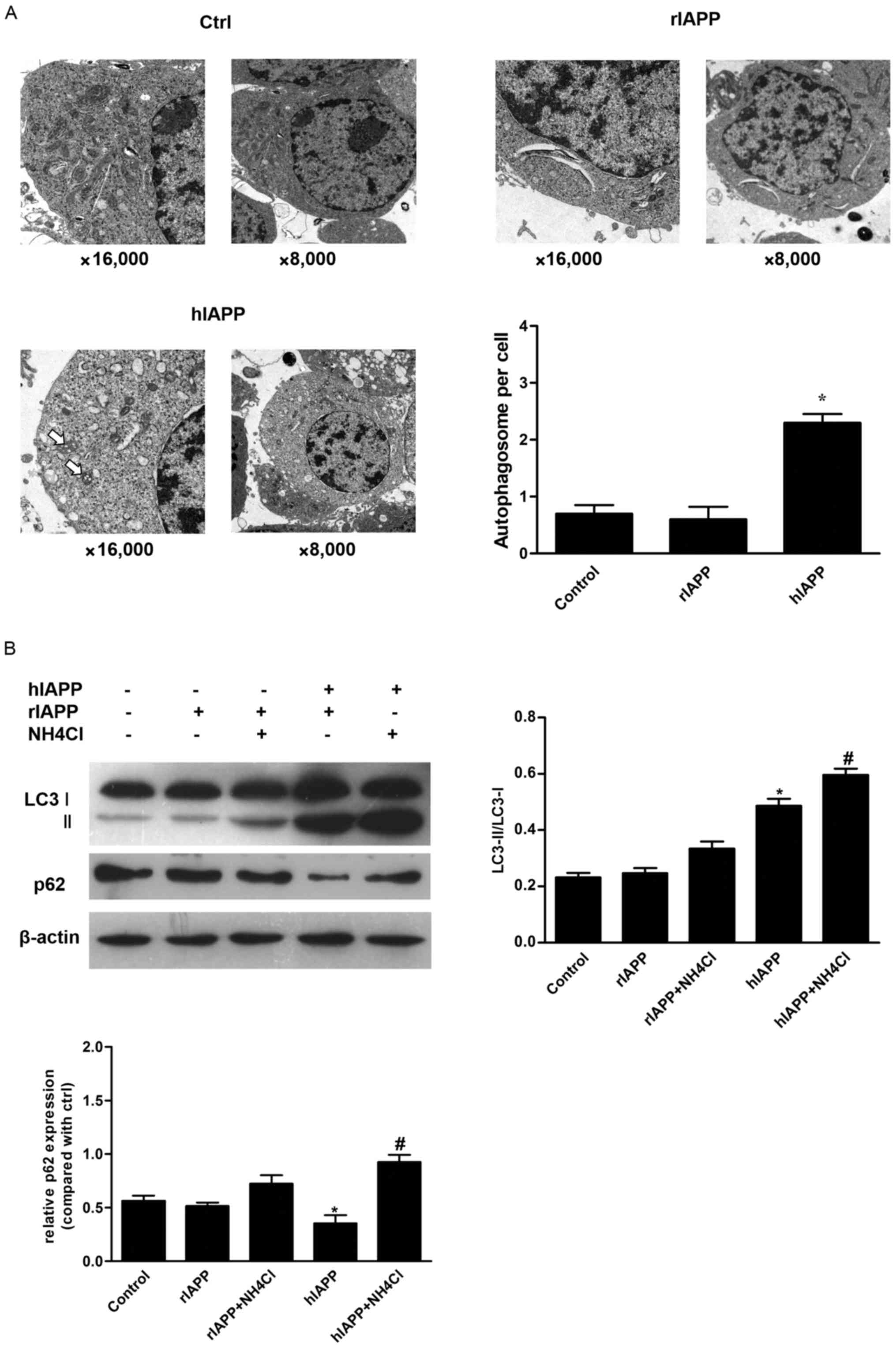
hIAPP induces autophagy in INS-1
cells
To verify the effect of hIAPP on autophagy in INS-1
cells, INS-1 cells were incubated in normal medium or medium
containing hIAPP (10 µM) or rIAPP (10 µM) for 24 h. Consistent with
a previous report (25), TEM
observation demonstrated a significantly increased number of
autophagosomes in hIAPP-treated INS-1 cells, compared with the
control. However, cells treated with rIAPP exhibited no significant
differences in the number of autophagosomes compared with the
control group (Fig. 1A). Autophagy
is a dynamic process and the number of autophagosomes at a certain
time point depends on a balance between autophagosome generation
and degradation (26). Increase of
autophagosome number may be either result from a decrease in
lysosome degradation or enhanced autophagosome formation (26).
Subsequently, western blot analysis was used to
evaluate autophagic markers LC3 and p62. LC3 is processed into a
soluble form LC3-I and subsequently LC3-I is converted to LC3-II,
which is specifically located in autophagic membrane (27). Therefore, the conversion of LC3-I
to LC3-II may reflect level of autophagy (28). P62 is incorporated into
autophagosomes by binding to LC3 and is degraded by autophagy
(28). Therefore, the amount of
p62 exhibits a negative association with autophagic activity. INS-1
cells were treated with rIAPP (10 µM) or hIAPP (10 µM), with or
without NH4Cl for 24 h and the corresponding vehicle
(DMSO <0.1%) was used as control. Treatment with hIAPP resulted
in a significant increase of LC3-II expression. NH4Cl is
a lysosomal inhibitor, which could inhibit the degradation of
autophagosomes (Fig. 1B).
Co-incubation of NH4Cl and hIAPP could further increase
LC3-II expression (Fig. 1B). By
contrast, the level of p62 was decreased in INS-1 cells treated
with hIAPP. Combined treatment with hIAPP and NH4Cl
resulted in accumulation of p62 (Fig.
1B). All above results support the hypothesis that hIAPP
induces autophagy in INS-1 cells.
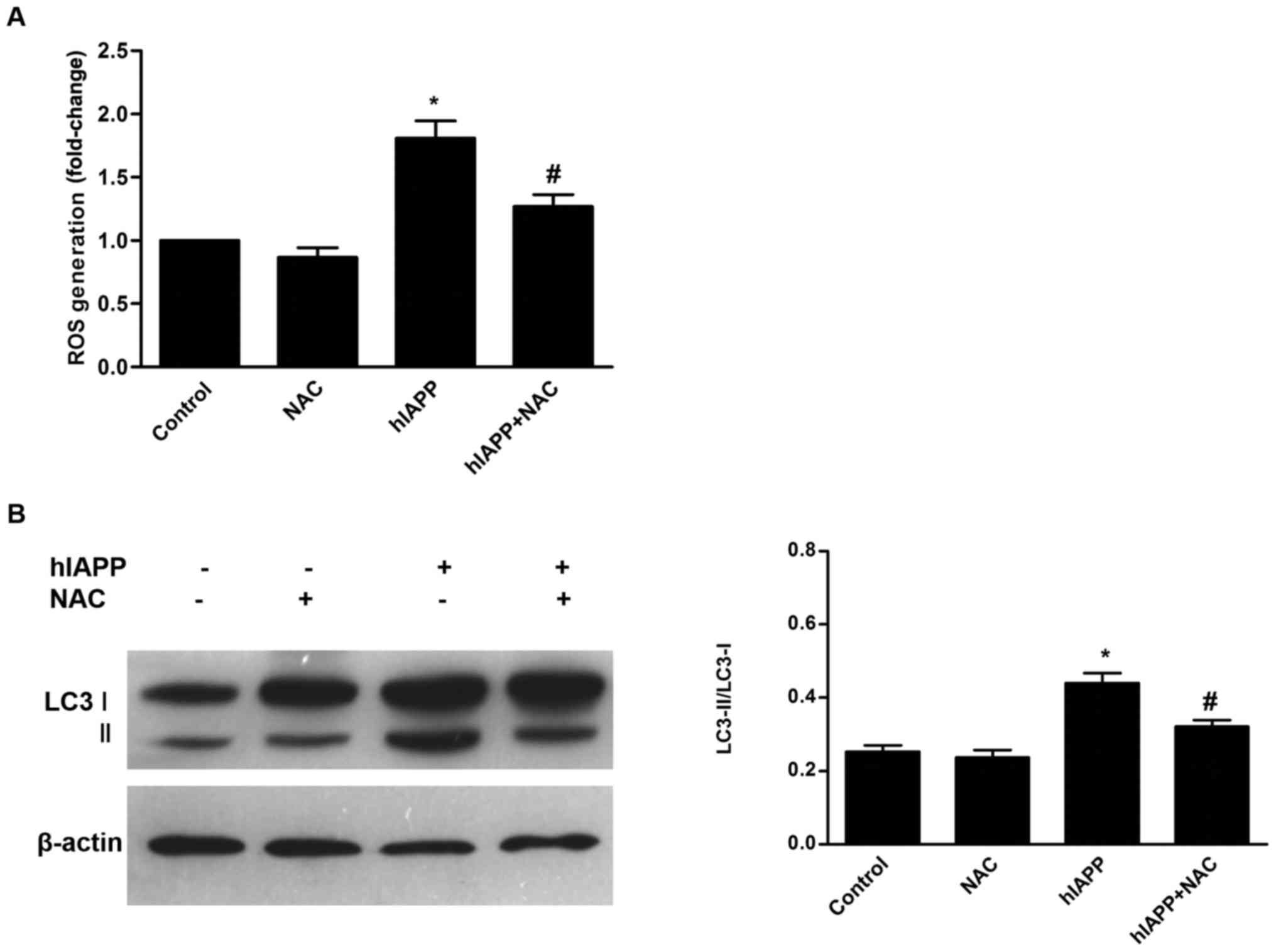
Involvement of ROS in hIAPP-stimulated
autophagy
Previous studies have demonstrated that ROS are
upstream modulators of autophagy (15,29).
To determine the effect of hIAPP on ROS generation by INS-1 cells,
INS-1 cells were treated with NAC (20 mM), hIAPP (10 µM), or NAC
(20 mM) and hIAPP (10 µM) for 24 h, and corresponding vehicle (DMSO
<0.1%) was used as control. Intracellular ROS level was
determined by DCFHDA assay. hIAPP enhanced intracellular ROS
generation in INS-1 cells, compared with the control (Fig. 2A). Antioxidant NAC alone did not
alter ROS levels compared with the control groups. Co-treatment
with NAC significantly inhibited ROS accumulation in hIAPP-treated
INS-1 cells, compared with the hIAPP group (P<0.01; Fig. 2A). To determine the effect of
hIAPP-induced oxidative stress on autophagy in INS-1 cells, western
blots were used and probed for LC3-II expression. NAC alone did not
alter LC3-II expression, but it significantly suppressed
hIAPP-induced LC3-II expression (Fig.
2B). These results confirmed that oxidative stress serves a
role in hIAPP-induced autophagy.
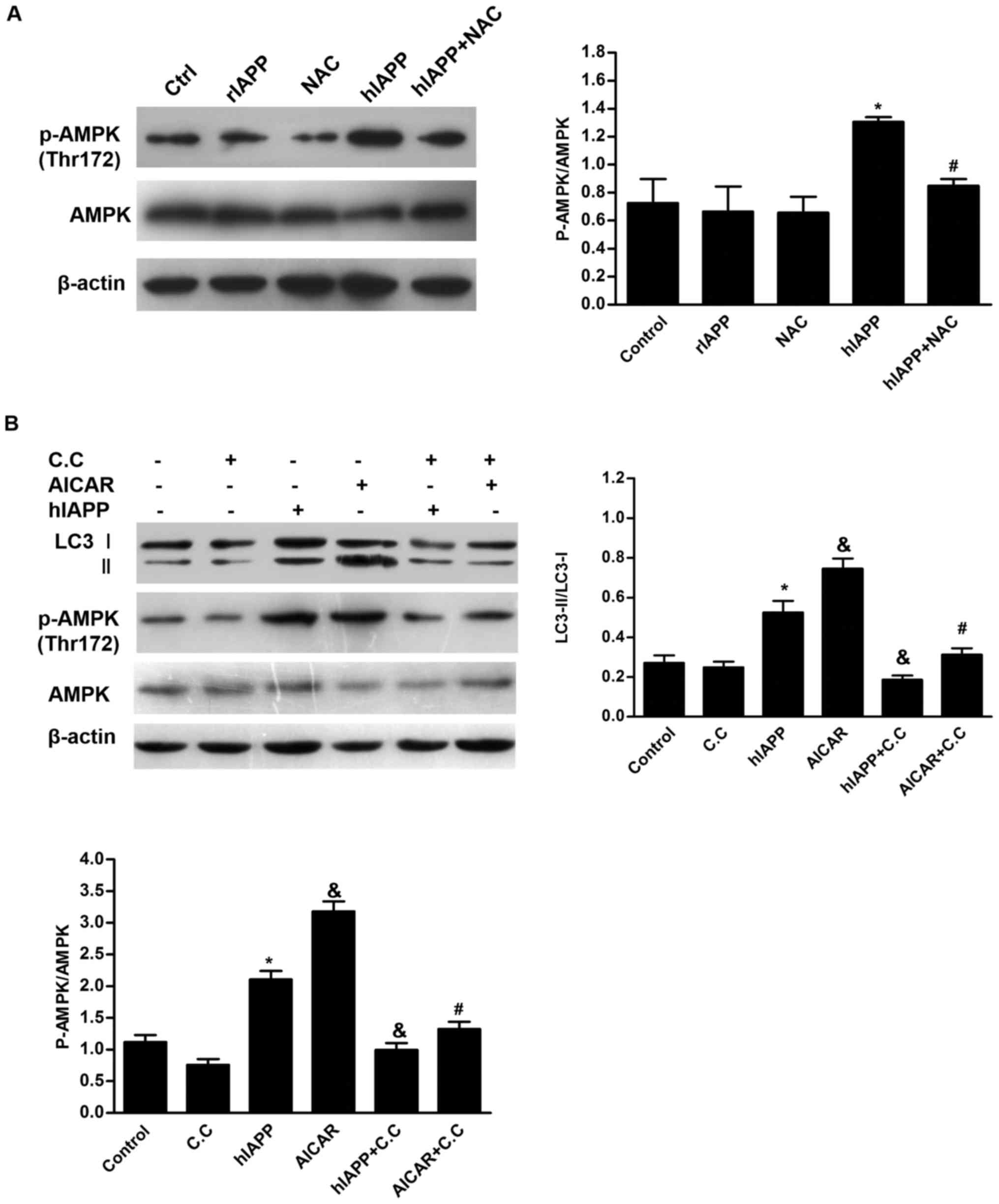
Effects of AMPK signaling pathway on
hIAPP-induced autophagy in INS-1 cells
Although connections between oxidative stress and
autophagy have been extensively studied (30,31),
the signaling pathway that links these processes in β cells has not
been examined in detail. Previous studies have suggested that
overproduction of ROS induces AMPK activation (32), activation of unc-51 like autophagy
activating kinase 1 and inhibition of mTOR complex 1 by AMPK,
leading to initiation of autophagy (33,34).
Western blots demonstrated that treatment with NAC or rIAPP alone
did not alter the level of AMPK phosphorylation (Fig. 3A). However, treatment with hIAPP
led to enhancement of AMPK phosphorylaiton at Thr172. These results
indicated that AMPK signaling pathway was activated by hIAPP in
INS-1 cells. However, this effect was partly inhibited by
co-culture with NAC (Fig. 3A),
suggesting that ROS may be involved in hIAPP-induced AMPK
activation. Similar to hIAPP, AMPK activator AICAR also enhanced
LC3-I to LC3-II conversion in INS-1 cells (Fig. 3B). However, both hIAPP- and
AICAR-induced LC3-II turnover was inhibited by AMPK inhibitor
compound C. These results suggested that AMPK serves a role in
regulation of autophagy in hIAPP-treated INS-1 cells.
Activation of autophagy through AMPK
signaling attenuates hIAPP-induced cytotoxicity and oxidative
injury in INS-1 cells
Inhibitor of autophagy 3-MA was used to study the
effects of autophagy on hIAPP-induced cytotoxicity and oxidative
damage. Treatment of INS-1 cells with 10 µM hIAPP for 24 h resulted
a significant decrease in cell viability compared with the control
(Fig. 4A). Light microscopy
observation also demonstrated that treatment with hIAPP resulted in
morphological alterations compared with the control group,
including oval shape, shrinkage, blebbing and detachment from the
culture dish. 3-MA alone exhibited minimal effect on cell survival.
However, 3-MA significantly enhanced hIAPP-induced cell viability
loss and the extent of above morphological alterations of INS-1
cells. The levels of ROS and MDA were measured as markers of
oxidative stress. Consistent with previous studies (13,35),
treatment of INS-1 cells with hIAPP resulted in elevated levels of
ROS and MDA compared with the control group (Fig. 4B). Co-treatment with 3-MA further
enhanced intracellular ROS and MDA generation in hIAPP-treated
cells.
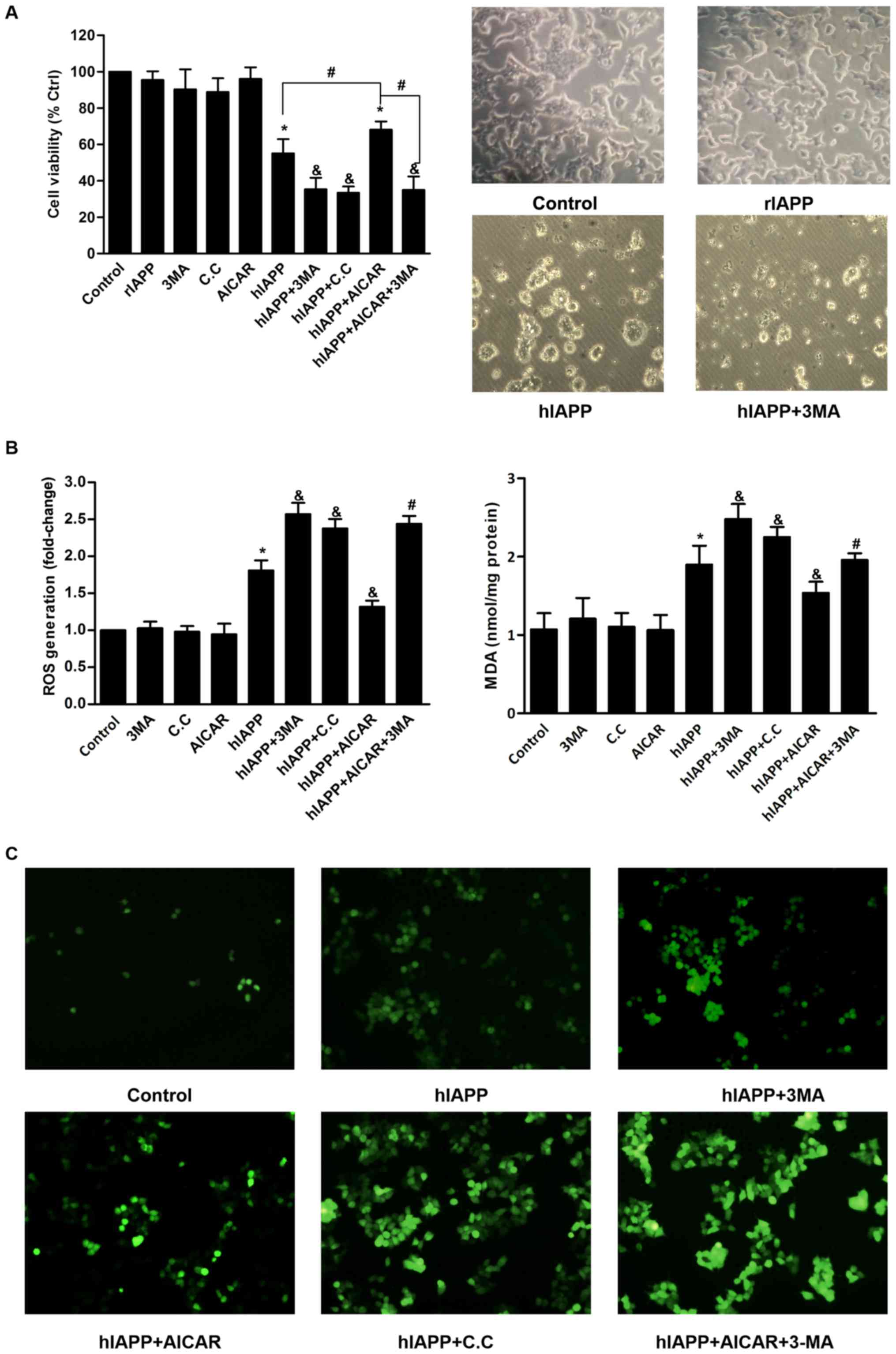 | Figure 4.Activation of autophagy through AMPK
signaling attenuates hIAPP-induced cytotoxicity and oxidative
injury. (A) Cell viability was determined by MTT assay. All data
are expressed as the mean ± standard deviation from five
independent experiments. *P<0.05 vs. the control group.
&P<0.05 vs. the hIAPP group.
#P<0.05. The morphology of INS-1 cells was observed
and images were captured under a microscope (magnification, ×160).
Representative morphologic images were presented. (B) Intracellular
ROS and MDA were determined according to the manufacturer's
protocol using a spectrophotometer. Intracellular levels of ROS
were expressed as relative ratio of control (valued as 1). All data
are expressed as the mean ± standard deviation from three
independent experiments. *P<0.05 vs. the control group.
&P<0.05 vs. the hIAPP group.
#P<0.05 vs. the hIAPP+AICAR group. (C) Intracellular
ROS levels were also observed by fluorescence microscopy
(magnification, ×100). The green fluorescence of DCF represents the
levels of intracellular ROS. AMPK, adenosine 5′-phosphate-activated
protein kinase; AICAR, AMPK activator
5-aminoimidazole-4-carboxamide1-β-D-ribofuranoside; MDA,
malondialdehyde; 3-MA, 3-methyladenine; ROS, reactive oxygen
species; hIAPP, human islet amyloid polypeptide; rIAPP, rodent
islet amyloid polypeptide; ctrl, control; C.C, compound C. |
Subsequently, the present study investigated the
effects of chemical modulation of autophagy through AMPK signaling
on hIAPP-induced cytotoxicity and oxidative damage. INS-1 cells
were treated with hIAPP in the presence or absence of compound C or
AICAR. Compound C or AICAR exhibited no effect on cell viability or
oxidative stress in INS-1 cells (Fig.
4). Pretreatment with compound C caused greater extent of cell
death and elevated levels of ROS and MDA in hIAPP-treated INS-1
cells (Fig. 4). By contrast,
pretreatment of INS-1 cells with AICAR partially prevented
hIAPP-induced cytotoxicity and oxidative stress (Fig. 4). However, these protective effects
of AICAR were abolished by the presence of 3MA (Fig. 4B). The effects of different
treatments on ROS generation in INS-1 cells were also visualized by
fluorescent microscopy. The above result findings were further
confirmed by the DCF fluorescence of each group (Fig. 4C). Together these results suggested
that chemical activation of AMPK in INS-1 cells promotes survival
against hIAPP-indcuced oxidative damage and this effect likely
occurs through induction of autophagy.
Discussion
Islet amyloid deposition is a characteristic
histopathological feature of patients with T2DM (18). The main component of islet amyloid
is hIAPP. Evidence suggests that hIAPP contributes to β cell
dysfunction and death in T2DM (4–7).
Autophagy is a self-digestion system used to maintain cellular
homeostasis through degradation and recycling of cellular
components (16). Dysregulated
autophagy has been suggested to be involved in the development of β
cell failure associated with T2DM (20). It has been reported that autophagy
serves a role in hIAPP-induced toxicity of β cells (25), but the underlying mechanism and
cellular regulatory pathway of hIAPP-induced autophagy remains to
be completely elucidated. The present study investigated how hIAPP
induces autophagy and the effects of chemical modulation of
autophagy through the AMPK pathway on the oxidative injury induced
by hIAPP in β cells. In agreement with previous studies (23,25),
the present results demonstrated that hIAPP promotes autophagy in
INS-1 cells. Electron micrographs indicated that INS-1 cells
following treatment with hIAPP exhibited an increased number of
autophagosomes compared with control cells. Treatment with hIAPP
also enhanced the conversion of LC3-I to LC3-II and was accompanied
by decreased expression of p62 in INS-1 cells.
Subsequently, the molecular mechanism of
hIAPP-induced autophagy was investigated. It has been previously
indicated that enhanced oxidative stress is associated with the
extent of islet amyloid deposition and β cell mass in T2DM
(4). ROS may act as initiators for
hIAPP-induced cytotoxicity in β cells (14,35).
It is also commonly accepted that ROS induce autophagy (36). To confirm the role of oxidative
stress in the induction of hIAPP-induced autophagy, INS-1 cells
were treated with hIAPP in the presence or absence of NAC for 24 h.
Consistent with previous studies (13,37),
the present study demonstrated that hIAPP significantly enhanced
ROS generation and this effect was attenuated by treatment with
antioxidant NAC. Treatment with NAC alone induced no effect on
LC3-II expression, but it partly inhibited LC3-II expression in
hIAPP-treated INS-1 cells. It can therefore be concluded that hIAPP
induces autophagy through generation of ROS.
ROS can activate a series of cell signaling pathways
involved in various cellular processes. The present understanding
of molecular mechanisms, signaling pathways and the redox reactions
regulated by autophagy is incomplete. AMPK is known as a sensor of
alterations in energy metabolism and redox state (38). Recent studies have demonstrated
that AMPK is a regulator of autophagy and can activate tuberous
sclersis complex 1/2, leading to inhibition of mTOR pathway and
initiation of autophagy (32,39,40).
However, there is also evidence indicating that AMPK may negatively
regulate autophagy in β cells (41), suggesting that regulation of
autophagy by AMPK depends on the cell type and environment. In the
present study, treatment with hIAPP significantly promoted AMPK
phosphorylation. Similar results were not obtained following
treatment with rIAPP, indicating that the induction of AMPK
phosphorylation is not the effect of IAPP monomers combining with
cellular receptors. Furthermore, it was observed that hIAPP-induced
AMPK phosphorylation was partly inhibited by co-treatment with NAC,
suggesting that AMPK activation may be induced by ROS. Inhibition
of AMPK by compound C reduced the amount of LC3-II in hIAPP-treated
INS-1 cells. AMPK activator AICAR could further promote LC3-II
expression. Based on these results, we hypothesized that AMPK is
major signaling pathway involved in activation of INS-1 cell
autophagy.
Autophagy can induce both positive and negative
effects and although it is generally regarded as a protector
against stressors, excessive autophagy can also lead to
non-apoptotic cell death. Oxidative stress is a mediator of
hIAPP-induced cytotoxicity in β cells (4,13,14).
In the present study, treatment of INS-1cells with hIAPP also
enhanced intracellular ROS and MDA generation. Accumulation of ROS
can cause DNA damage, protein denaturation and lipid peroxidation
(42). MDA is an end products of
lipid peroxidation and it can alter the structure and function of
the cell membrane and inhibit cellular metabolism, leading to
cytotoxicity (42). Therefore, MDA
is commonly used as a marker for oxidative stress (42). Because of relatively low levels of
antioxidants, including superoxide dismutase (SOD) and glutathione
peroxidase (GSH-Px), β cells are susceptible to oxidative damage
(43). ROS have been proposed as
initiators of hIAPP-induced toxicity. Damaged organelles, including
mitochondrion and endoplasmic reticulum are the main sources of ROS
during cellular stress (17).
Autophagy can selectively remove these obsolete organelles in order
to limit ROS amplification (15).
Oxidized damaged proteins, which are toxic to β cells are also
degraded by autophagy. In certain cases, the ROS scavenger catalase
also undergoes protein degradation via autophagy and inhibition of
autophagy reduces accumulation of ROS and rescues cells from death
(23,24). Therefore the effect of autophagy on
oxidative stress may be altered under different pathological
conditions. In the present study, it was demonstrated that
inhibition of autophagy or AMPK exacerbates oxidative stress and
renders INS-1 cells more susceptible to hIAPP-induced toxicity. By
contrast, stimulating autophagy by AMPK activator AICAR alleviated
hIAPP-induced cell death and oxidative stress in INS-1 cells. The
protective effect of AICAR was abolished by co-treatment with 3-MA.
Therefore, chemical activation of autophagy through AMPK may limit
hIAPP-induced oxidative injury in INS-1 cells.
In conclusion, the present study demonstrated that
hIAPP promotes autophagy via ROS mediated AMPK signaling pathway in
INS-1 cells. Autophagy may act as an antioxidative mechanism to
antagonize hIAPP-induced cytotoxicity in β cells. Chemical
activation of autophagy through AMPK signaling attenuates
hIAPP-induced oxidative injury in INS-1 cells. AMPK is the target
of numerous agents including certain hypoglycemic drugs (44,45).
Although the present study limited to in vitro experiments,
similar mechanisms may act in hIAPP-mediated cellular dysfunction
of human β cells. These results also suggest that pharmacological
modulation of autophagy through AMPK may be a therapeutic target to
conserve β cell mass in the initiation and progression of T2DM.
Acknowledgements
The authors would like to gratefully acknowledge
Professor Baoli Wang at the Institute of Endocrinology at Tianjin
Medical University Metabolic Disease Hospital for providing the
facilities to perform this study.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. H0713:81241030) and
the Tianjin Basic and Frontier Technology Research Program (grant
no. M0102).
Availability of data and materials
The datasets used and analyzed during the current
study are available from corresponding author on reasonable
request.
Authors' contributions
GX and TZ conceived and designed the study. GX, XL,
YJ, JZ and JX performed the experiments. GX and TZ wrote the paper.
TZ reviewed and edited the manuscript. All authors read and
approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Cooper GJ, Willis AC, Clark A, Turner RC,
Sim RB and Reid KB: Purification and characterization of a peptide
from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl
Acad Sci USA. 84:8628–8632. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kahn SE, D'Alessio DA, Schwartz MW,
Fujimoto WY, Ensinck JW, Taborsky GJ Jr and Porte D Jr: Evidence of
cosecretion of islet amyloid polypeptide and insulin by beta-cells.
Diabetes. 39:634–638. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lukinius A, Wilander E, Westermark GT,
Engström U and Westermark P: Co-localization of islet amyloid
polypeptide and insulin in the B cell secretory granules of the
human pancreatic islets. Diabetologia. 32:240–244. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Brien TD, Butler PC, Westermark P and
Johnson KH: Islet amyloid polypeptide: A review of its biology and
potential roles in the pathogenesis of diabetes mellitus. Vet
Pathol. 30:317–332. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Westermark P, Andersson A and Westermark
GT: Islet amyloid polypeptide, islet amyloid, and diabetes
mellitus. Physiol Rev. 91:795–826. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kloppel G, Löhr M, Habich K, Oberholzer M
and Heitz PU: Islet pathology and the pathogenesis of type 1 and
type 2 diabetes mellitus revisited. Surv Synth Pathol Res.
4:110–125. 1985.PubMed/NCBI
|
|
7
|
Costes S, Langen R, Gurlo T, Matveyenko AV
and Butler PC: β-Cell failure in type 2 diabetes: A case of asking
too much of too few? Diabetes. 62:327–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lorenzo A, Razzaboni B, Weir GC and
Yankner BA: Pancreatic islet cell toxicity of amylin associated
with type-2 diabetes mellitus. Nature. 368:756–760. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gurlo T, Ryazantsev S, Huang CJ, Yeh MW,
Reber HA, Hines OJ, O'Brien TD, Glabe CG and Butler PC: Evidence
for proteotoxicity in beta cells in type 2 diabetes: Toxic islet
amyloid polypeptide oligomers form intracellularly in the secretory
pathway. Am J Pathol. 176:861–869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Westermark GT and Westermark P: Importance
of aggregated islet amyloid polypeptide for the progressive
beta-cell failure in type 2 diabetes and in transplanted human
islets. Exp Diabetes Res. 2008:5283542008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Czarna M and Jarmuszkiewicz W: Role of
mitochondria in reactive oxygen species generation and removal;
relevance to signaling and programmed cell death. Postepy Biochem.
52:145–156. 2006.(In Polish). PubMed/NCBI
|
|
12
|
Xin A, Mizukami H, Inaba W, Yoshida T,
Takeuchi YK and Yagihashi S: Pancreas atrophy and islet amyloid
deposition in patients with elderly-onset type 2 diabetes. J Clin
Endocrinol Metab. 102:3162–3171. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li XL, Xu G, Chen T, Wong YS, Zhao HL, Fan
RR, Gu XM, Tong PC and Chan JC: Phycocyanin protects INS-1E
pancreatic beta cells against human islet amyloid
polypeptide-induced apoptosis through attenuating oxidative stress
and modulating JNK and p38 mitogen-activated protein kinase
pathways. Int J Biochem Cell Biol. 41:1526–1535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Konarkowska B, Aitken JF, Kistler J, Zhang
S and Cooper GJ: Thiol reducing compounds prevent human
amylin-evoked cytotoxicity. FEBS J. 272:4949–4959. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang YP, Liang ZQ, Gu ZL and Qin ZH:
Molecular mechanism and regulation of autophagy. Acta Pharmacol
Sin. 26:1421–1434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wen X, Wu J, Wang F, Liu B, Huang C and
Wei Y: Deconvoluting the role of reactive oxygen species and
autophagy in human diseases. Free Radic Biol Med. 65:402–410. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hull RL, Westermark GT, Westermark P and
Kahn SE: Islet amyloid: A critical entity in the pathogenesis of
type 2 diabetes. J Clin Endocrinol Metab. 89:3629–3643. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morita S, Sakagashira S, Shimajiri Y,
Eberhardt NL, Kondo T, Kondo T and Sanke T: Autophagy protects
against human islet amyloid polypeptide-associated apoptosis. J
Diabetes Investig. 2:48–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ebato C, Uchida T, Arakawa M, Komatsu M,
Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al:
Autophagy is important in islet homeostasis and compensatory
increase of beta cell mass in response to high-fat diet. Cell
Metab. 8:325–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Las G, Serada SB, Wikstrom JD, Twig G and
Shirihai OS: Fatty acids suppress autophagic turnover in β-cells. J
Biol Chem. 286:42534–42544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rubinsztein DC, DiFiglia M, Heintz N,
Nixon RA, Qin ZH, Ravikumar B, Stefanis L and Tolkovsky A:
Autophagy and its possible roles in nervous system diseases, damage
and repair. Autophagy. 1:11–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oh SH, Kim YS, Lim SC, Hou YF, Chang IY
and You HJ: Dihydrocapsaicin (DHC), a saturated structural analog
of capsaicin, induces autophagy in human cancer cells in a
catalase-regulated manner. Autophagy. 4:1009–1019. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu L, Wan F, Dutta S, Welsh S, Liu Z,
Freundt E, Baehrecke EH and Lenardo M: Autophagic programmed cell
death by selective catalase degradation. Proc Natl Acad Sci USA.
103:4952–4957. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shigihara N, Fukunaka A, Hara A, Komiya K,
Honda A, Uchida T, Abe H, Toyofuku Y, Tamaki M, Ogihara T, et al:
Human IAPP-induced pancreatic β cell toxicity and its regulation by
autophagy. J Clin Invest. 124:3634–3644. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pant K, Saraya A and Venugopal SK:
Oxidative stress plays a key role in butyrate-mediated autophagy
via Akt/mTOR pathway in hepatoma cells. Chem Biol Interact.
273:99–106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smuder AJ, Sollanek KJ, Nelson WB, Min K,
Talbert EE, Kavazis AN, Hudson MB, Sandri M, Szeto HH and Powers
SK: Crosstalk between autophagy and oxidative stress regulates
proteolysis in the diaphragm during mechanical ventilation. Free
Radic Biol Med. 115:179–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu S, Sun Y and Li Z: Resveratrol
protects Leydig cells from nicotine-induced oxidative damage
through enhanced autophagy. Clin Exp Pharmacol Physiol. Nov
22–2017.(Epub ahead of print).
|
|
32
|
She C, Zhu LQ, Zhen YF, Wang XD and Dong
QR: Activation of AMPK protects against hydrogen peroxide-induced
osteoblast apoptosis through autophagy induction and NADPH
maintenance: New implications for osteonecrosis treatment? Cell
Signal. 26:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zraika S, Hull RL, Udayasankar J,
Aston-Mourney K, Subramanian SL, Kisilevsky R, Szarek WA and Kahn
SE: Oxidative stress is induced by islet amyloid formation and
time-dependently mediates amyloid-induced beta cell apoptosis.
Diabetologia. 52:626–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gorowara S, Sapru S and Ganguly NK: Role
of intracellular second messengers and reactive oxygen species in
the pathophysiology of V. cholera O139 treated rabbit ileum.
Biochim Biophys Acta. 1407:21–30. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li X, Ma L, Zheng W and Chen T: Inhibition
of islet amyloid polypeptide fibril formation by
selenium-containing phycocyanin and prevention of beta cell
apoptosis. Biomaterials. 35:8596–8604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schimmack G, Defronzo RA and Musi N:
AMP-activated protein kinase: Role in metabolism and therapeutic
implications. Diabetes Obes Metab. 8:591–602. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Feng Z, Zhang H, Levine AJ and Jin S: The
coordinate regulation of the p53 and mTOR pathways in cells. Proc
Natl Acad Sci USA. 102:8204–8209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu J, Wu JJ, Yang LJ, Wei LX and Zou DJ:
Rosiglitazone protects against palmitate-induced pancreatic
beta-cell death by activation of autophagy via 5′-AMP-activated
protein kinase modulation. Endocrine. 44:87–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han D, Yang B, Olson LK, Greenstein A,
Baek SH, Claycombe KJ, Goudreau JL, Yu SW and bKim EK: Activation
of autophagy through modulation of 5′-AMP-activated protein kinase
protects pancreatic beta-cells from high glucose. Biochem J.
425:541–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jia Z and Misra HP: Reactive oxygen
species in in vitro pesticide-induced neuronal cell (SH-SY5Y)
cytotoxicity: Role of NFkappaB and caspase-3. Free Radic Biol Med.
42:288–298. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Robertson RP: Chronic oxidative stress as
a central mechanism for glucose toxicity in pancreatic islet beta
cells in diabetes. J Biol Chem. 279:42351–42354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Osman I, Fairaq A and Segar L:
Pioglitazone attenuates injury-induced neointima formation in mouse
femoral artery partially through the activation of AMP-activated
protein kinase. Pharmacology. 100:64–73. 2017. View Article : Google Scholar : PubMed/NCBI
|