Introduction
Obesity, characterized by an excessive increase in
body weight that results in excessive fat accumulation, is
increasing at a high rate in all age groups (1,2).
Obesity is associated with significant metabolic disturbances and
leads to severe pathology in a number of organs, including the
heart (3). Excessive fat intake
limits metabolic flexibility for the mitochondrial production of
adenosine 5′-triphosphate, which may lead to cellular injury and
inflammation, and has been considered to be a major underlying
factor in the pathogenesis of a number of diseases (4). In the heart, lipid overload has toxic
effects and causes cardiac dysfunction (5). However, the mechanisms of
lipotoxicity are not yet completely understood.
Autophagy is an evolutionarily conserved mechanism
that maintains cellular and nutrient homeostasis by degrading
misfolded proteins and damaged organelles, and has been identified
as a major underlying factor in the pathogenesis of several chronic
diseases (6,7). Dysregulation of autophagy may
contribute to the development of cardiorenal metabolic syndrome
including insulin resistance (8),
hypertension and maladaptive immune modulation (9). A recent study focused on the role of
autophagy in cardiovascular diseases and demonstrated that the role
of autophagy is different in different cells and situations
(10). Nevertheless, it remains
unclear how a high-fat diet (HFD) influences the level of autophagy
and the role it serves in obesity-associated cardiomyopathy.
Furthermore, mitophagy as a special mode of autophagy is essential
for the homeostasis of a healthy network of functioning
mitochondria, and how it influences the hearts of HFD mice remains
unclear. In the present study, a HFD was used to set up an obesity
animal model to investigate the role of autophagy and mitophagy in
the heart.
Materials and methods
Animal care and feeding
All the experimental procedures were approved by the
Animal Care and Use Committee of Renmin Hospital of Wuhan
University (Wuhan, China), and were in accordance with the Guide
for the Care of Laboratory Animals published by the US National
Institutes of Health (publication no. 85–23, revised in 1996). The
8 week-old male C57BL/6 mice (n=30; weighing 22–24 g) purchased
from the Institute of Laboratory Animal Science, Chinese Academy of
Medical Sciences & Peking Union Medical College (Beijing,
China) were randomly housed five mice/cage, given free access to
water, and fed either a HFD (60% kcal fat; cat. no. D12492;
Research Diets, Inc., New Brunswick, NJ, USA; n=15) or a normal fat
diet (10% kcal fat; cat. no. D12450B; Research Diets, Inc.; n=15)
for 42 weeks. Animals were housed in cages with a controlled
temperature (22–25°C) and humidity (50±5%) and a 12/12 h light/dark
cycle.
Hemodynamic analysis
During the hemodynamic measurement, a microtip
catheter transducer (SPR-839; Millar, Inc., Houston, TX, USA) was
inserted into the right carotid artery and advanced gradually into
the left ventricle under anesthesia with 1.5% isoflurane. The
signals were continuously recorded using a Millar Pressure-Volume
system (MPVS-400; Millar, Inc.) and analyzed using PVAN data
analysis software (version 3.5; Millar, Inc.). The cardiac output,
end-diastolic pressure, end-diastolic volume and Tau_w (left
ventricular relaxation time constant) were measured by pressure
volume (PV) loop.
Measurement of random blood glucose
and glucose tolerance test
Random blood glucose was measured once using a
Relion Ultima diabetic glucometer (Abbott Diabetes Care, Abbott
Park, IL, USA) using blood from the tail vein from normal chow (NC)
(n=5) or HFD non-fasting mice (n=5).
Intraperitoneal glucose tolerance tests (IPGTT) were
performed once at 50 weeks of age in all mice following 42 weeks of
NC or HFD. Prior to the IPGTTs, mice were fasted for 6 h and then
given a bolus of glucose (1.5 g glucose/kg body weight dissolved in
sterile saline) by intraperitoneal injection. Blood samples were
collected from the tail vein and assessed for glucose concentration
prior to injection (0 min) and at 30, 60, 90, 120 and 150 min
post-injection using a Relion Ultima diabetic glucometer (Abbott
Diabetes Care; n=7 per group).
Blood collection
Mice were anesthetized with 1.5% isoflurane and the
mouse orbital sinus was punctured with a capillary glass tube prior
to the blood being collected (~400 µl/mouse) into a 1.5 ml
Eppendorf tube. The samples were then centrifuged at 1,007 × g for
20 min at 4°C, and the serum was collected and stored at −80°C for
use in further experiments.
Triglyceride (TG) and cholesterol
(CHO) detection
The serum TG and CHO of non-fasting mice were
measured using the TG (cat. no. A110-1) and CHO (cat. no. A111-1)
assay kits, which was purchased from Nanjing Jiancheng
Bioengineering Institute (Nanjing, China). All kits were used
according to the manufacturer's protocol. The absorbance values at
510 nm were detected with a two-color infrared imaging system
(Odyssey; LI-COR Biosciences).
Tissue protein extraction
Heart tissue extracts were collected from the mice
by homogenizing the left ventricular samples in lysis buffer (50 mM
Tris-HCl, 0.5% NP-40, 250 mM NaCl, 5 mM EDTA and 50 mM NaF).
Subsequently, the protein concentration was measured with a
bicinchoninic acid (BCA) protein assay kit using a microplate
reader (Synergy HT; BioTek Instruments, Inc., Winooski, VT, USA).
The proteins were subsequently collected and stored at −80°C for
use in further experiments.
Mitochondria extraction
To determine the mitophagy level of mitochondria by
western blotting, the isolation of mitochondria from cardiac
tissues was performed using the Tissue Mitochondria Isolation kit
(Beyotime Institute of Biotechnology, Beijing, China; cat. no.
C3006). First, a fresh 50-mg left ventricular sample was washed
once with PBS and placed in an ice bath in 500 µl pre-cooled PBS
for 3 min, followed by centrifugation at 600 × g for 20 sec at 4°C,
and the supernatant was discarded. Next, cardiac tissue was
digested with 400 µl pre-cooled trypsin digestion solution for 20
min and centrifuged at 600 × g for 20 sec at 4°C. Subsequently, the
tissue was resuspended with 100 µl mitochondrial separation reagent
A and centrifuged at 11,000 × g for 10 min at 4°C, and the
supernatant was discarded. Finally, 150 µl mitochondrial lysate was
used to lyse the mitochondria and extract the mitochondrial
protein. The protein concentration was measured using the BCA
protein assay kit with a microplate reader (Synergy HT; BioTek
Instruments, Inc.).
Western blot analysis
Heart tissue protein (80 µg) or cell protein (50 µg)
were separately loaded onto 12, 10 or 8% SDS-PAGE gels and
subsequently transferred onto Immobilon-FL transfer membranes (cat.
no. IPFL00010; EMD Millipore, Billerica, MA, USA) in transfer
buffer at 200 mA. The membranes were blocked with 5% non-fat milk
at room temperature for 1 h and then incubated overnight at 4°C
with specific antibodies. Anti-GAPDH (1:1,000; cat. no. 2118),
C/EBP-homologous protein (CHOP; (1:500; cat. no. 2895),
microtubule-associate proteins 1A/1B light chain 3-like protein
(LC3)-II/I (1:500; cat. no. 12741), autophagy-related protein 7
(Atg7; 1:1,000; cat. no. 2613s) and phosphorylated
(p)-sequesteosome-1/(P62; 1:500; cat. no. 13121s) were purchased
from Cell Signaling Technology, Inc., (Danvers, MA, USA). Beclin-1
(1:500; cat. no. ab55878), parkin (1:1,000; cat. no. ab15954),
mitofusin2 (1:1,000; cat. no. ab56889), serine/threonine-protein
kinase PINK1 mitochondrial (PINK1; 1:1,000; cat. no. ab186303) and
voltage-dependent anion-selective channel protein 1 (VDAC1;
1:1,000; cat. no. ab191440) were purchased from Abcam (Cambridge,
UK). Peroxisome proliferator-activated receptor-α (PPARα; 1:1,000;
cat. no. sc-9000), x-box-binding protein-1 (XBP-1; 1:500; cat. no.
sc-7160) and glucose-regulated protein 78 (GRP78; 1:500; cat. no.
sc-13968) were purchased from Santa Cruz Biotechnology, Inc.,
(Dallas, TX, USA). Subsequently, the membranes were incubated with
IRDye 800CW conjugated goat anti-mouse IgG (cat. no. P/N 925–32210;
1:1,250; LI-COR Biosciences, Lincoln, NE, USA) and goat anti-rabbit
IgG (cat. no. P/N 925–32211; 1:1,250; LI-COR Biosciences) for 1 h
at room temperature. The blots were subsequently probed using
Odyssey Software (version 3.0; LI-COR Biosciences).
Scanning electron microscopy
(SEM)
Freshly isolated heart tissues were fixed with 2.5%
glutaraldehyde in phosphate buffer (pH 7.0) at 4°C for 8 h and then
delivered to the electron microscopy facility of the Renmin
Hospital of Wuhan University for post-processing. Briefly,
following three washes in phosphate buffer (pH 7.4) for 10 min at
4°C, tissues were fixed for 90 min in a 1.0% OsO4
solution at 4°C, prepared with the phosphate buffer (pH 7.4).
Tissues were then rehydrated in a descending alcohol series and
sputter coated with silver ions. Finally, SEM observations were
performed using a scanning electron microscope (TM-3000; Hitatchi,
Ltd., Tokyo, Japan). Micrographs were captured in a blinded manner
at magnifications of ×3,000, ×5,000 and ×8,000.
Cell culture and treatment
H9C2 cells, purchased from the Cell Bank of Type
Culture Collection of Chinese Academy of Sciences (Shanghai,
China), were cultured in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal
bovine serum (Chinese Academy of Sciences, Shanghai, China) at a
temperature of 37°C and 5% CO2. The cells
(1×106 cells/well) were transfected in 6-well plates and
allowed to adhere for 24 h. Cells were left untreated or were
treated with 400 µM palmitate (PA; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 24 h at a temperature of 37°C. To regulate
autophagy, the autophagy inhibitor bafilomycin A1 (Baf A1; 50 nM;
Sigma-Aldrich; Merck KGaA) was added for 1 h prior to the addition
of 400 µM PA and incubation for 24 h. Cellular proteins were
extracted for the detection of autophagy using western blot
according to the aforementioned protocol. Lysis buffer (50 µl) was
added to each well, and proteins were extracted according to the
aforementioned protocol.
Monomeric cherry (mCherry)-green
fluorescent protein (GFP)-LC3 adenovirus transfection and confocal
microscopy
A plasmid-encoded tandem mCherry-GFP-LC3 construct
adenovirus was purchased for the quantification of autophagic
maturation from ViGene Biosciences (Rockville, MD, USA). Cells
(5×105/ml; 500 µl/well) were seeded onto a 24-well plate
with chamber slides placed below and allowed to adhere for 24 h.
Following the addition of 5.0×10−3 mg/ml ADV-HR to
improve the efficiency of transfection, H9C2 cells were
simultaneously transfected with mCherry-GFP-LC3 adenovirus (both
ViGene Biosciences Inc., Rockville, MD, USA; 1.0×1010
plaque forming units/ml; 0.5 µl/well) according to the
manufacturer's protocol and cultured at 37°C and 5% CO2
for 2 h. The medium was replaced with fresh complete culture medium
and the cells were incubated for another 24 h prior to the addition
of Baf A1. Following 1 h, the cells were untreated or treated with
400 µM PA for 24 h. Following removal of the culture media and
washing with 1% Triton X-100 in PBS with 0.1% Tween-20 for 15 min
at room temperature, the cells were stained with 300 nM
4′,6-diamidino-2-phenylindole dihydrochloride (Invitrogen; Thermo
Fisher Scientific, Inc.) for 5 min at room temperature. Images were
obtained using an Olympus confocal microscope (Olympus Corporation,
Tokyo, Japan), with a magnification of ×400. A minimum of 10 fields
of view were observed per sample.
Statistical analysis
All results are presented as the mean ± standard
error of mean. Unpaired two-tailed Student's t-test was used to
determine differences between the means of the HFD and NC groups.
One-way analysis of variance (ANOVA) was used to analyze multiple
groups and Bonferroni's post-hoc testing was employed following
ANOVA for testing significant differences between groups. Glucose
tolerance was assessed by calculating the area under the curve. All
statistical analyses were performed using SPSS 17.0 software (SPSS,
Inc., Chicago, IL, USA). Figures were compiled using GraphPad Prism
6.02 software (GraphPad software, Inc., La Jolla, CA, USA). All
experiments were repeated in triplicates. P<0.05 was considered
to indicate a statistically significant difference.
Results
HFD causes pathological obesity in
mice
Male mice were fed either NC or a HFD for 42 weeks
starting at 8 weeks of age. The long-term consumption of a HFD
caused a significantly increased body and heart weight compared
with the NC group (P<0.05; Fig. 1A
and B). Additionally, HFD-fed mice exhibited a significant
increase in body fat and lean body mass percentage compared with
the NC diet (P<0.05; Fig. 1C).
The levels of TG, CHO and glucose were significantly increased,
which indicated there was a metabolic disturbance in the HFD mice
(P<0.05; Fig. 1D-F).
Furthermore, the HFD-exposed mice exhibited an impaired glucose
tolerance level (Fig. 1E and
F).
HFD causes cardiac dysfunction in
mice
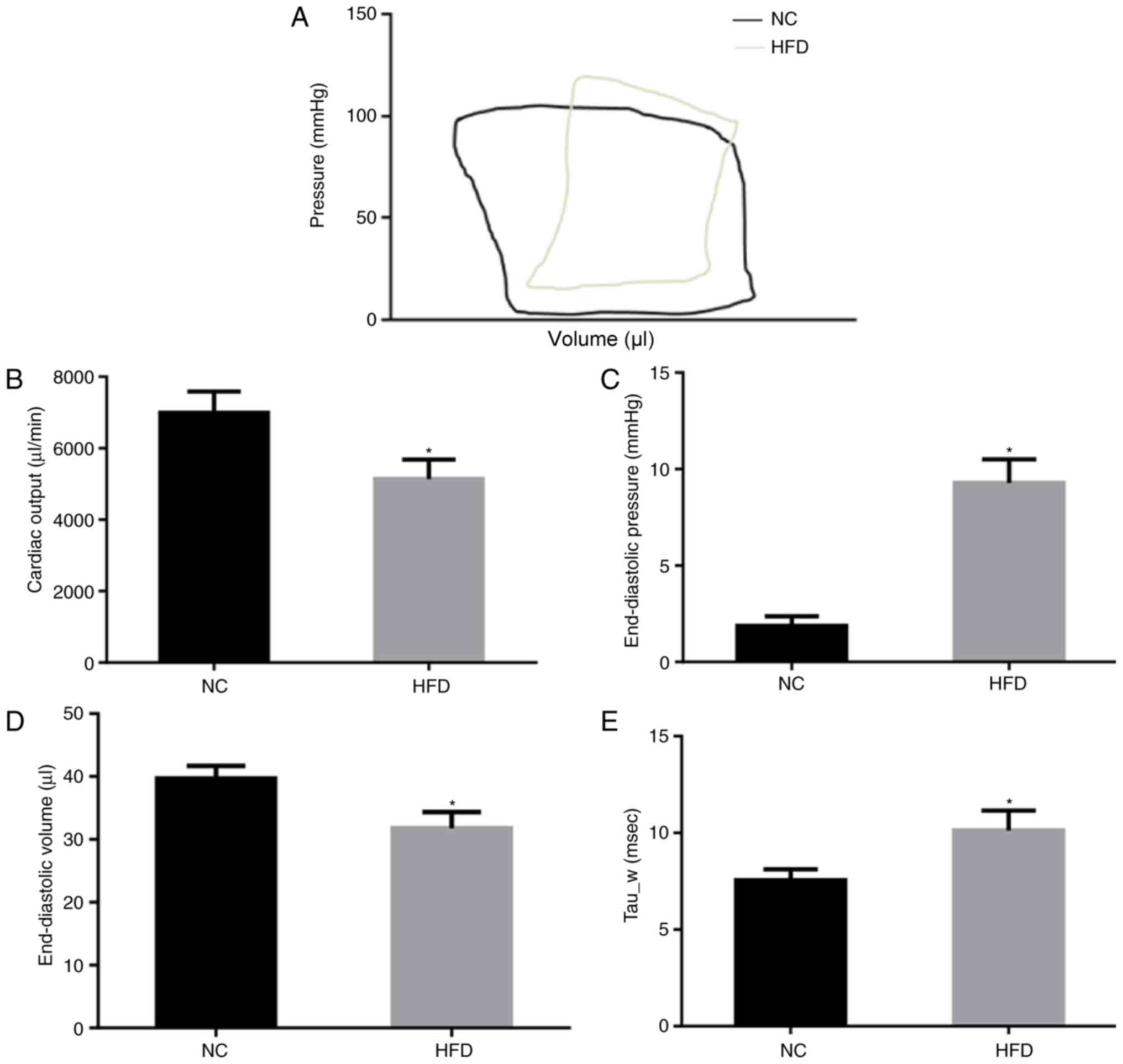
The cardiac function of NC and HFD mice was
evaluated by PV loop (Fig. 2A)
following 42 weeks of feeding. The results of the PV loop analysis
revealed that HFD damaged systolic and diastolic cardiac functions.
In the HFD mice, cardiac output was significantly decreased
(P<0.05; Fig. 2B), indicating
systolic dysfunction. In addition, the end-diastolic pressure
significantly increased (P<0.05; Fig. 2C) although end-diastolic volume
decreased (Fig. 2D) in the HFD
mice compared with the NC mice, indicating significant cardiac
diastolic dysfunction. Tau_w, a further indicator of diastolic
function of the heart, was significantly increased in HFD mice
(P<0.05; Fig. 2E). These
results suggested that HFD induced cardiac dysfunction of systole
and diastole.
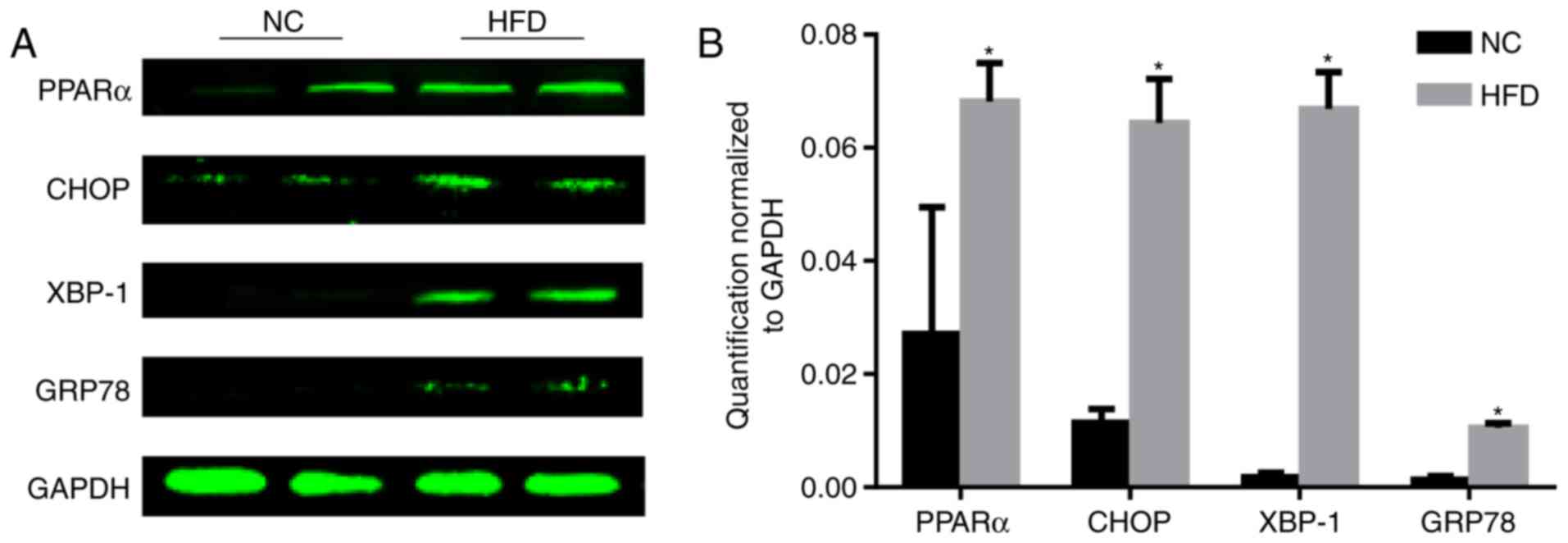
ERS is upregulated during obesity
PPARα is associated with dyslipidemia and
cardiovascular disease (11). The
level of PPARα was tested, and the results demonstrated that the
level of PPARα significantly increased following HFD feeding
compared with the NC group (P<0.05; Fig. 3). As the reduction of PPARα
expression may inhibit ERS (12),
the level of ERS was tested. During ER stress, the level of CHOP
expression is elevated and its role in programmed cell death of
ER-stressed cells is correlated with its role in promoting protein
synthesis and oxidative stress inside the ER (13). GRP78, additionally termed binding
immunoglobulin protein, is a heat shock protein 70 molecular
chaperone located in the lumen of the ER that binds newly
synthesized proteins as they are translocated into the ER. When
protein folding is disturbed inside the ER, GRP78 synthesis is
increased (14). XBP-1 is a
well-conserved component of the unfolded protein response, which is
crucial for glucose homeostasis and lipid metabolism (15). A HFD increased the protein
expression of CHOP, XBP-1 and GRP78 (Fig. 3), indicating activation of ERS.
HFD-induced obesity impairs
autophagy
According to a previous study using hepatocytes, ERS
may trigger ER-associated degradation and autophagy, which can
clear unfolded proteins and restore protein homeostasis (16). Therefore the influence of a HFD on
autophagy was tested. To study the role of autophagy in HFD-induced
cardiac dysfunction, the expression of autophagy-associated
proteins in the heart was analyzed. During autophagy, LC3-I is
converted to LC3-II through lipidation by the ubiquitin-like system
involving Atg7. This process allows for LC3 to become associated
with autophagic vesicles, whose membranes are marked by P62
(17), and levels of LC3-I and P62
may be decreased when autophagic flux is unobstructed. Beclin-1 is
critical to autophagy as it may stimulate autophagy when
overexpressed in mammalian cells (18). In the present study, HFD induced a
significant upregulation of Atg7, beclin-1 and P62, although it
downregulated the level of LC3-II/I compared with the NC group.
These imbalances in protein expression involved in autophagy
confirmed that autophagy was impaired in the HFD-fed mice (Fig. 4A and B). Consistent with the
results of the western blotting, electron microscopy demonstrated
an increased number of lipid droplets and autophagosomes in HFD-fed
mice compared with the NC group (Fig.
4C and D), which indicated that there was no barrier to the
formation of autophagosomes. However, there were fewer
autolysosomes in HFD-fed mice, which indicated that there was
inhibition of autophagosome degradation. These results may explain
the cause of the increase in P62.
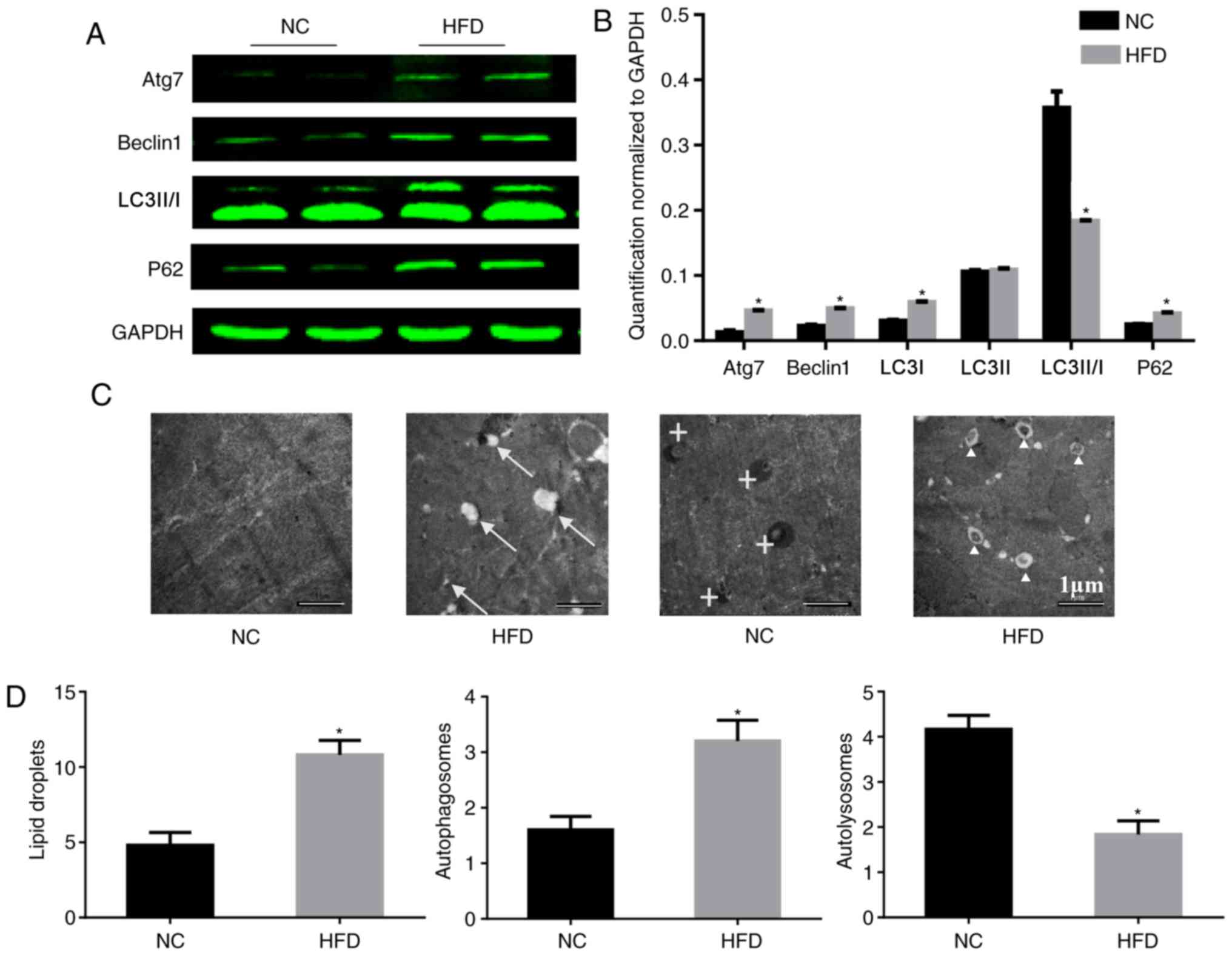 | Figure 4.HFD reduces the level of autophagy in
heart tissue. (A) Protein expression of Atg7, beclin-1, LC3-II/I,
P62 and GAPDH, and (B) quantification. n=4 per group. (C)
Representative electron micrographs of the cardiac muscle from the
septum identified lipid droplets in only the HFD mice, and there
were more autophagosomes existing in the HFD mice. Original
magnification, ×5,000. The arrows mark lipid droplets around
cardiomyocytes, crosses mark autolysosomes and triangles mark
autophagosomes. (D) Quantitative analysis of lipid droplets,
autophagosomes and autolysosomes in the images. n=2 per group.
*P<0.05 vs. respective NC. NC, normal chow; HFD, high fat diet;
Atg7, autophagy-related protein 7; LC3-II/-I, MAP1 light chain
3-like protein III/I; P62, sequesteosome-1. |
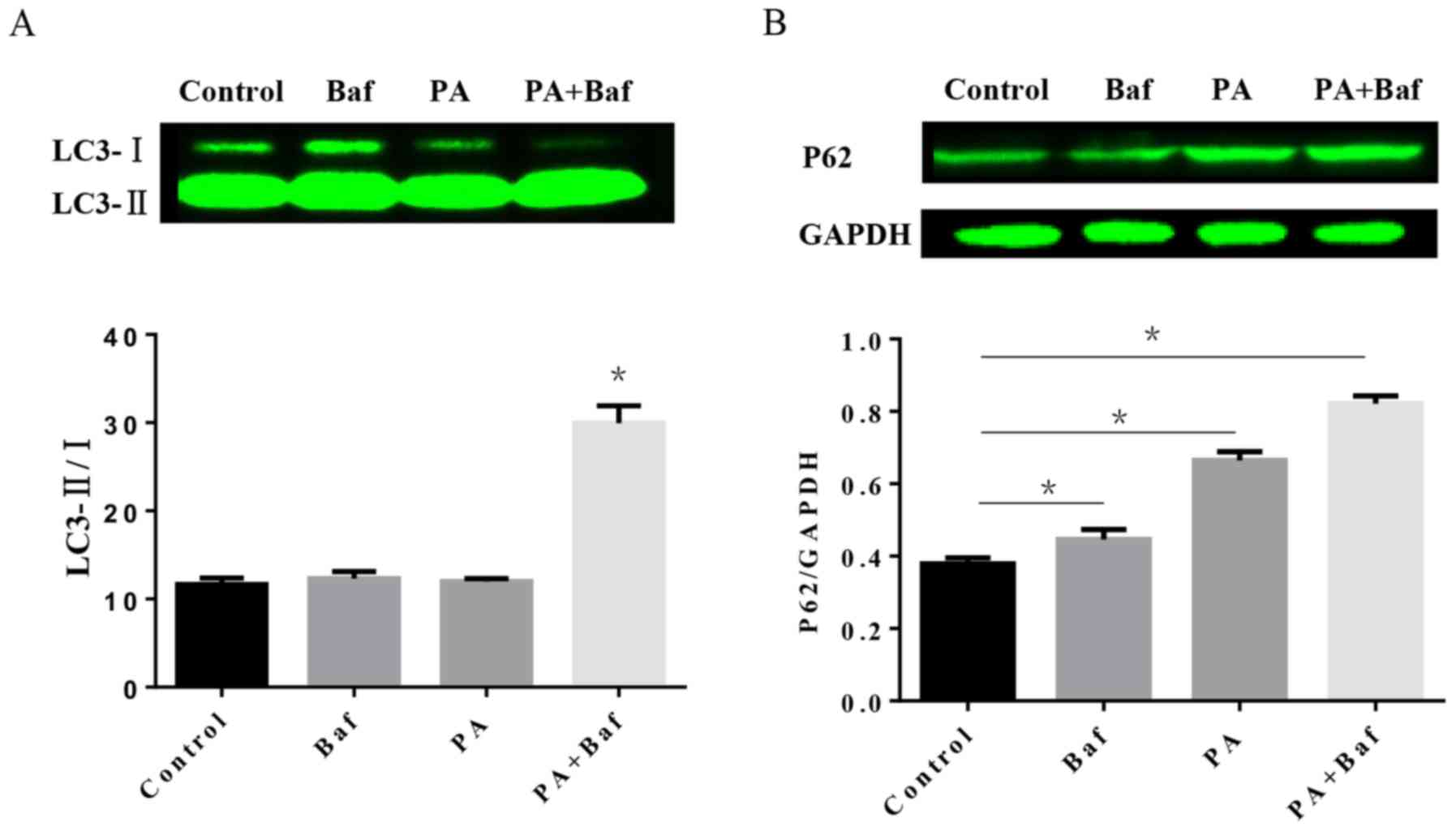
Impairment of autophagic flux in H9C2
cells treated with PA
To investigate the autophagic process involved in
HFD, the protein expression of LC3 and P62 was tested by western
blotting in vitro. The results of the western blotting
demonstrated that PA, a primary product of fatty acid synthase, may
increase LC3-II/I in the presence of Baf A1 (Fig. 5A), an autophagic inhibitor which
prevents the maturation of autophagic vesicles by inhibiting the
fusion between lysosomes and autolysosomes. Furthermore, P62
expression was altered when treated with PA, Baf A1 and the two
together (Fig. 5B).
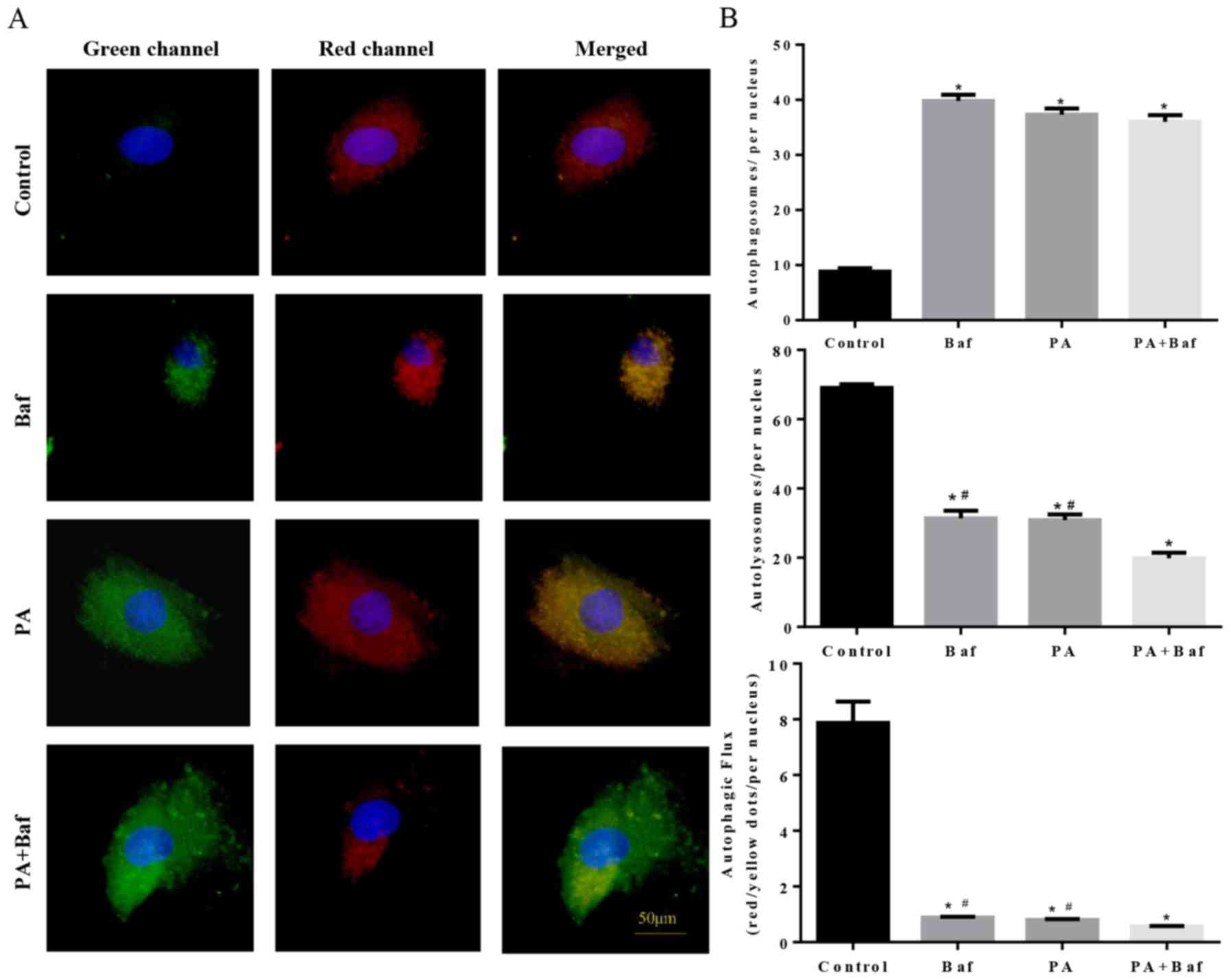
To clarify whether autophagic flux was influenced by
PA, mCherry-GFP-LC3, a tandem fluorescent indicator was used. Since
the green fluorescence of the fusion protein is sensitive to the
acidic environment of lysosomes and is quickly quenched in
autolysosomes, only red fluorescence was detectable in the
autolysosomes. Therefore, when lysosomal degradation was inhibited
by treating the cells with Baf A1, an accumulation of green puncta
was observed (Fig. 6A and B),
which is consistent with PA-induced cells. Notably, an increase in
green puncta was observed in Baf A1+PA treated cells compared with
the other groups. These results suggested that PA inhibited
autophagic flux via suppression of lysosomal degradation.
HFD-induced obesity impairs the
regulation of mitochondrial quality control
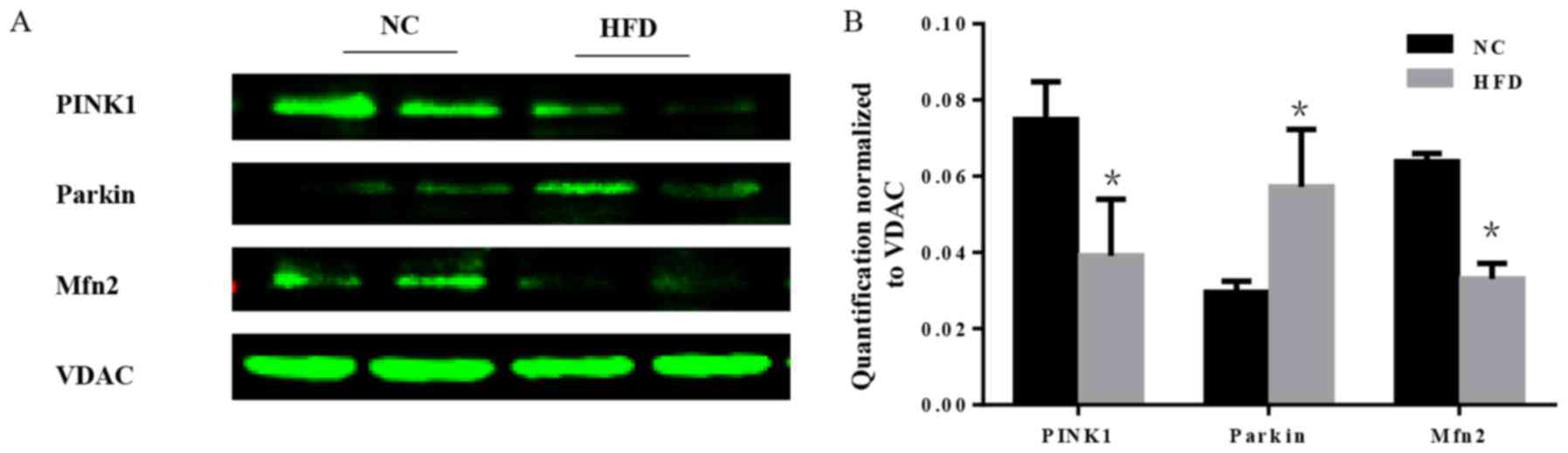
Mitochondria are essential organelles that serve
important roles in cellular energy metabolism. The mitochondrial
quality control systems that have evolved to repair or eliminate
dysfunctional mitochondria are of importance (19). Furthermore, mitochondrial quality
surveillance systems attenuate potentially damaging processes that
occur in the cytosol and ER (20).
As a part of mitochondrial quality surveillance systems, mitophagy
may selectively engulf and clear mitochondria, thus mitophagy is
essential for the homeostasis of a healthy network of functioning
mitochondria. In the course of mitophagy, PINK1 and the E3
ubiquitin ligase parkin act as synergistic mediators (21). In response to mitochondrial
depolarization, PINK1 accumulates on the mitochondrial outer
membrane, which subsequently recruits parkin from the cytosol and
activates its ubiquitin ligase activity to eliminate damaged
mitochondria (22). Furthermore,
mitochondrial fusion is a part of mitochondrial quality
surveillance systems, and mitofusin 2 (Mfn2) is a mitochondrial
membrane protein that is involved in mitochondrial fusion in
mammalian cells (23). The
expression level of PINK1, parkin and Mfn2 was tested. The results
demonstrated that there was a significant increase in parkin, and a
significant decrease in PINK1 and Mfn2 in HFD mice compared with
the NC group (P<0.05; Fig. 7),
which indicated that the mitochondrial quality surveillance system
was dysfunctional.
Discussion
In the present study mice were fed with a HFD for 42
weeks to investigate the influence of obesity on the heart. During
the in vivo study, a HFD was observed to induce obesity and
lead to metabolic abnormalities and cardiac function deterioration.
In addition, it was also demonstrated that ERS was increased in
long-term HFD mice. Furthermore, autophagy and mitophagy in the
obese mouse heart were dysfunctional.
Over the past few decades, the incidence of obesity
has been increasing worldwide. It has been demonstrated that the
intake of a high level of fat is associated with increased
cardiovascular disease risks (24,25),
and lipotoxicity is detrimental to numerous cell types, including
myocytes, hepatocytes and β cells (26,27).
A number of studies have investigated lipotoxicity in
cardiomyocytes (28–30). Although a similar phenomenon of
decreased autophagy as a result of exposure to a HFD has been
demonstrated in other cell types, including proximal tubule cells,
embryonic stem cells and neurons (26,31,32),
there have been few reports investigating the association between
autophagy and obesity. In the present study mice were exposed to a
HFD similar to that of obese humans to investigate the role that
autophagy serves.
The mechanism through which HFD-induced obesity
impairs cardiac function may be multifactorial. The results of the
present study demonstrated that the ER served an important role in
this process, which is consistent with another study (33). However, the results of the present
study indicated that the level of autophagy was decreased in HFD
mice, which may be due to the metabolic ER sensor that suppresses
the alternative activation of macrophages in obesity (34), as the density of macrophages is
associated with autophagy (35).
However, the results of the present study were in contrast to the
study by Kong et al (36),
which instead demonstrated that ERS enhances the level of
autophagy. This contradiction may due to the long-term HFD
weakening the effect of ERS on macrophages.
In the present study, autophagy was aberrant in
HFD-fed mice. An increase in the expression of Atg7 and beclin-1
was observed. However, a HFD decreased the expression of LC3-II/I
and increased the expression of P62. Although beclin-1 may
stimulate autophagy at an early stage, this does not indicate that
HFD-fed mice exhibit an improved autophagic system, as autophagy is
followed by fusion with lysosomes and the degradation of the
contents. Consistent with the results of the electron microscopy,
autophagosomes increased and autolysosomes decreased in the heart
tissues of HFD-fed mice, which indicated that the formation of
autophagosomes was normal or even increased in HFD-fed mice
compared with the NC mice; however, the autophagic flux was
suppressed in HFD-fed mice. To confirm the results of the animal
experiments, H9C2 cells were treated with PA and Baf A1 in
vitro. The results demonstrated that PA+Baf increased the
LC3-II/I ratio and P62 compared with the control at the protein
level. Notably, adding PA or Baf A1 alone did not influence the
level of LC3-II/I, however, when administered in combination, the
level of LC3-II/I was significantly enhanced. In addition, the
level of P62 was significantly increased following treatment with
Baf A1, PA or Baf A1+PA. This suggested that PA and Baf A1 may
exhibit a synergistic effect resulting in the inhibition of fusion
between lysosomes and autolysosomes. In addition, the
mCherryGFP-LC3 adenovirus transfection demonstrated that PA was
able to induce an accumulation of autophagosomes, but autolysosomes
were decreased in H9C2, consistent with the treatment with Baf.
This indicated that the administration of PA caused a marked
decrease in autolysosome density, and was associated with the
fluency of autophagic flux. PA-induced impairment of autophagic
flux was increased when Baf A1 was added. These results indicated
that the accumulation of autophagosomes induced by the HFD was due
to inhibition of autophagosome degradation. Furthermore, it may be
a compensatory mechanism under long-term HFD for the increasing
expression levels of Atg7, Beclin-1, LC3-I and P62
autophagy-associated proteins. There are an increasing number of
studies revealing novel information on autophagy, and the
understanding of autophagy in the heart is altering. Upregulation
of autophagy in cardiomyocytes is considered to be an adaptive
response to stress, including lipopolysaccharide exposure,
ischemia/reperfusion and overload-induced cardiac dysfunction,
while suppressing the process accelerates heart failure (37,38).
On the contrary, downregulation of autophagy may be a sign of
functional aberration due to a number of pathological disorders in
cardiovascular disease (39,40).
However, the role of autophagy in HFD-induced cardiac injury is not
well elucidated. Studies have demonstrated that mice receiving a
HFD for >16 weeks may exhibit an altered cardiac autophagic
response (41,42), and HFD consumption for 24 weeks
leads to the impairment of autophagic flux, primarily due to the
disruption of autophagosome degradation (43). Furthermore, a HFD study performed
to investigate cardiac function revealed that autophagy was
decreased in mice at a time interval of 44 weeks of HFD
administration compared with 24 weeks, and that mice who has been
administered a HFD for 44 weeks exhibited markedly increased
myocardial fibrosis and cardiac hypertrophy (44). Based on these studies and the aim
to investigate the role of autophagy in a longer period of time
feeding of high fat diet, following the guidelines of Canadian
Council on Animal Care (CCAC) on choosing an appropriate endpoint
(45,46), a time interval of 42 weeks was
chosen in the present study to represent the humane endpoint, and
the animals did not present any signs of pain/suffering/discomfort
prior to this time interval. The present study indicated that the
duration of HFD consumption may be one of the factors that affect
the autophagic response in the heart. The decreased autophagic flux
may due to the impaired activity of lysosomal proteolysis.
Mitochondria are critical for cardiomyocyte survival
and the maintenance of normal cardiac function. There is evidence
demonstrating that mitochondrial dysfunction is an important
contributor to the function of heart (47). In addition, impaired autophagic
flux suggests compromised mitophagy. A previous study has
identified that mitophagy appears to be regulated by three distinct
signaling pathways, including FUN14 domain-containing protein 1,
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 and the
PINK1/parkin signaling pathways. However, under stress conditions,
the PINK1/parkin signaling pathway is the main pathway responsible
for clearing dysfunctional mitochondria (48). PINK1 is ubiquitously expressed and
localized to the mitochondria. It is essential for the cell to
maintain PINK1 expression at a certain level. In neurons, genetic
ablation of PINK1 leads to mitochondrial dysfunction (49). Furthermore, PINK1 possesses a
distinct, non-redundant function in the surveillance and
maintenance of cardiac tissue homeostasis (50). In the present study a decrease in
PINK1 was observed, indicating mitochondrial dysfunction and
mitophagy blockage in HFD mice. Although the present study detected
an increase in parkin, it does not affect mitophagy in the absence
of PINK1 (51). Mitochondria are
highly dynamic, either undergoing fission/fusion to form small
individual units or interconnecting with each other for rapid
responses to mitochondrial damages. Considering that mitochondrial
fusion prevents mitophagy and may therefore protect myocytes
against excessive degradation of mitochondria (52), the level of Mfn2, a mitochondrial
membrane protein, which is expressed abundantly in the heart and is
a mediator of mitochondrial fusion, was tested. In addition, it is
downstream of PINK1. The results demonstrated a decrease in Mfn2 in
the mitochondria of obese mice, which demonstrated that HFD may
damage the mitochondrion through the inhibition of mitophagy and
mitochondrial fusion. However, the activation of other pathways
that can induce mitophagy and other mediators acting on parkin,
except PINK1, were not analyzed in the present study.
In addition, the hemodynamic measurements of a total
of 7 mice per group were investigated; however, one mouse succumbed
to mortality in the normal fat diet group (mortality rate, 14.29%),
whereas two mice succumbed to mortality in the HFD group (mortality
rate, 28.57%) following the termination of hemodynamic measurements
which is consistent with the findings of Mattson (53). Reasons for such mortalities we
speculated are mainly because this intravascular process is
invisible and it may lead to the rupture of blood vessels or the
heart. In addition, Calligaris et al (54) revealed that mice fed with a
high-fat diet for a prolonged duration of time presented reduced
aortic vasoconstriction and reduced contractile response, which
suggested that HFD may affect vascular function and increase the
difficulty of PV measurement in HFD mice.
In conclusion, a HFD has been demonstrated to induce
dyslipidemia and cardiac dysfunction. These effects may be mediated
by the upregulation of ERS, and the downregulation of autophagy and
mitophagy in the HFD hearts. However, an investigation into how to
reduce the dyslipidemia and dysfunction caused by HFD were not
included in the present study. Nevertheless, the results of the
present study indicated that autophagy and mitophagy served an
important role in HFD-induced cardiomyopathy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81270303, 81470516
and 81530012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, Z-PW, YY, NZ and Y-GJ performed the study,
analyzed and interpreted the data, and wrote the manuscript. YC, NZ
and Q-ZT conceived the hypothesis and participated in the
experimental design. YC, Z-PW, YY and C-XW participated in the data
interpretation and manuscript drafting. All authors approved final
version of the manuscript.
Ethics approval and consent to
participate
All the experimental procedures were approved by the
Animal Care and Use Committee of Renmin Hospital of Wuhan
University, and were in accordance with the Guide for the Care of
Laboratory Animals published by the US National Institutes of
Health (publication no. 85-23, revised in 1996).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanley AJ, Harris SB, Gittelsohn J,
Wolever TM, Saksvig B and Zinman B: Overweight among children and
adolescents in a Native Canadian community: Prevalence and
associated factors. Am J Clin Nutr. 71:693–700. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schell LM and Gallo MV: Overweight and
obesity among North American Indian infants, children, and youth.
Am J Hum Biol. 24:302–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
von Sarnowski B, Putaala J, Grittner U,
Gaertner B, Schminke U, Curtze S, Huber R, Tanislav C, Lichy C,
Demarin V, et al: Lifestyle risk factors for ischemic stroke and
transient ischemic attack in young adults in the stroke in Young
Fabry patients study. Stroke. 44:119–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mu Y, Yan WJ, Yin TL, Zhang Y, Li J and
Yang J: Diet-induced obesity impairs spermatogenesis: A potential
role for autophagy. Sci Rep. 7:434752017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deng X, Xie Y and Zhang A: Advance of
autophagy in chronic kidney diseases. Ren Fail. 39:306–313. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rezabakhsh A, Cheraghi O, Nourazarian A,
Hassanpour M, Kazemi M, Ghaderi S, Faraji E, Rahbarghazi R, Avci
ÇB, Bagca BG and Garjani A: Type 2 diabetes inhibited human
mesenchymal stem cells angiogenic response by over-activity of the
autophagic pathway. J Cell Biochem. 118:1518–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu X and Ren J: Unmasking the janus faces
of autophagy in obesity-associated insulin resistance and cardiac
dysfunction. Clin Exp Pharmacol Physiol. 39:200–208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Quan W, Jung HS and Lee MS: Role of
autophagy in the progression from obesity to diabetes and in the
control of energy balance. Arch Pharm Res. 36:223–229. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Kong F, Pan Q, Du Y, Ye J, Zheng F,
Li H and Zhou J: Autophagy protects against cholesterol-induced
apoptosis in pancreatic β-cells. Biochem Biophys Res Commun.
482:678–685. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Starobinets H, Ye J, Broz M, Barry K,
Goldsmith J, Marsh T, Rostker F, Krummel M and Debnath J: Antitumor
adaptive immunity remains intact following inhibition of autophagy
and antimalarial treatment. J Clin Invest. 126:4417–4429. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Contreras AV, Torres N and Tovar AR:
PPAR-α as a key nutritional and environmental sensor for metabolic
adaptation. Adv Nutr. 4:439–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van der Krieken SE, Popeijus HE, Mensink
RP and Plat J: Link between ER-stress, PPAR-alpha activation and
BET inhibition in relation to apolipoprotein A-I transcription in
HepG2 cells. J Cell Biochem. 118:2161–2167. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lewy TG, Grabowski JM and Bloom ME: BiP:
Master regulator of the unfolded protein response and crucial
factor in flavivirus biology. Yale J Biol Med. 90:291–300.
2017.PubMed/NCBI
|
|
15
|
Sha H, He Y, Yang L and Qi L: Stressed out
about obesity: IRE1α-XBP1 in metabolic disorders. Trends Endocrinol
Metab. 22:374–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Sun RQ, Camera D, Zeng XY, Jo E,
Chan SM, Herbert TP, Molero JC and Ye JM: Endoplasmic reticulum
stress up-regulates Nedd4-2 to induce autophagy. FASEB J.
30:2549–2556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guan H, Yang H, Yang M, Yanagisawa D,
Bellier JP, Mori M, Takahata S, Nonaka T, Zhao S and Tooyama I:
Mitochondrial ferritin protects SH-SY5Y cells against H2O2-induced
oxidative stress and modulates α-synuclein expression. Exp Neurol.
291:51–61. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Braun RJ and Westermann B: With the help
of MOM: Mitochondrial contributions to cellular quality control.
Trends Cell Biol. 27:441–452. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bernardini JP, Lazarou M and Dewson G:
Parkin and mitophagy in cancer. Oncogene. 36:1315–1327. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao C, Chen Z, Xu X, An X, Duan S, Huang
Z, Zhang C, Wu L, Zhang B, Zhang A, et al: Pink1/Parkin-mediated
mitophagy play a protective role in cisplatin induced renal tubular
epithelial cells injury. Exp Cell Res. 350:390–397. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fang D, Yan S, Yu Q, Chen D and Yan SS:
Mfn2 is required for mitochondrial development and synapse
formation in human induced pluripotent stem Cells/hiPSC derived
cortical neurons. Sci Rep. 6:314622016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Adachi C, Yamanaka-Okumura H, Katayama T,
Taketani Y and Takeda E: Single vegetable meal content equivalence
as an alternative to fat for satiety: A randomised trial in
Japanese women. Asia Pac J Clin Nutr. 25:478–486. 2016.PubMed/NCBI
|
|
25
|
De Lorenzo A, Bernardini S, Gualtieri P,
Cabibbo A, Perrone MA, Giambini I and Di Renzo L: Mediterranean
meal versus Western meal effects on postprandial ox-LDL, oxidative
and inflammatory gene expression in healthy subjects: A randomized
controlled trial for nutrigenomic approach in cardiometabolic risk.
Acta Diabetol. 54:141–149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takabatake Y, Yamamoto T and Isaka Y:
Stagnation of autophagy: A novel mechanism of renal lipotoxicity.
Autophagy. 13:775–776. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hao T, Zhang H, Li S and Tian H:
Glucagon-like peptide 1 receptor agonist ameliorates the insulin
resistance function of islet β cells via the activation of
PDX-1/JAK signaling transduction in C57/BL6 mice with high-fat
diet-induced diabetes. Int J Mol Med. 39:1029–1036. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng P, Xie Z, Yuan Y, Sui W, Wang C, Gao
X, Zhao Y, Zhang F, Gu Y, Hu P, et al: Plin5 alleviates myocardial
ischaemia/reperfusion injury by reducing oxidative stress through
inhibiting the lipolysis of lipid droplets. Sci Rep. 7:425742017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sakamoto A, Saotome M, Hasan P, Satoh T,
Ohtani H, Urushida T, Katoh H, Satoh H and Hayashi H:
Eicosapentaenoic acid ameliorates palmitate-induced lipotoxicity
via the AMP kinase/dynamin-related protein-1 signaling pathway in
differentiated H9c2 myocytes. Exp Cell Res. 351:109–120. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qin H, Zhang Y, Wang R, Du X, Li L and Du
H: Puerarin suppresses Na+-K+-ATPase-mediated systemic inflammation
and CD36 expression, and alleviates cardiac lipotoxicity in vitro
and in vivo. J Cardiovasc Pharmacol. 68:465–472. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rhee JS, Saben JL, Mayer AL, Schulte MB,
Asghar Z, Stephens C, Chi MM and Moley KH: Diet-induced obesity
impairs endometrial stromal cell decidualization: A potential role
for impaired autophagy. Hum Reprod. 31:1315–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E and
Tanaka K: Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li SJ, Liu CH, Chu HP, Mersmann HJ, Ding
ST, Chu CH, Wang CY and Chen CY: The high-fat diet induces
myocardial fibrosis in the metabolically healthy obese minipigs-The
role of ER stress and oxidative stress. Clin Nutr. 36:760–767.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J,
Shao M, Zhao F, He S, Yang L, et al: The metabolic ER stress sensor
IRE1α suppresses alternative activation of macrophages and impairs
energy expenditure in obesity. Nat Immunol. 18:519–529. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu X and Ren J: Macrophage migration
inhibitory factor (MIF) knockout preserves cardiac homeostasis
through alleviating Akt-mediated myocardial autophagy suppression
in high-fat diet-induced obesity. Int J Obes (Lond). 39:387–396.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kong FJ, Wu JH, Sun SY and Zhou JQ: The
endoplasmic reticulum stress/autophagy pathway is involved in
cholesterol-induced pancreatic β-cell injury. Sci Rep. 7:447462017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang J, Zhao P, Quan N, Wang L, Chen X,
Cates C, Rousselle T and Li J: The endotoxemia cardiac dysfunction
is attenuated by AMPK/mTOR signaling pathway regulating autophagy.
Biochem Biophys Res Commun. 492:520–527. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hashemzaei M, Heravi Entezari R, Rezaee R,
Roohbakhsh A and Karimi G: Regulation of autophagy by some natural
products as a potential therapeutic strategy for cardiovascular
disorders. Eur J Pharmacol. 802:44–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao D, Wang W, Wang H, Peng H, Liu X, Guo
W, Su G and Zhao Z: PKD knockdown inhibits pressure
overload-induced cardiac hypertrophy by promoting autophagy via
AKT/mTOR pathway. Int J Biol Sci. 13:276–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ceylan-Isik AF, Kandadi MR, Xu X, Hua Y,
Chicco AJ, Ren J and Nair S: Apelin administration ameliorates high
fat diet induced cardiac hypertrophy and contractile dysfunction. J
Mol Cell Cardiol. 63:4–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cui M, Yu H, Wang J, Gao J and Li J:
Chronic caloric restriction and exercise improve metabolic
conditions of dietary-induced obese mice in autophagy correlated
manner without involving AMPK. J Diabetes Res. 2013:8527542013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hsu HC, Chen CY, Lee BC and Chen MF:
High-fat diet induces cardiomyocyte apoptosis via the inhibition of
autophagy. Eur J Nutr. 55:2245–2254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Z, Li L, Zhao H, Peng S and Zuo Z:
Chronic high fat diet induces cardiac hypertrophy and fibrosis in
mice. Metabolism. 64:917–925. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Canadian Council on Animal Care: Guide to
the care and use of experimental animals. Ottawa ON, CCAC. 1:(2nd
Edn). 2121993.
|
|
46
|
Hamm TE: Proposed institutional animal
care and use committee guidelines for death as an endpoint in
rodent studies. Contemporary Topics Lab Animal Sci. 34:69–71.
1995.
|
|
47
|
Aroor AR, Mandavia C, Ren J, Sowers JR and
Pulakat L: Mitochondria and oxidative stress in the cardiorenal
metabolic syndrome. Cardiorenal Med. 2:87–109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rüb C, Wilkening A and Voos W:
Mitochondrial quality control by the Pink1/Parkin system. Cell
Tissue Res. 367:111–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wood-Kaczmar A, Gandhi S, Yao Z, Abramov
AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E,
et al: PINK1 is necessary for long term survival and mitochondrial
function in human dopaminergic neurons. PLoS One. 3:e24552008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Billia F, Hauck L, Konecny F, Rao V, Shen
J and Mak TW: PTEN-inducible kinase 1 (PINK1)/Park6 is
indispensable for normal heart function. Proc Natl Acad Sci USA.
108:9572–9577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vives-Bauza C, Zhou C, Huang Y, Cui M, de
Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of Parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:378–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gomes LC, Di Benedetto G and Scorrano L:
During autophagy mitochondria elongate, are spared from degradation
and sustain cell viability. Nat Cell Biol. 13:589–598. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mattson DL: Long-term measurement of
arterial blood pressure in conscious mice. Am J Physiol.
274:R564–R570. 1998.PubMed/NCBI
|
|
54
|
Calligaris SD, Lecanda M, Solis F, Ezquer
M, Gutiérrez J, Brandan E, Leiva A, Sobrevia L and Conget P: Mice
long-term high-fat diet feeding recapitulates human cardiovascular
alterations: An animal model to study the early phases of diabetic
cardiomyopathy. PLoS One. 8:e609312013. View Article : Google Scholar : PubMed/NCBI
|