Introduction
Chronic nasal and sinus inflammatory injury is
closely associated with the development of chronic obstructive
pulmonary disease (COPD) (1).
Furthermore, clinical studies have also confirmed that patients
with nasal and sinus inflammation have a greater risk of
cardiovascular disease due to chronic airway remodeling and airflow
limitations (2). Additionally,
anti-inflammatory therapy has been demonstrated to alleviate upper
airway symptoms such as rhinorrhea, nasal obstruction and sneezing
in patients with COPD (3,4). Thus, this information illustrates
that sinonasal inflammation is the pathogenesis responsible for
airway damage and COPD development. Accordingly, reducing the
excessive inflammatory response and increasing the resistance of
nasal epithelial cells to inflammation-induced damage are vital for
slowing or preventing COPD progression.
In response to inflammatory injury, Toll-like
receptors (TLRs), particularly TLR4, have been demonstrated as the
primary downstream effectors of inflammatory responses (5,6).
Increased TLR4 expression induces cellular oxidative stress and
calcium overload, leading to nasal epithelial cell death (7). Subsequently, TLR4 also promotes
transforming growth factor-β and matrix metalloproteinase 9
expression (8,9), the regulators of tissue fibrosis,
resulting in airway remodeling. Notably, TLR4 inhibitors exert
beneficial effects on nasal epithelial cells under chronic
inflammatory injury (10). This
evidence indicates that TLR4 modulation is vital for preserving
nasal epithelial cell function and reducing airway damage. However,
the underlying mechanism by which TLR4 induces nasal epithelial
cell damage remains unknown.
Previous studies have demonstrated that mitochondria
are the primary target of inflammatory injury (11,12).
Activated TLR4, induced by the inflammatory response, promotes
mitochondrial membrane potential collapse and mitochondrial energy
disorder (10,13). Furthermore, the damaged
mitochondria ultimately induce caspase-9-associated apoptotic
signaling (14,15), leading to nasal epithelial cell
death. Recently, mitophagy has become a research hotspot (16). Mitophagy, a selective type of
autophagy for mitochondria, can remove mitochondria via lysosomal
degradation (17). Moderate
mitophagy has been demonstrated to reduce cellular apoptosis via
the timely removal of damaged mitochondria (18,19).
By contrast, excessive mitophagy aggravates the cellular damage via
aberrant mitochondrial degradation (20,21).
Previous studies have demonstrated that mitophagy contributes to
endothelial cell apoptosis in ischemia/reperfusion-triggered
inflammatory injury (22,23). Furthermore, in cancer cells,
mitophagy activation impairs tumor migration via reducing energy
production and generating reactive oxygen species (ROS)-associated
oxidative stress (24). Based on
this knowledge, it is important to investigate whether mitophagy is
involved in inflammatory injury in nasal epithelial cells.
In addition to mitophagy, phosphatase and tensin
homolog (PTEN) is the intrinsic defender of nasal and sinus
inflammatory injury (25,26). PTEN functions as a tumor suppressor
involving in the regulation of the cell growth and differentiation
(27). Previous studies have
demonstrated that PTEN upregulation provides a survival advantage
to nasal epithelial cells in asthma (28). Furthermore, pharmacological PTEN
inhibition amplifies acute kidney injury (29,30),
regulates inflammation-induced migration of myelocytes in zebrafish
(31), and impacts nuclear
factor-κB inflammatory pathways (32). This information confirms that PTEN
is associated with inflammatory response. Notably, whether PTEN can
regulate TLR4-associated inflammatory injury in nasal epithelial
cells via mitophagy is unknown. The aim of the present study was to
investigate the role of PTEN in inflammatory-associated injury of
nasal epithelial cells, with a focus on mitophagy and the
TLR4-c-Jun kinase (JNK)-Bcl2-interacting protein 3 (Bnip3)
signaling pathway.
Materials and methods
Cell experiments and treatment
The human nasal epithelial cell line (RPMI 2650)
used in the present study was purchased from the Chinese Academy of
Sciences Cell Bank (Shanghai, China). The cells were incubated with
L-Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) at 37°C/5%
CO2. TNFα (0–20 ng/ml; Selleck Chemicals, Houston, TX,
USA) was added into the medium for 12 h to induce inflammatory
injury in vitro according to a previous study (33). To inhibit mitophagy,
3-methyladenine (3-MA; 10 mM, Selleck Chemicals) was applied for
~45 min in nasal epithelial cell line (1×106) (34). To activate mitophagy, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 5 µM, Selleck
Chemicals) was applied for ~30 min in nasal epithelial cell line
(1×106).
Immunofluorescence staining
The samples were first washed with cold
phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for
30 min at room temperature, and then permeabilized in 0.1% Triton
X-100 for 10 min at 4°C. Subsequently, 10% goat serum albumin
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was
used to block the samples for 1 h at room temperature.
Subsequently, samples were incubated with primary antibodies
overnight at 4°C (35). After
three rinses in PBS, secondary antibodies (Alexa Fluor 488 donkey
anti-rabbit secondary antibodies (1:1,000; cat. no. A-21206;
Invitrogen; Thermo Fisher Scientific, Inc.) were added to the
samples for 1 h at room temperature (34). The following primary antibodies
were used in the present study: PTEN (1:5,000; cat. no. ab31392),
mitochondrial import receptor subunit TOM20 homolog (mitochondrial
marker; 1:1,000; cat. no. ab78547), lysosome-associated membrane
glycoprotein 1 (lysosome marker, 1:1,000; cat. no. ab24170), and
HtrA serine peptidase 2 (1:500; HtrA2/Omi; cat. no. ab32092; all
Abcam, Cambridge, UK). Images were observed with an inverted
microscope (magnification, ×40; BX51; Olympus Corporation, Tokyo,
Japan). Image-Pro Plus 4.5 software (Media Cybernetics, Inc.,
Rockville, MD, USA) was used to quantify the immunofluorescence
according to a previous study (36). Mitophagy is the result of fusion
between mitochondria and lysosome. The green mitochondria locate
with red lysosome would generate the orange mitophagy. Then, the
number of orange dot was measured to quantify the number of
mitophagy (37).
Western blotting
Cells were lysed in Laemmli Sample Buffer (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Proteins were isolated and
concentrations were determined using the Bicinchoninic Acid Protein
Assay kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol (38).
Total protein (40–60 µg) was separated by 12–15% SDS-PAGE.
Following electrophoresis, the proteins were transferred to a
polyvinylidene fluoride membrane (Roche Applied Science, Penzberg,
Germany) (39). Bands were
detected using an enhanced chemiluminescence substrate (Applygen
Technologies, Inc., Beijing, China). Membranes were blocked with 5%
nonfat dried milk in Tris-buffered saline containing 0.05% Tween-20
(TBST) for 2 h at room temperature. Band intensities were
normalized to the respective internal standard signal intensity
(β-actin; 1:2,000; cat. no. ab8226 and GAPDH; 1:2,000; cat. no.
ab9485; both Abcam) (40). The
experiment was repeated three times. The primary antibodies used in
the study were as follows: Sequestome-1 (p62; 1:1,000; cat. no.
ab56416), Bnip3 (1:1,000; cat. no. ab109362), Bcl-associated X
protein (Bax; 1:1,000; cat. no. ab32503; all Abcam), pro-caspase-3
(1:1,000; cat. no. 9662; Cell Signaling Technology, Inc., Danvers,
MA, USA), caspase-9 (1:1,000; cat. no. ab32539; Abcam), Beclin-1
(1:1,000; cat. no. 3495), cleaved caspase-3 (1:1,000; cat. no.
9664; both Cell Signaling Technology, Inc.), TLR4 (1:1,000; cat.
no. ab13556; Abcam), microtubule-associated protein light chain
(LC)3II (1:1,000; cat. no. 3868; Cell Signaling Technology, Inc.),
PTEN (1:1,000; cat. no. ab31392; Abcam), Bcl2 (1:1,000; cat. no.
3498; Cell Signaling Technology, Inc.), Bad (1:1,000; cat. no.
ab90435), CXCR4 (1:1,000; cat. no. ab1670), CXCR7 (1:1,000; cat.
no. ab38089; all Abcam), t-JNK (1:1,000; cat. no. 4672), p-JNK
(1:1,000; cat. no. 9251) and autophagy protein 5 (Atg5; 1:1,000;
cat. no. 12994; all Cell Signaling Technology, Inc.). The second
antibodies used in the present study were as follows: Horseradish
peroxidase-conjugated secondary antibodies (1:2,000; cat. nos. 7076
and 7074; Cell Signaling Technology, Inc.) for 1 h at room
temperature. Band intensities were normalized to the respective
internal standard signal intensity (β-actin or GAPDH) using
Quantity One Software (version 4.6.2; Bio-Rad Laboratories,
Inc.).
Flow cytometric analysis of cellular
ROS
To observe the cellular ROS levels, flow cytometric
analysis was used. In brief, nasal epithelial cell
(1×106) was washed with PBS, and the ROS probe (5 mg/ml;
dihydroethidium; Molecular Probes; Thermo Fisher Scientific, Inc.)
was incubated with the cells for ~30 min at 37°C in the dark.
Subsequently, the cells were washed with PBS to remove the ROS
probe. Following this, the cells were digested with 0.25%
pancreatin (41). Following
resuspension in PBS, the cells were immediately analyzed using a
flow cytometer (Sysmex Partec GmbH, Görlitz, Germany). The
quantification of cellular ROS was performed per 10,000 cells in
each group, and the data were analyzed with Flowmax software
(Sysmex Partec, Version 2.3, Germany) (42).
Mitochondria permeability transition
pore (mPTP) opening assay and ATP production
mPTP opening is an early event in mitochondrial
apoptosis (43). In the present
study, mPTP opening was measured via tetramethylrhodamine ethyl
ester fluorescence. The nasal epithelial cell (1×106)
was washed with PBS approximately three times and then were loaded
with tetramethylrhodamine ethyl ester. The baseline fluorescence of
tetramethylrhodamine ethyl ester was recorded. Following 30 min,
the tetramethylrhodamine ethyl ester fluorescence was recorded
again. According to a previous study (44), the mPTP opening rate was determined
when the fluorescence intensity was decreased to half of the
baseline fluorescence intensity. ATP production was detected to
reflect mitochondrial function. First, the samples were washed with
cold PBS approximately three times. Subsequently, the samples were
lysed in Laemmli Sample Buffer, and the luciferase-based ATP assay
kit (cat. no. S0026B, Beyotime Institute of Biotechnology, Haimen,
China) was used. ATP production was measured via a microplate
reader (45).
JC-1 staining and isolation of
mitochondrial-enriched fraction
The JC-1 assay was used to investigate mitochondrial
potential. Briefly, the MitoProbe™ JC-1 assay kit (Thermo Fisher
Scientific Inc.) was applied to cells (1×106) at 37°C in
the dark for 15–20 min. Subsequently, PBS was used to wash the
cells three times. Finally, mitochondrial potential was determined
using a fluorescence microscope and the images were captured
(46). To isolate the
mitochondrial fraction in order to analyze the expression
associated with mitochondrial HtrA2/Omi, cells (1×106)
were washed with cold PBS and incubated on ice in lysis buffer
(Beyotime Institute of Biotechnology) for 30 min. Then,
mitochondria were isolated using a commercial kit (cat. no. C3601,
Beyotime Institute of Biotechnology) according to the previous
study (47).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to isolate total RNA from cells
(24). Subsequently, the Reverse
Transcription kit (Kaneka Eurogentec S.A., Seraing, Belgium) was
applied to transcribe RNA (1 µg in each group) into cDNA at room
temperature (~25°C) for 30 min. The qPCR was performed with primers
and matched probes from the Universal Fluorescence-labeled Probe
Library (Roche Applied Science) using SYBR™ Green PCR Master Mix
(Thermo Fisher Scientific, Inc.) (48). The primers used in the present
study were as follows: PTEN forward, 5′-GACTGGCCCAGTGTTCTTCGCTTC-3′
and reverse, 5′-GCTTCTGACAGAAGGAAAGCCAA-3′; and GAPDH forward,
5′-GCTACAGCTTCACCACCACA-3′ and reverse,
5′-GCCATCTCTTGCTCGAAGTC-3′). The cycling conditions were as
follows: 95°C for 8 min, followed by 35 cycles of 95°C for 10 sec
and 72°C for 12 sec, for telomere PCR. Fold change of PTEN mRNA
expression was normalized by GAPDH as an internal control.
RNA interference assay
In the present study, to inhibit TLR4 expression,
small interference (si)RNA against TLR4 was used. The siRNA
sequences were as follows: siRNA sense strand,
5′-GCTACTGTAGGAUAGTAU-3′ and antisense strand,
3′-TCTTCUUAGCTGCATAAU-5′. The siRNA was designed and purchased from
Yangzhou Ruibo Biotech Co., Ltd. (Yangzhou, China). To transfect
the siRNA, Opti-Minimal Essential Medium (Invitrogen; Thermo Fisher
Scientific, Inc.) was incubated with nasal epithelial cell
(1×106) for at least 24 h. Subsequently,
Lipofectamine® 2000 transfection reagent (Thermo Fisher
Scientific, Inc.) was used according to the manufacturer's protocol
to perform siRNA transfection (70 nM/well of siRNA) (49). Following transfection for 36–48 h,
cells were lysed, and the proteins were isolated to measure the
TLR4 expression via western blotting.
Cell migration assay
For the cell migration assay, Transwell units with
an 8 µm pore size polycarbonate filter were used. Cells
(~1×105) were seeded in the upper chamber of the
Transwell units with 1% FBS. The lower chamber was filled with 600
µl of L-DMEM supplemented with 1% FBS. After incubating at 37°C for
12 h, the medium was removed, and cells were fixed with 3.7%
paraformaldehyde for ~10 min (48). The cells in the upper chamber were
removed by a cotton swab. Subsequently, the migrated cells were
stained with 0.1% crystal violet for 15 min at room temperature.
Subsequently, the samples were observed under a digital microscope
system (IX81; Olympus Corporation). The images were captured, and
the migrated cells were recorded in at least five fields (50).
Lactate dehydrogenase (LDH) assay and
caspase-3/-9 activity detection
LDH is released into the medium when cellular
membranes rupture (51). To
evaluate the LDH level in the medium, an LDH Release Detection kit
(Beyotime Institute of Biotechnology) was used. To analyze changes
in caspase-3 and caspase-9, caspase-3/-9 activity kits (Beyotime
Institute of Biotechnology) were used according to the
manufacturer's protocol (52). To
analyze caspase-3 activity, 5 µl DEVD-p-NA substrate (4 mM, 200 µM
final concentration) was added to the samples for 2 h at 37°C. To
measure caspase-9 activity, 5 µl LEHD-p-NA substrate (4 mM, 200 µM
final concentration) was added to the nasal epithelial cell
(1×106) for 1 h at 37°C. Subsequently, the wavelength at
400 nm was recorded via a microplate reader to reflect the
caspase-3 and caspase-9 activities (53).
Bromodeoxyuridine (BrdU) assay
To evaluate cellular proliferation, a BrdU assay
(Guangzhou RiboBio Co., Ltd., Guangzhou, China) was used according
to the method of a previous study (54). Firstly, cells with or without PTEN
overexpression were fixed in 4% paraformaldehyde at 4°C for 30 min,
followed by permeabilization with 0.5% Triton X-100 for ~20 min at
room temperature. Subsequently, samples were incubated in 2 N HCl
solution for 30 min at 37°C to unmask the antigens, followed by a
neutralization step with 0.1 M sodium tetraborate at room
temperature for 30 min. Subsequently, a BrdU antibody (1:200; cat.
no. ab8152; Abcam) was incubated with the samples overnight 4°C.
Following three rinses in PBS, a secondary antibody (1:1,000; cat.
no. A-21206; Invitrogen; Thermo Fisher Scientific, Inc.) was added
to the samples for 1 h at room temperature (55). Finally, the cells were stained with
DAPI (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 5 min at
room temperature to identify the nuclei. Subsequently, the samples
were observed under a fluorescence microscope. Images were
captured, and the number of BrdU-positive cells was measured via
counting at least three random separate fields.
MTT and terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assays
MTT experiments were performed in 96-well plates.
Nasal epithelial cell (1×106) was washed 3 times with
PBS at room temperature, and 50 µl MTT reagent was added to each
well. The samples were subsequently incubated for 4 h at 37°C in a
humid atmosphere containing 5% CO2. The MTT solution was
removed, 200 µl dimethyl sulfoxide was added to each sample, and
the samples were incubated for 10 min at room temperature (13,56).
Following the addition of Sorensen's buffer, the absorbance at the
wavelength of 570 nm was determined. To detect DNA fragmentation in
the cell nuclei (a marker of apoptosis in testicular tissue), a
TUNEL assay for nasal epithelial cell (1×106) was
performed using an In Situ Cell Death Detection kit (Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's protocol (57).
DAPI (5 mg/ml) was used to label the nuclei (at room temperature
for ~30 min) (58).
Construction of adenovirus for PTEN
overexpression
The pDC315-PTEN vector was designed and purchased
from Vigene Biosciences, Inc. (Rockville, MD, USA). Briefly, the
plasmid (3.0 µg per 1×104 cells/well) was transfected
into 1×106 293 cells (CRL-1573™; American Type Culture
Collection, Manassas, VA, USA) using Lipofectamine 2000®
(Invitrogen; 110 Thermo Fisher Scientific, Inc.). When the cells
detached from the plates, the medium supernatant was collected.
Subsequently, the viral supernatant was identified and amplified to
obtain adenovirus-PTEN. Subsequently, 100 multiplicity of infection
adenovirus-PTEN was transduced into the cells to overexpress PTEN
(59). Following the transfection
of nasal epithelial cells with Ad-PTEN, the overexpression
efficiency of Ad-PTEN-transfected nasal epithelial cell was
evaluated via western blotting and an immunofluorescence assay
using PTEN antibody. The western blotting and immunofluorescence
were performed as aforementioned. PTEN fluorescence was observed
under an inverted microscope (excitation wavelength, 550 nm;
magnification, ×40; BX51; Olympus Corporation).
Statistical analysis
All data are expressed as the mean + standard
deviation. Statistical analyses were performed using SPSS software
(version 17.0; SPSS, Inc., Chicago, IL, USA). The results from more
than two groups were evaluated by one-way analysis of variance
followed by the least significant difference test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Regaining PTEN expression increases
human nasal epithelial cell survival in the context of TNFα-induced
inflammatory injury
TNFα was used to induce an inflammatory injury, and
nasal epithelial cell viability was detected via MTT assay. As
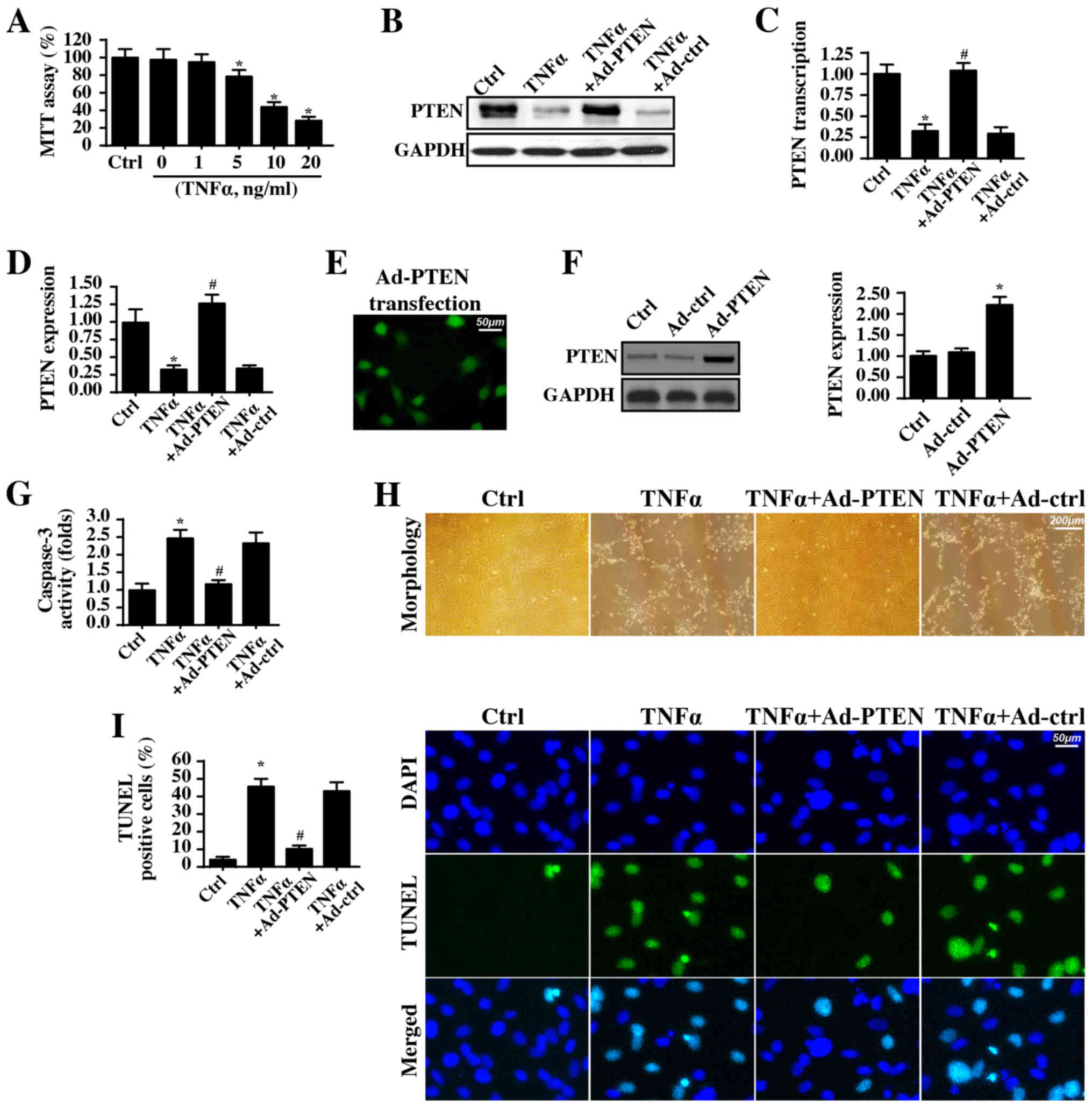
illustrated in Fig. 1A, the TNFα
incubation dose-dependently reduced cellular viability. Notably,
the minimum cytotoxic concentration of TNFα was 5 ng/ml. These
results were similar to a previous study (60). Therefore, based on this data and
previous findings (60), a dosage
of 5 ng/ml TNFα for ~12 h was used for the subsequent experiments.
Additionally, an alteration in PTEN levels was observed following
the inflammatory injury. Compared with that of the control cells,
PTEN expression was significantly decreased in response to TNFα
treatment (Fig. 1B-D). To explain
the causal role of PTEN in TNFα-induced inflammatory injury, PTEN
was overexpressed in TNFα-treated nasal epithelial cells. The
overexpression efficiency in Ad-PTEN-transfected cells was verified
via immunofluorescence assay (Fig.
1E) and western blotting (Fig.
1F). Notably, PTEN overexpression significantly reduced
caspase-3 activity compared with that in TNFα-treated cells
(Fig. 1G), indicating that PTEN
overexpression improved nasal epithelial cell survival in the
context of TNFα-induced inflammatory injury. The cell morphology
with TNFα treatment is presented in Fig. 1H and PTEN overexpression preserved
cellular organization and normal cell morphology upon TNFα
exposure. To provide more direct, evidence for the anti-apoptotic
effect of PTEN in inflammatory injury, a TUNEL assay was conducted.
Compared with the number of positive cells in the control group,
the TNFα treatment increased the number of TUNEL-positive cells
(Fig. 1I), and this effect was
inhibited by PTEN overexpression. In summary, these data suggested
that PTEN overexpression prevented nasal epithelial cell apoptosis
during a TNFα-induced inflammatory injury.
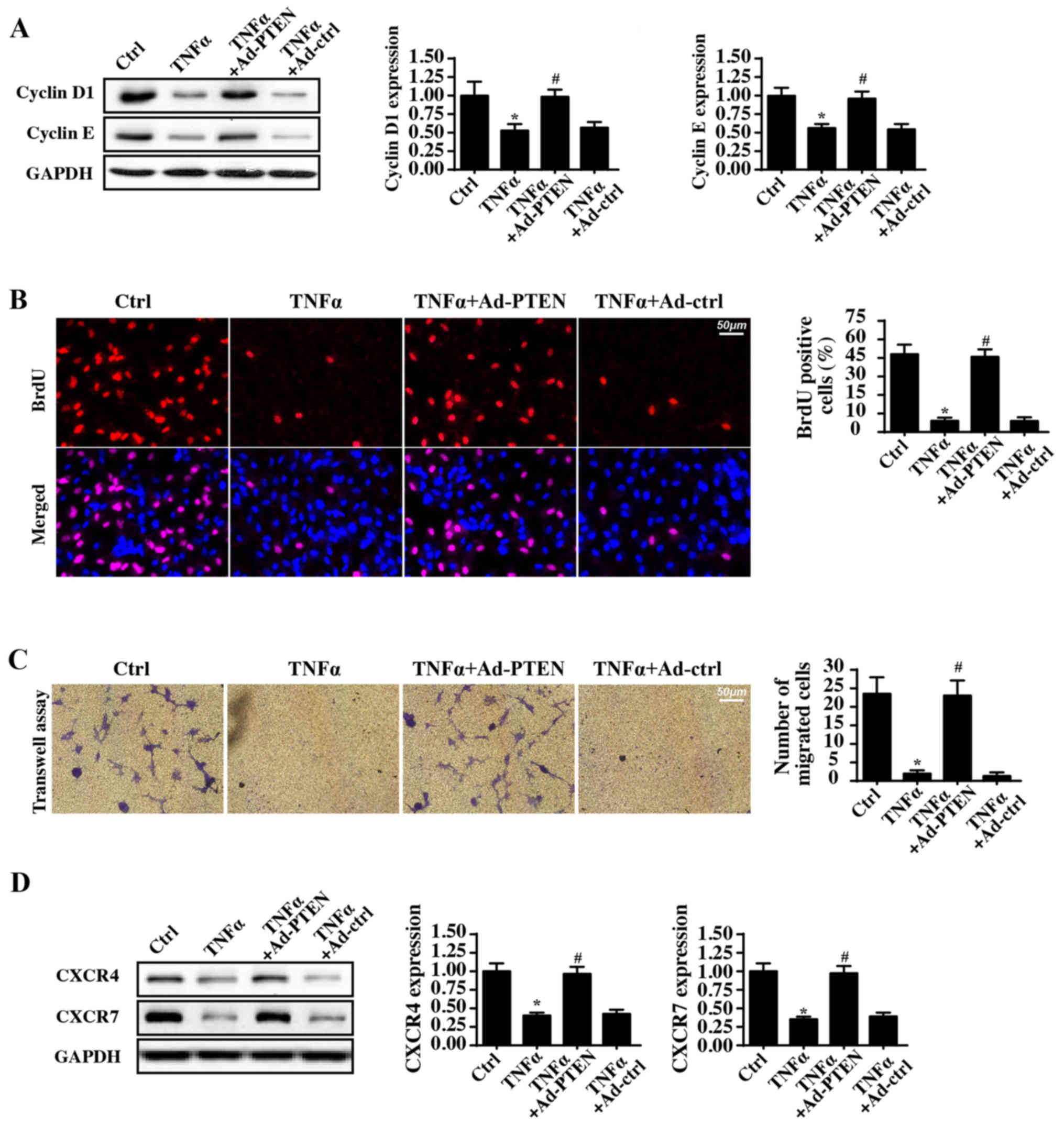
PTEN reverses the proliferation and
migration of nasal epithelial cells during inflammatory injury
In addition to cellular apoptosis, the protective
role of PTEN in cellular proliferation and migration, which are
vital for airway repair in response to the inflammatory injury, was
also observed. Cyclin D1 and Cyclin E expression levels were
significantly decreased in response to TNFα treatment, whereas PTEN
overexpression reversed this effect (Fig. 2A). Regarding cellular
proliferation, Cyclin D1 and Cyclin E interact with each other and
generate the cyclin-dependent kinase (Cdk)4/6-cyclin D and/or
Cdk2-cyclin E complexes, which accelerate the transition from the
G0/G1 to S stage (38).
Subsequently, to observe the number of cells at S-phase, BrdU
staining was used. Compared with the control group, TNFα incubation
significantly reduced the proportion of BrdU-positive cells
(Fig. 2B). In comparison, PTEN
overexpression reversed this decrease in the number of
BrdU-positive cells.
Subsequently, the cellular migratory response was
also observed. In Transwell assays, the number of migrated cells
was decreased by TNFα treatment and was increased to normal levels
with PTEN overexpression (Fig.
2C). These data suggested that during TNFα-mediated
inflammatory injury, PTEN overexpression increased cellular
migration. In addition, the chemotactic factor C-X-C chemokine
receptor type (CXCR)4/7 expression level was also measured.
Notably, the enriched CXCR4/7 expression was inhibited by the TNFα
treatment (Fig. 2D). By contrast,
PTEN overexpression restored the intracellular CXCR4/7 levels.
Collectively, this information indicated that PTEN is necessary for
nasal epithelial cell proliferation and migration during
inflammatory injury.
Overexpression of PTEN alleviates
mitochondrial damage
Recent studies have suggested that mitochondria are
a potential target in inflammatory injury (11,61).
Mitochondrial damage leads to energy metabolism disorders (62). Moreover, inflammatory injury also
activates mitochondrial apoptosis, resulting in cellular death
(63,64). Due to energy deficiencies and
apoptosis activation, nasal epithelial cells are unable to survive,
migrate and proliferate under inflammatory injury conditions
(65). Therefore, whether PTEN
protected nasal epithelial cells via maintaining mitochondrial
homeostasis was investigated. First, cellular ROS levels were
detected via flow cytometry. When compared with the control group,
TNFα treatment produced excessive cellular ROS levels (Fig. 3A). Bu contrast, PTEN overexpression
almost completely reversed the ROS production induced by TNFα. As a
consequence of the cellular oxidative stress, TNFα treatment
mediated the opening of the mPTP, and this effect was inhibited by
PTEN overexpression (Fig. 3B).
mPTP opening is believed to be the primary initiating factor for
the mitochondrial death pathway via facilitating HtrA2/Omi leakage
from the mitochondria into the cytoplasm/nucleus (66). Immunofluorescence assays of
HtrA2/Omi levels demonstrated low levels of HtrA2/Omi expression in
the cytoplasm/nucleus (Fig. 3C).
By contrast, TNFα treatment promoted HtrA2/Omi expression in the
cytoplasm. Furthermore, western blotting also demonstrated that
TNFα treatment increased, whereas PTEN overexpression reduced, the
expression of cytoplasmic HtrA2/Omi (Fig. 3D), indicating that TNFα-mediated
HrtA2/Om2 leakage into cytoplasm may be repressed by PTEN
overexpression. Finally, alterations in the proteins associated to
the mitochondrial death pathway were investigated. Bax and Bad
expression levels were both upregulated, whereas the expression of
protective Bcl2 was downregulated in response to the TNFα treatment
(Fig. 3D). Bcl2 interacts with and
inhibits Bax in order to limit excessive mPTP opening (67). TNFα treatment also increased
caspase-9 and caspase-3 expression (Fig. 3D), indicative of mitochondrial
death pathway activation. However, PTEN overexpression corrected
the imbalance between Bcl2 and Bax expression and thus alleviated
caspase-3 and caspase-9 expression. These data indicated that PTEN
was capable of repressing TNFα-induced mitochondrial injury.
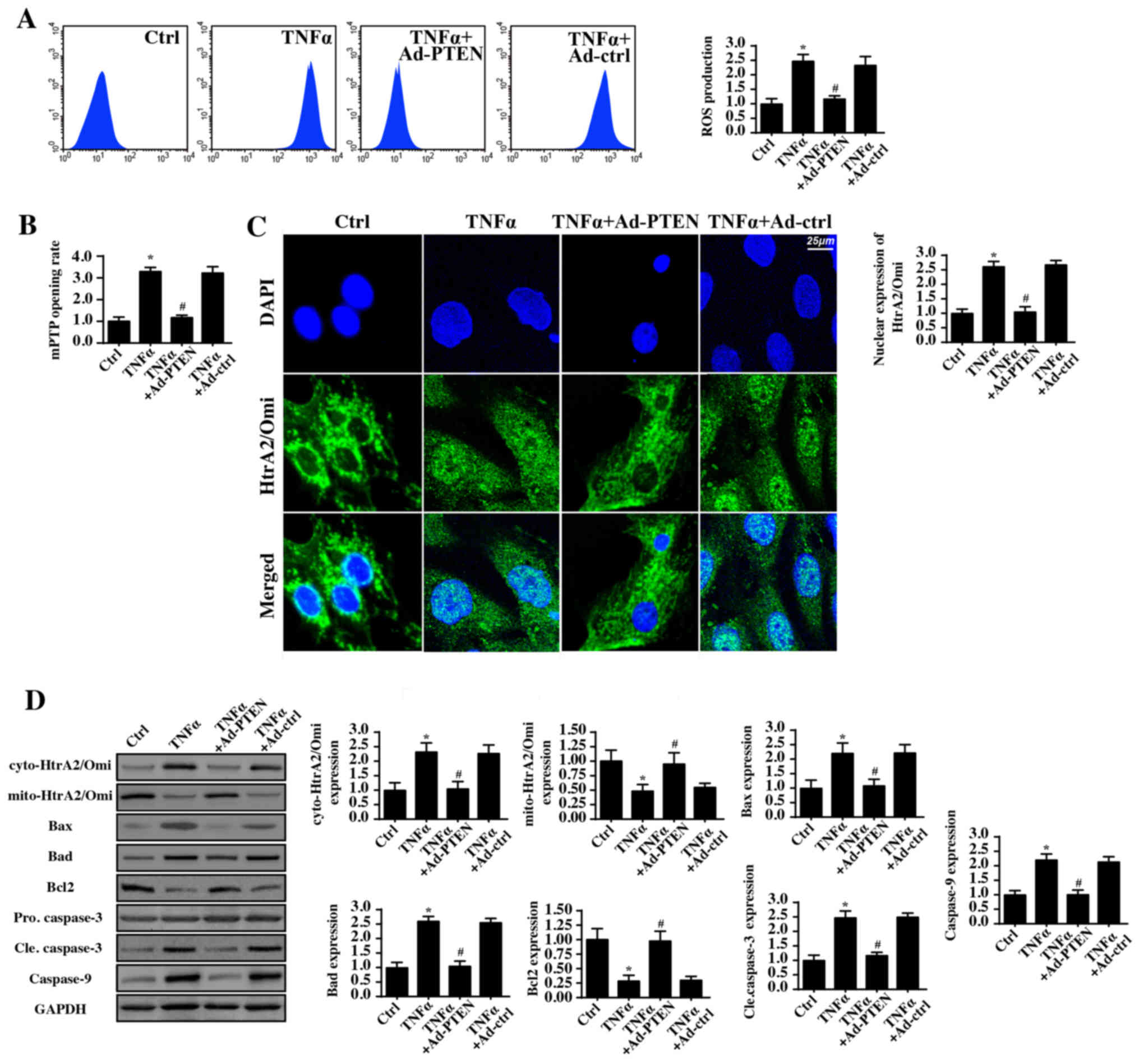 | Figure 3.PTEN overexpression protects
mitochondria against TNFα-mediated inflammatory injury. (A) The
alterations in cellular ROS levels were detected via flow
cytometry. (B) The mPTP opening rate. (C) Mitochondrial HtrA2/Omi
expression was diffused, as detected via immunofluorescence. Scale
bar, 25 µm. (D) Western blotting was used to evaluate the
expression of mitochondrial apoptotic proteins following TNFα
treatment with or without PTEN overexpression. *P<0.05 vs. Ctrl
group; #P<0.05 vs. TNFα group. Ctrl, control; TNFα,
tumor necrosis factor α; Ad, adenovirus; PTEN, phosphatase and
tensin homolog; ROS, reactive oxygen species; mPTP; mitochondria
permeability transition pore; HtrA2/Omi, HtrA serine peptidase 2;
cyto, cytoplasmic; mito, mitochondrial; Bax, Bcl-associated X
protein; Bad, Bcl-2-associated death promoter; Bcl2, B-cell
lymphoma. The experiment was repeated three times. |
PTEN reduces mitophagy activity
Previous studies from several researchers have
demonstrated that mitochondrial injury results from the mitophagy,
a type of autophagy selective for mitochondria. Moderate mitophagy
removes poorly structured mitochondria (59). In contrast, excessive mitophagy
induces mitochondrial dysfunction via aberrant mitochondrial
degradation (22). To test whether
PTEN protected mitochondrial function via inhibiting mitophagy,
mitophagy activity was investigated via western blotting. Compared
to the levels in the control group, TNFα treatment significantly
increased the mitochondrial LC3II, Beclin1, p62 and Atg5 levels
(Fig. 4A). By contrast, PTEN
overexpression significantly alleviated these mitophagy parameters.
These data indicated that PTEN inhibited TNFα-mediated mitophagy
activation.
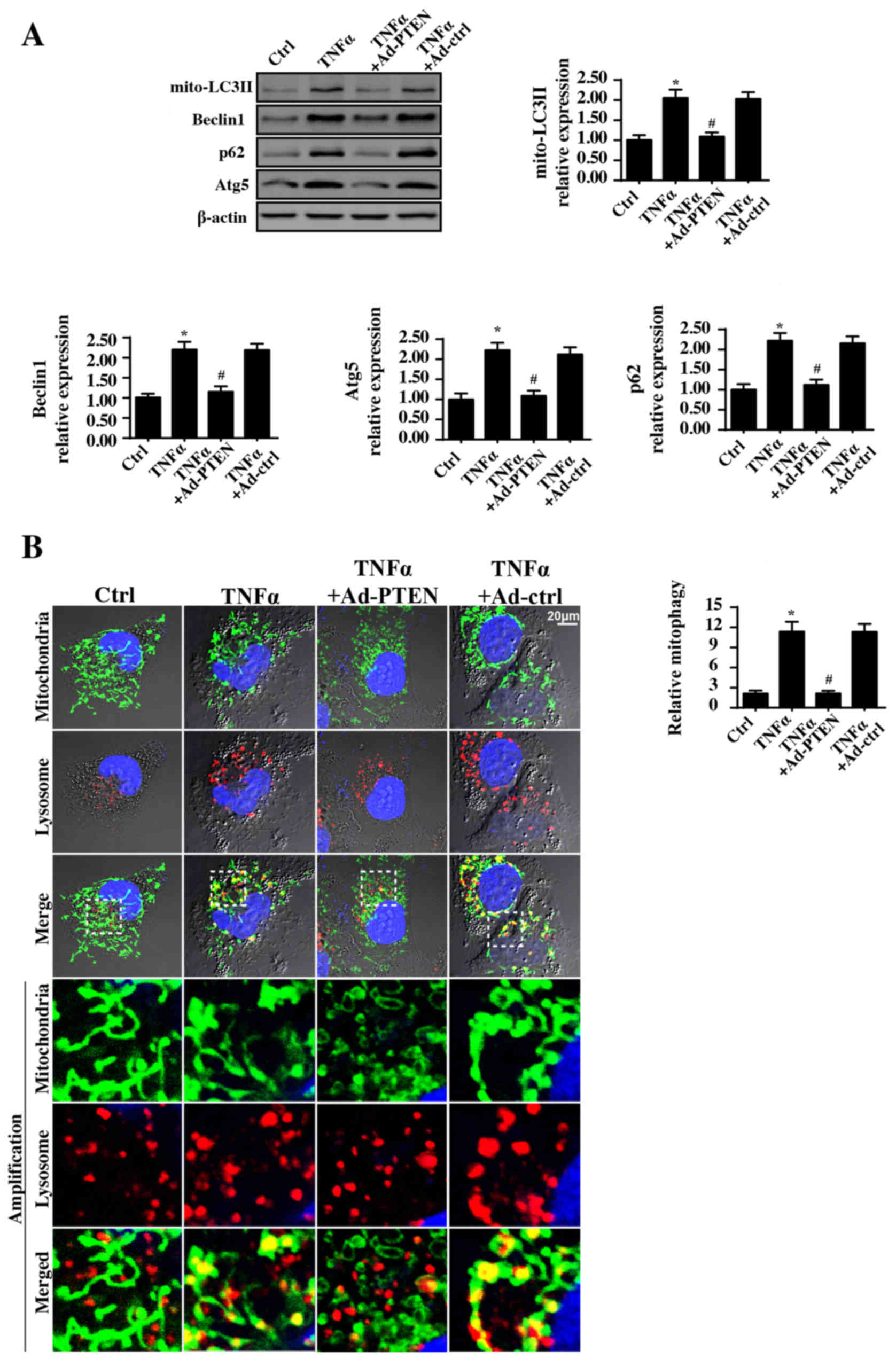 | Figure 4.PTEN overexpression inhibits
mitophagy activity. (A) Western blotting was used to observe
alterations in the mitophagy markers. (B) Immunofluorescence assays
of mitophagy was carried out via co-staining of mitochondria and
lysosomes and the extent of mitophagy was recorded. Mitophagy is
the result of fusion between mitochondria and lysosome. The green
mitochondria locates with red lysosome would generate the orange
mitophagy. Then, the number of orange dots were measured to
quantify the number of mitophagy. Scale bar, 20 µm.*P<0.05 vs.
Ctrl group; #P<0.05 vs. TNFα group. Ctrl, control;
TNFα, tumor necrosis factor α; Ad, adenovirus; PTEN, phosphatase
and tensin homolog; mito, mitochondrial; LC3II,
microtubule-associated protein light chain 3 II; p62, Sequestome-1;
Atg5, autophagy protein 5. The experiment was repeated three
times. |
Furthermore, to provide direct evidence for the
inhibitory role of PTEN in mitophagy modulation, immunofluorescence
assay of mitophagy via mitochondria and lysosome co-staining was
performed. Compared with the control group, TNFα promoted fusion
between mitochondria and lysosomes (Fig. 4B), indicative of mitophagy
activation. By contrast, PTEN overexpression reduced the overlap
between mitochondrial and lysosomal staining, suggestive of
mitophagy inhibition.
Mitophagy inhibition induced by PTEN
overexpression provides mitochondrial protection
To determine whether mitophagy inhibition by PTEN
overexpression was responsible for the mitochondrial protection,
mitophagy activity in PTEN-overexpressing cells was reactivated via
FCCP, a mitophagy activator. Meanwhile, mitophagy activity was also
inhibited via 3-MA in TNFα-treated cells, which was used as the
negative control group. Subsequently, mitochondrial damage was
detected via staining with JC-1, a mitochondrial
potential-sensitive dye. Compared with that of the control cells,
TNFα treatment reduced the mitochondrial potential (Fig. 5A), and this effect was inhibited by
3-MA or PTEN overexpression, suggesting that mitophagy inhibition
reversed mitochondrial function. By contrast, mitophagy activation
in PTEN-overexpressing cells disrupted the mitochondrial potential.
In addition, the cellular oxidative stress level was also
investigated via ROS staining. Compared with that of the control
group, the cellular ROS level was increased in the TNFα-treated
cells but was decreased to normal levels with 3-MA treatment or
with PTEN overexpression (Fig.
5B). Notably, mitophagy activation reversed the inhibitory
effects of PTEN-overexpression on ROS overproduction. Furthermore,
it was also detected caspase-9 activity, which is activated in
response to mitochondrial damage. Notably, caspase-9 activity was
upregulated following TNFα treatment. In contrast, 3-MA treatment
or PTEN overexpression significantly reduced caspase-9 activity.
However, mitophagy reactivation by FCCP in PTEN-overexpressing
cells again increased caspase-9 activity (Fig. 5C). Finally, the changes in cellular
ATP production were also investigated. Compared with the ATP
generated in the control group, TNFα treatment reduced ATP
production, and this effect was reversed by 3-MA treatment or PTEN
overexpression. Notably, mitophagy activation rescued this decline
in cellular ATP production (Fig.
5D). In summary, these data indicated that PTEN overexpression
protected mitochondrial homeostasis via suppressing mitophagy
activity.
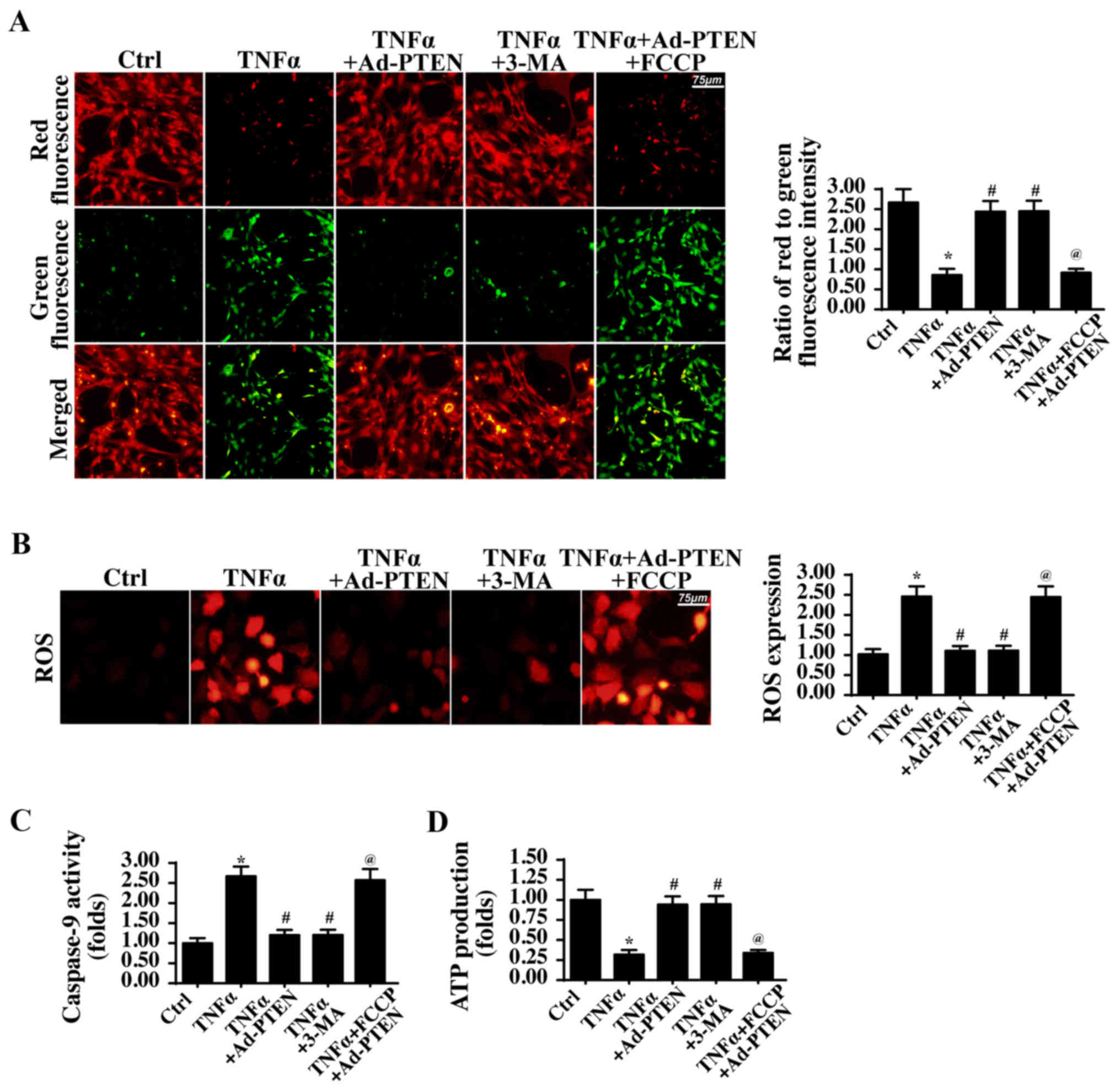 | Figure 5.PTEN-regulated mitophagy influences
mitochondrial function. (A) Mitochondrial potential was observed
via JC1 staining. Red fluorescence indicated a normal mitochondrial
potential, whereas green fluorescence suggested a damaged
mitochondrial potential. Scale bar, 75 µm. (B) The cellular ROS
level was measured using a dihydroethidium probe. Scale bar, 75 µm.
(C) Caspase-9 activity was detected to reflect mitochondrial
apoptosis during mitophagy activation and/or inactivation. (D) ATP
production was evaluated via ELISA. *P<0.05 vs. Ctrl group;
#P<0.05 vs. TNFα group; @P<0.05 vs.
TNFα+Ad-PTEN group. Ctrl, control; TNFα, tumor necrosis factor α;
Ad, adenovirus; PTEN, phosphatase and tensin homolog; 3-MA,
3-methyladenine; FCCP, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone; ROS, ROS, reactive
oxygen species; ATP, adenosine triphosphate. The experiment was
repeated three times. |
PTEN regulates mitophagy via
repressing the TLR4-JNK-Bnip3 pathway
To determine the mechanism by which PTEN inhibits
mitophagy, the JNK-Bnip3 pathway was investigated. Previous reports
have demonstrated that mitophagy is activated via the JNK-Bnip3
axis in fatty liver disease (59),
colorectal cancer (68),
hepatocellular carcinoma (24) and
cardiac reperfusion injury (23).
Accordingly, whether PTEN modified mitophagy via the JNK-Bnip3
pathway was investigated. Additionally, to determine whether TLR4
was involved in the JNK-Bnip3 pathway activation, TLR4 expression
we knocked down using siRNA (Fig.
6A). In the present study, it was demonstrated that JNK and
Bnip3 were both activated in response to the TNFα treatment, via
western blotting (Fig. 6B). By
contrast, PTEN overexpression significantly reduced JNK
phosphorylation levels and Bnip3 expression in TNFα-treated
cells.
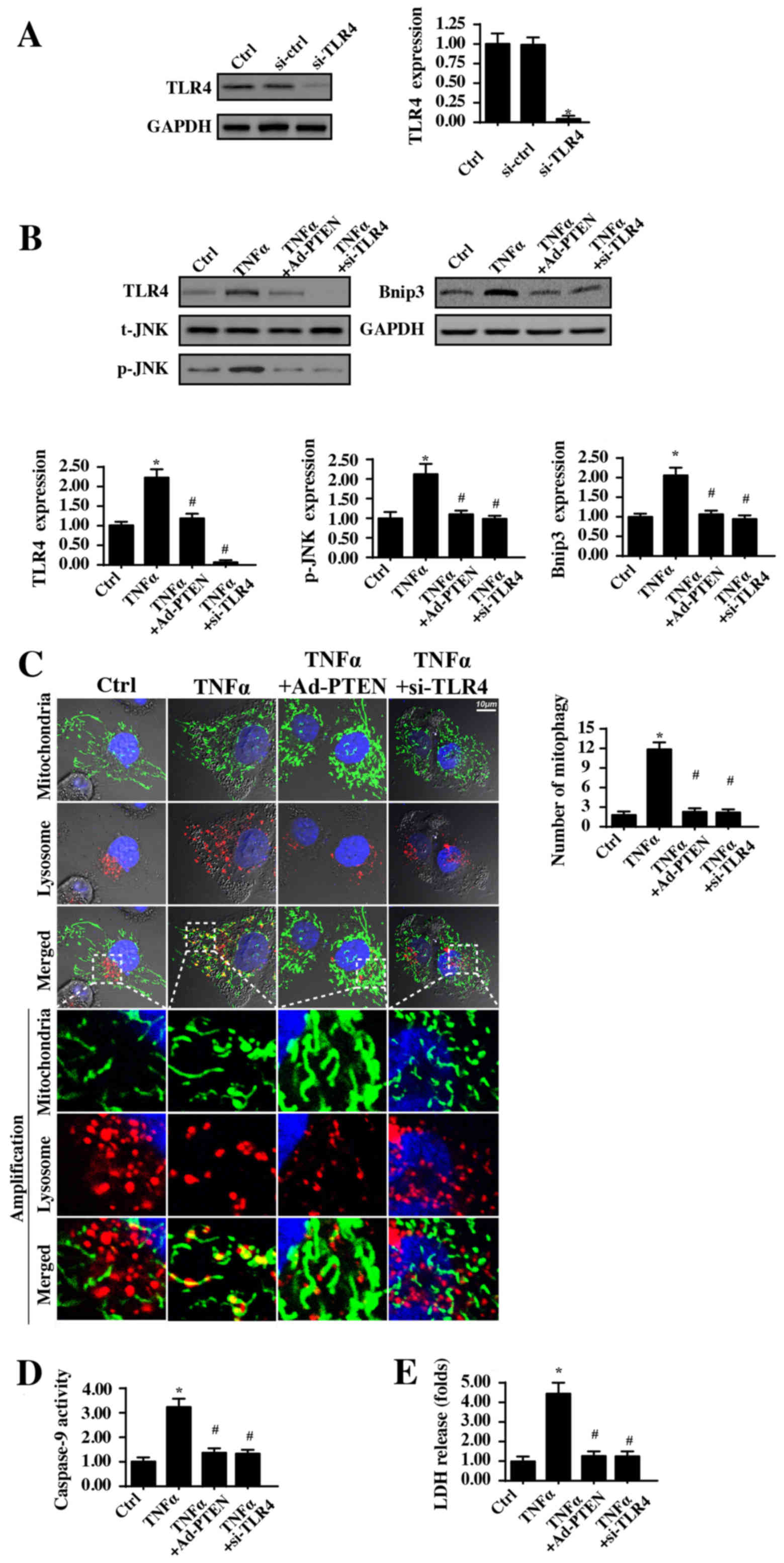 | Figure 6.PTEN controls mitophagy activity via
the TLR4-JNK-Bnip3 pathway. (A) TLR4 siRNA was used to reduce TLR4
expression. (B) Western blotting was used to analyze the expression
of TLR4, phosphorylated JNK and Bnip3 with or without PTEN
overexpression and TLR4 silencing. (C) Immunofluorescence assays of
mitophagy were carried out via co-staining mitochondria (TOM20
homolog) and lysosomes (lysosome-associated membrane glycoprotein
1). The boxed area under each image represents the amplification of
the white square and the amount of mitophagy was recorded. Scale
bar, 10 µm. (D) Caspase-9 activity was detected to reflect
mitochondrial apoptosis when TLR4 expression was knocked down. (E)
LDH release assays were carried to measure cell death when TLR4
expression was silenced. *P<0.05 vs. Ctrl group or si-ctrl
group; #P<0.05 vs. TNFα group. Ctrl, control; si,
small interfering RNA; TLR4, Toll-like receptor 4; TNFα, tumor
necrosis factor α; Ad, adenovirus; PTEN, phosphatase and tensin
homolog; t, total; JNK, c-Jun kinase; p, phosphorylated; Bnip3,
Bcl2-interacting protein 3; LDH, lactate dehydrogenase. The
experiment was repeated three times. |
To investigate the upstream signaling responsible
for the JNK-Bnip3 pathway activation by TNFα, TLR4 expression was
measured. Notably, TLR4 expression was increased in response to the
TNFα treatment, and this trend was strongly inhibited by PTEN
overexpression (Fig. 6B). Notably,
following knockdown of TLR4 expression in TNFα-treated cells, JNK
phosphorylation levels and Bnip3 expression were decreased
(Fig. 6B). These data indicated
that TLR4 functioned upstream of JNK-Bnip3 axis activation.
Additionally, whether TLR4 was also associated with mitophagy
activation, an immunofluorescence assay of mitophagy was conducted.
As demonstrated in Fig. 6C,
TNFα-mediated mitochondria-lysosome fusion was inhibited by TLR4
silencing or PTEN overexpression, suggesting that TLR4 was
necessary for TNFα-induced mitophagy activation. Furthermore,
whether TLR4 was also required for TNFα-associated mitochondrial
damage and cellular death, caspase-9 activity was evaluated, and
the LDH release assay was conducted. TLR4 knockdown significantly
reduced caspase-9 activity (Fig.
6D) and LDH release (Fig. 6E);
similar results were also obtained in PTEN-overexpressing cells. In
summary, the present data indicated that TNFα activated mitophagy
via the TLR4-JNK-Bnip3 pathway, leading to mitochondrial damage and
cellular death. However, PTEN overexpression inhibited the
TLR4-JNK-Bnip3-mitophagy axis, maintaining mitochondrial
homeostasis and providing a survival advantage for nasal epithelial
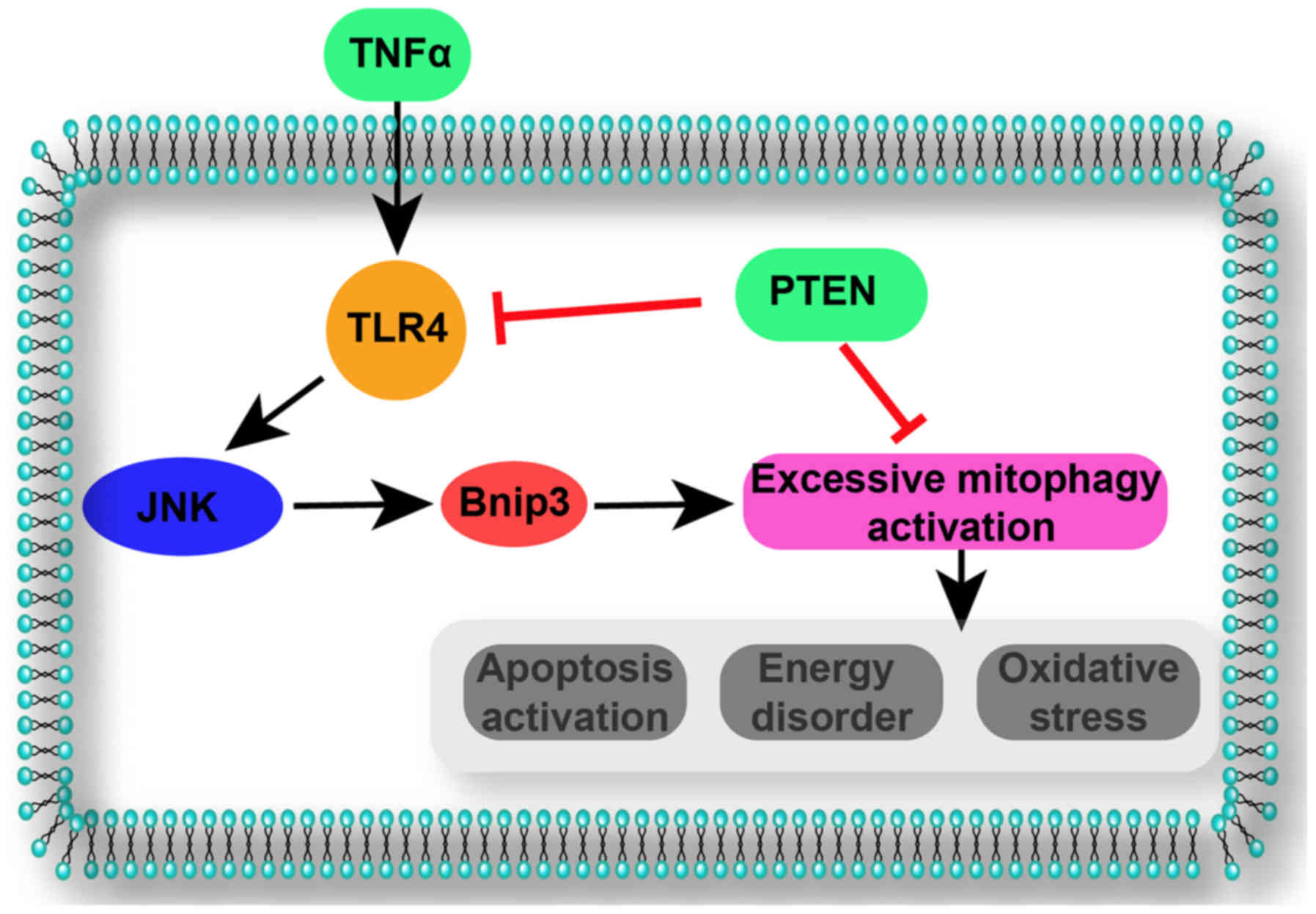
cells under TNFα-induced inflammatory injury (Fig. 7).
Discussion
In the present study, the following were
demonstrated: i) PTEN was downregulated in nasal epithelial cells
under TNFα-induced inflammatory injury; ii) regaining PTEN
expression alleviated inflammation-mediated cellular apoptosis,
proliferation arrest and migration inhibition; iii)
mechanistically, PTEN overexpression attenuated mitochondrial
dysfunction via repression of mitophagy activity; iv)
inflammation-activated mitophagy resulted in aberrant mitochondrial
degradation, leading to a reduced energy supply and apoptosis
activation; and v) PTEN overexpression abrogated lethal mitophagy
via blocking the TLR4-JNK-Bnip3 pathway, maintaining mitochondrial
homeostasis and promoting cellular survival in the context of
inflammatory injury. Based on the above findings, the mechanism
underlying inflammatory injury-induced apoptosis in nasal
epithelial cells was investigated; this pathogenesis was associated
with PTEN downregulation, mitophagy upregulation and TLR4-JNK-Bnip3
pathway activation. To the best of our knowledge, this is the first
study to describe the role of PTEN-associated mitophagy in nasal
epithelial cell inflammatory injury.
Several studies have reported that COPD development
is associated with chronic nasal and sinus inflammation (69). In addition, anti-inflammation
therapy is effective for alleviating upper airway symptoms such as
rhinorrhea, nasal obstruction and sneezing (70). Therefore, this information
confirmed that chronic nasal and sinus inflammatory injury is the
primary pathogenesis for COPD progression. Accordingly, identifying
the molecular basis underlying nasal and sinus inflammatory
injuries may uncover potential therapeutic targets for clinical
practice. In response to TNFα stimulation, PTEN expression was
downregulated in nasal epithelial cells. However, reintroduction of
PTEN expression rescued nasal epithelial cell viability, growth and
mobility. Accordingly, these data identified PTEN as an endogenous
defender against inflammation-mediated cellular damage in nasal
epithelial cells. Previous studies have also illustrated the
beneficial role of PTEN in acute kidney injury (28), diabetes (71), cardiomyopathy (72) and chronic fatty liver disease
(73). Therefore, an approach to
reverse PTEN activity could be considered as adjuvant therapy to
enhance anti-inflammatory treatments in patients with chronic nasal
and sinus inflammation, but this needs to be investigated
further.
Functional studies have demonstrated that
inflammation induced nasal epithelial cell death via mitophagy
(74). In response to TNFα
stimulation in the current study, the TLR4-JNK-Bnip3 pathway was
upregulated and was accompanied by increased mitophagy activity.
Excessive mitophagy results in aberrant mitochondrial degradation,
leading to a shortage in the energy supply and mitochondrial
apoptosis activation (59,63). Notably, several studies have
reported the protective role of mitophagy in acute cardiac ischemia
reperfusion injury (19), diabetes
(75) and non-alcoholic fatty
liver disease (59). These studies
noted that moderate mitophagy activation removed the damaged
mitochondria in a timely manner and maintained mitochondrial
homeostasis. Notably, a report has argued that mitophagy is harmful
to cell viability (62). Excessive
mitophagy activation impaired liver cancer migration and promoted
endothelial mitochondrial apoptosis (22,24).
This discrepancy may result from the different mitophagy regulatory
signaling pathways. Several studies have confirmed that various
mitophagy activation pathways distinctly influence mitophagy
function and cell fate (59,76).
Notably, Bnip3-associated mitophagy is lethal for cardiomyocytes
(23,77), liver cancer (24) and colorectal cancer (68), which is similar to the present
findings. These findings hinted that modulating Bnip3-mediated
mitophagy is vital for nasal epithelial cell survival under
inflammatory injury.
Furthermore, the TLR4-JNK-Bnip3 pathway seemed to be
responsible for mitophagy activation under inflammatory injury,
in vitro. However, PTEN overexpression had the ability to
block the TLR4-JNK-Bnip3 pathway. Notably, a previous study
reported the inhibitory effects of PTEN overexpression on TLR4 or
JNK expression (24). Accordingly,
to the best of our knowledge, this is the first study to establish
the regulatory role of PTEN in the TLR4-JNK-Bnip3 pathway in
chronic nasal and sinus inflammation. Therefore, the present data
provided more information about the association between PTEN and
the TLR4-JNK-Bnip3 pathway.
Collectively, the present data investigated the
molecular mechanism underlying inflammation-associated nasal
epithelial cell damage. Inflammation-induced PTEN downregulation
resulted in TLR4-JNK-Bnip3-mitophagy pathway activation, which
eventually amplified the cellular death signals in nasal epithelial
cells. However, rescuing PTEN activity blocked the TLR4-JNK-Bnip3
pathway and halted mitophagy, favoring nasal epithelial cell
survival in inflammatory injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML, SW and XY made substantial contributions to the
concept and design of the present study, performance of
experiments, data analysis and interpretation, and manuscript
writing.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lv H, Liu Q, Wen Z, Feng H, Deng X and Ci
X: Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute
lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox
Biol. 12:311–324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng KJ, Bao YY and Zhou SH: The role of
hypoxia inducible factor in nasal inflammations. Eur Rev Med
Pharmacol Sci. 20:5067–5076. 2016.PubMed/NCBI
|
|
3
|
Bergmark RW and Pynnonen M: Diagnosis and
first-line treatment of chronic sinusitis. JAMA. 318:2344–2345.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith KA, Waypa GB and Schumacker PT:
Redox signaling during hypoxia in mammalian cells. Redox Biol.
13:228–234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blackburn NJR, Vulesevic B, McNeill B,
Cimenci CE, Ahmadi A, Gonzalez-Gomez M, Ostojic A, Zhong Z,
Brownlee M, Beisswenger PJ, et al: Methylglyoxal-derived advanced
glycation end products contribute to negative cardiac remodeling
and dysfunction post-myocardial infarction. Basic Res Cardiol.
112:572017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Griffiths HR, Gao D and Pararasa C: Redox
regulation in metabolic programming and inflammation. Redox Biol.
12:50–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brasacchio D, Alsop AE, Noori T, Lufti M,
Iyer S, Simpson KJ, Bird PI, Kluck RM, Johnstone RW and Trapani JA:
Epigenetic control of mitochondrial cell death through
PACS1-mediated regulation of BAX/BAK oligomerization. Cell Death
Differ. 24:961–970. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu P, Wang J, Hong F, Wang S, Jin X, Xue
T, Jia L and Zhai Y: Melatonin prevents obesity through modulation
of gut microbiota in mice. J Pineal Res. 62:2017. View Article : Google Scholar
|
|
9
|
Yin Y, Li F, Li S, Cai J, Shi J and Jiang
Y: TLR4 influences hepatitis B virus related hepatocellular
carcinoma by regulating the Wnt/β-catenin pathway. Cell Physiol
Biochem. 42:469–479. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du GQ, Shao ZB, Wu J, Yin WJ, Li SH, Wu J,
Weisel RD, Tian JW and Li RK: Targeted myocardial delivery of GDF11
gene rejuvenates the aged mouse heart and enhances myocardial
regeneration after ischemia-reperfusion injury. Basic Res Cardiol.
112:72017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galley HF, McCormick B, Wilson KL, Lowes
DA, Colvin L and Torsney C: Melatonin limits paclitaxel-induced
mitochondrial dysfunction in vitro and protects against
paclitaxel-induced neuropathic pain in the rat. J Pineal Res.
63:2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kozlov AV, Lancaster JR Jr, Meszaros AT
and Weidinger A: Mitochondria-meditated pathways of organ failure
upon inflammation. Redox Biol. 13:170–181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han L, Wang H, Li L, Li X, Ge J, Reiter RJ
and Wang Q: Melatonin protects against maternal obesity-associated
oxidative stress and meiotic defects in oocytes via the
SIRT3-SOD2-dependent pathway. J Pineal Res. 63:2017. View Article : Google Scholar :
|
|
14
|
Zhou H, Yang J, Xin T, Li D, Guo J, Hu S,
Zhou S, Zhang T, Zhang Y, Han T and Chen Y: Exendin-4 protects
adipose-derived mesenchymal stem cells from apoptosis induced by
hydrogen peroxide through the PI3K/Akt-Sfrp2 pathways. Free Radic
Biol Med. 77:363–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T,
Ma Q, Han T, Zhang Y, Tian F and Chen Y: Liraglutide protects
cardiac microvascular endothelial cells against
hypoxia/reoxygenation injury through the suppression of the
SR-Ca(2+)-XO-ROS axis via activation of the
GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 95:278–292.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schock SN, Chandra NV, Sun Y, Irie T,
Kitagawa Y, Gotoh B, Coscoy L and Winoto A: Induction of
necroptotic cell death by viral activation of the RIG-I or STING
pathway. Cell Death Differ. 24:615–625. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu L, Feng D, Chen G, Chen M, Zheng Q,
Song P, Ma Q, Zhu C, Wang R, Qi W, et al: Mitochondrial
outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in
mammalian cells. Nat Cell Biol. 14:177–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S,
Zhang Y, Han T, Ren J, Cao F and Chen Y: Melatonin suppresses
platelet activation and function against cardiac
ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy
pathways. J Pineal Res. 63:2017. View Article : Google Scholar :
|
|
20
|
Chen L, Liu L, Li Y and Gao J: Melatonin
increases human cervical cancer HeLa cells apoptosis induced by
cisplatin via inhibition of JNK/Parkin/mitophagy axis. In Vitro
Cell Dev Biol Anim. 54:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nunez-Gomez E, Pericacho M, Ollauri-Ibáñez
C, Bernabéu C and López-Novoa JM: The role of endoglin in
post-ischemic revascularization. Angiogenesis. 20:1–24. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:2017. View Article : Google Scholar :
|
|
23
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu
S, Zhu P, Wang W and Zhou H: Yap promotes hepatocellular carcinoma
metastasis and mobilization via governing
cofilin/F-actin/lamellipodium axis by regulation of
JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14:59–71. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jia M, Chen X, Liu J and Chen J: PTEN
promotes apoptosis of H2O2-injured rat nasal epithelial cells
through PI3K/Akt and other pathways. Mol Med Rep. 17:571–579.
2018.PubMed/NCBI
|
|
26
|
Ackermann M, Kim YO, Wagner WL, Schuppan
D, Valenzuela CD, Mentzer SJ, Kreuz S, Stiller D, Wollin L and
Konerding MA: Effects of nintedanib on the microvascular
architecture in a lung fibrosis model. Angiogenesis. 20:359–372.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gadicherla AK, Wang N, Bulic M,
Agullo-Pascual E, Lissoni A, De Smet M, Delmar M, Bultynck G,
Krysko DV, Camara A, et al: Mitochondrial Cx43 hemichannels
contribute to mitochondrial calcium entry and cell death in the
heart. Basic Res Cardiol. 112:272017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang N, Zhang H, Cai X and Shang Y:
Epigallocatechin-3-gallate inhibits inflammation and
epithelialmesenchymal transition through the PI3K/AKT pathway via
upregulation of PTEN in asthma. Int J Mol Med. 41:818–828.
2018.PubMed/NCBI
|
|
29
|
Zhou J, Zhong J, Lin S, Huang Z, Chen H,
Tang S, Yang C and Fan Y: Inhibition of PTEN activity aggravates
post renal fibrosis in mice with ischemia reperfusion-induced acute
kidney injury. Cell Physiol Biochem. 43:1841–1854. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang
D, Wang J, Qin Y, Liu Y, Tang C, et al: The mitochondria-targeted
antioxidant MitoQ ameliorated tubular injury mediated by mitophagy
in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11:297–311.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dong ZW, Ren CG, Xia Y, Su D, Du TT, Fan
HB, Yuan H, Wang L, Dong M, Li WC, et al: Pten regulates
homeostasis and inflammation-induced migration of myelocytes in
zebrafish. J Hematol Oncol. 7:172014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vahed Zununi S, Barzegari A, Saadat Rahbar
Y, Goreyshi A and Omidi Y: Leuconostoc mesenteroides-derived
anticancer pharmaceuticals hinder inflammation and cell survival in
colon cancer cells by modulating NF-κB/AKT/PTEN/MAPK pathways.
Biomed Pharmacother. 94:1094–1100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu S, Wang X, Geng P, Tang X, Xiang L, Lu
X, Li J, Ruan Z, Chen J, Xie G, et al: Melatonin regulates PARP1 to
control the senescence-associated secretory phenotype (SASP) in
human fetal lung fibroblast cells. J Pineal Res. 63:2017.
View Article : Google Scholar
|
|
34
|
Dufour F, Rattier T, Shirley S, Picarda G,
Constantinescu AA, Morlé A, Zakaria AB, Marcion G, Causse S,
Szegezdi E, et al: N-glycosylation of mouse TRAIL-R and human
TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ.
24:500–510. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang
H, Tang J, Li H, Feng M, Deng P, et al: Melatonin prevents abnormal
mitochondrial dynamics resulting from the neurotoxicity of cadmium
by blocking calcium-dependent translocation of Drp1 to the
mitochondria. J Pineal Res. 60:291–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Alghanem AF, Wilkinson EL, Emmett MS,
Aljasir MA, Holmes K, Rothermel BA, Simms VA, Heath VL and Cross
MJ: RCAN1.4 regulates VEGFR-2 internalisation, cell polarity and
migration in human microvascular endothelial cells. Angiogenesis.
20:341–358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Glab JA, Doerflinger M, Nedeva C, Jose I,
Mbogo GW, Paton JC, Paton AW, Kueh AJ, Herold MJ, Huang DC, et al:
DR5 and caspase-8 are dispensable in ER stress-induced apoptosis.
Cell Death Differ. 24:944–950. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S,
Li Y, Zhou H and Chen Y: Ripk3 promotes ER stress-induced
necroptosis in cardiac IR injury: A mechanism involving calcium
overload/XO/ROS/mPTP pathway. Redox Biol. 16:157–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han J, Weisbrod RM, Shao D, Watanabe Y,
Yin X, Bachschmid MM, Seta F, Janssen-Heininger YMW, Matsui R, Zang
M, et al: The redox mechanism for vascular barrier dysfunction
associated with metabolic disorders: Glutathionylation of Rac1 in
endothelial cells. Redox Biol. 9:306–319. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou W, Yu L, Fan J, Wan B, Jiang T, Yin
J, Huang Y, Li Q, Yin G and Hu Z: Endogenous parathyroid hormone
promotes fracture healing by increasing expression of BMPR2 through
cAMP/PKA/CREB pathway in mice. Cell Physiol Biochem. 42:551–563.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iggena D, Winter Y and Steiner B:
Melatonin restores hippocampal neural precursor cell proliferation
and prevents cognitive deficits induced by jet lag simulation in
adult mice. J Pineal Res. 62:2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou H, Li D, Shi C, Xin T, Yang J, Zhou
Y, Hu S, Tian F, Wang J and Chen Y: Effects of Exendin-4 on bone
marrow mesenchymal stem cell proliferation, migration and apoptosis
in vitro. Sci Rep. 5:128982015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Couto JA, Ayturk UM, Konczyk DJ, Goss JA,
Huang AY, Hann S, Reeve JL, Liang MG, Bischoff J, Warman ML and
Greene AK: A somatic GNA11 mutation is associated with extremity
capillary malformation and overgrowth. Angiogenesis. 20:303–306.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang
Z, Li Z, Feng X, Hao J, Zhang K, et al: Melatonin synergizes the
chemotherapeutic effect of 5-fluorouracil in colon cancer by
suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J Pineal
Res. 62:2017. View Article : Google Scholar
|
|
45
|
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D,
Zhou H and Chen Y: Melatonin protected cardiac microvascular
endothelial cells against oxidative stress injury via suppression
of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by
activation of MAPK/ERK signaling pathway. Cell Stress Chaperones.
23:101–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Banerjee K, Keasey MP, Razskazovskiy V,
Visavadiya NP, Jia C and Hagg T: Reduced FAK-STAT3 signaling
contributes to ER stress-induced mitochondrial dysfunction and
death in endothelial cells. Cell Signal. 36:154–162. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garcia-Niño WR, Correa F,
Rodriguez-Barrena JI, León-Contreras JC, Buelna-Chontal M,
Soria-Castro E, Hernández-Pando R, Pedraza-Chaverri J and Zazueta
C: Cardioprotective kinase signaling to subsarcolemmal and
interfibrillar mitochondria is mediated by caveolar structures.
Basic Res Cardiol. 112:152017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou H, Yang J, Xin T, Zhang T, Hu S, Zhou
S, Chen G and Chen Y: Exendin-4 enhances the migration of
adipose-derived stem cells to neonatal rat ventricular
cardiomyocyte-derived conditioned medium via the phosphoinositide
3-kinase/Akt-stromal cell-derived factor-1α/CXC chemokine receptor
4 pathway. Mol Med Rep. 11:4063–4072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu
P, Ma Q, Tian F and Chen Y: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
Ischemia/reperfusion injury via induction of mROS-mediated
cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP
opening. J Am Heart Assoc. 6:pii: e005328. 2017. View Article : Google Scholar
|
|
50
|
Zhou H, Wang S, Zhu P, Hu S, Chen Y and
Ren J: Empagliflozin rescues diabetic myocardial microvascular
injury via AMPK-mediated inhibition of mitochondrial fission. Redox
Biol. 15:335–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Murphy PS, Wang J, Bhagwat SP, Munger JC,
Janssen WJ, Wright TW and Elliott MR: CD73 regulates
anti-inflammatory signaling between apoptotic cells and
endotoxin-conditioned tissue macrophages. Cell Death Differ.
24:559–570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kalyanaraman B: Teaching the basics of
cancer metabolism: Developing antitumor strategies by exploiting
the differences between normal and cancer cell metabolism. Redox
Biol. 12:833–842. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Das N, Mandala A, Naaz S, Giri S, Jain M,
Bandyopadhyay D, Reiter RJ and Roy SS: Melatonin protects against
lipid-induced mitochondrial dysfunction in hepatocytes and inhibits
stellate cell activation during hepatic fibrosis in mice. J Pineal
Res. 62:2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang HH, Chen Y, Gao CY, Cui ZT and Yao
JM: Protective effects of MicroRNA-126 on human cardiac
microvascular endothelial cells against
hypoxia/reoxygenation-induced injury and inflammatory response by
activating PI3K/Akt/eNOS signaling pathway. Cell Physiol Biochem.
42:506–518. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sigala F, Efentakis P, Karageorgiadi D,
Filis K, Zampas P, Iliodromitis EK, Zografos G, Papapetropoulos A
and Andreadou I: Reciprocal regulation of eNOS, H2S and
CO-synthesizing enzymes in human atheroma: Correlation with plaque
stability and effects of simvastatin. Redox Biol. 12:70–81. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xu J, Wu Y, Lu G, Xie S, Ma Z, Chen Z,
Shen HM and Xia D: Importance of ROS-mediated autophagy in
determining apoptotic cell death induced by physapubescin B. Redox
Biol. 12:198–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hu SY, Zhang Y, Zhu PJ, Zhou H and Chen
YD: Liraglutide directly protects cardiomyocytes against
reperfusion injury possibly via modulation of intracellular calcium
homeostasis. J Geriatr Cardiol. 14:57–66. 2017.PubMed/NCBI
|
|
58
|
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S and
Sun C: Melatonin alleviates inflammasome-induced pyroptosis through
inhibiting NF-κB/GSDMD signal in mice adipose tissue. J Pineal Res.
63:2017. View Article : Google Scholar
|
|
59
|
Zhou H, Du W, Li Y, Shi C, Hu N, Ma S,
Wang W and Ren J: Effects of melatonin on fatty liver disease: The
role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and
mitophagy. J Pineal Res. 64:2018. View Article : Google Scholar
|
|
60
|
Jahandiez V, Cour M, Bochaton T, Abrial M,
Loufouat J, Gharib A, Varennes A, Ovize M and Argaud L: Fast
therapeutic hypothermia prevents post-cardiac arrest syndrome
through cyclophilin D-mediated mitochondrial permeability
transition inhibition. Basic Res Cardiol. 112:352017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang X, Wang H, Ni HM, Xiong A, Wang Z,
Sesaki H, Ding WX and Yang L: Inhibition of Drp1 protects against
senecionine-induced mitochondria-mediated apoptosis in primary
hepatocytes and in mice. Redox Biol. 12:264–273. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou H, Zhu P, Wang J, Zhu H, Ren J and
Chen Y: Pathogenesis of cardiac ischemia reperfusion injury is
associated with CK2α-disturbed mitochondrial homeostasis via
suppression of FUNDC1-related mitophagy. Cell Death Differ.
25:1080–1093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu
S, Ren J and Chen Y: NR4A1 aggravates the cardiac microvascular
ischemia reperfusion injury through suppressing FUNDC1-mediated
mitophagy and promoting Mff-required mitochondrial fission by CK2α.
Basic Res Cardiol. 113:232018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou H, Shi C, Hu S, Zhu H, Ren J and Chen
Y: BI1 is associated with microvascular protection in cardiac
ischemia reperfusion injury via repressing
Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. Apr
6–2018.(Epub ahead of print). View Article : Google Scholar
|
|
65
|
Zhou H, Li D, Zhu P, Ma Q, Toan S, Wang J,
Hu S, Chen Y and Zhang Y: Inhibitory effect of melatonin on
necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway
attenuates cardiac microvascular ischemia reperfusion injury. J
Pineal Res. e125032018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Le Cras TD, Mobberley-Schuman PS, Broering
M, Fei L, Trenor CC 3rd and Adams DM: Angiopoietins as serum
biomarkers for lymphatic anomalies. Angiogenesis. 20:163–173. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Niaudet C, Bonnaud S, Guillonneau M,
Gouard S, Gaugler MH, Dutoit S, Ripoche N, Dubois N, Trichet V,
Corre I and Paris F: Plasma membrane reorganization links acid
sphingomyelinase/ceramide to p38 MAPK pathways in endothelial cells
apoptosis. Cell Signal. 33:10–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li Q, Qi F, Meng X, Zhu C and Gao Y: Mst1
regulates colorectal cancer stress response via inhibiting
Bnip3-related mitophagy by activation of JNK/p53 pathway. Cell Biol
Toxicol. Oct 24–2017.(Epub ahead of print).
|
|
69
|
Kumar A, Kunal S and Shah A: Frequency and
effect of type 1 hypersensitivity in patients from India with
chronic obstructive pulmonary disease and associated upper airways
symptoms. Asia Pac Allergy. 7:199–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hong H, Tao T, Chen S, Liang C, Qiu Y,
Zhou Y and Zhang R: MicroRNA-143 promotes cardiac ischemia-mediated
mitochondrial impairment by the inhibition of protein kinase
Cepsilon. Basic Res Cardiol. 112:602017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yang S, Zhang J, Wang S, Shi J and Zhao X:
Knockdown of angiopoietin-like protein 2 ameliorates diabetic
nephropathy by inhibiting TLR4. Cell Physiol Biochem. 43:685–696.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Deng W, Wang Y, Long X, Zhao R, Wang Z,
Liu Z, Cao S and Shi B: miR-21 reduces hydrogen peroxide-induced
apoptosis in c-kit+ cardiac stem cells in vitro through
PTEN/PI3K/Akt signaling. Oxid Med Cell Longev. 2016:53891812016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang Z, Li N, Wang B and Lin J:
Nonalcoholic fatty liver disease progression in rats is accelerated
by splenic regulation of liver PTEN/AKT. Saudi J Gastroenterol.
21:232–238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Van Nostrand JL, Bowen ME, Vogel H, Barna
M and Attardi LD: The p53 family members have distinct roles during
mammalian embryonic development. Cell Death Differ. 24:575–579.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ligeza J, Marona P, Gach N, Lipert B,
Miekus K, Wilk W, Jaszczynski J, Stelmach A, Loboda A, Dulak J, et
al: MCPIP1 contributes to clear cell renal cell carcinomas
development. Angiogenesis. 20:325–340. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ and
Chen Y: Protective role of melatonin in cardiac
ischemia-reperfusion injury: From pathogenesis to targeted therapy.
J Pineal Res. 64:2018. View Article : Google Scholar
|
|
77
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|