Introduction
Stroke is a heterogeneous disease and is the leading
cause of mortality in the world. For patients with ischaemic
stroke, anticoagulant therapy has been the mainstay of treatment to
prevent recurrent ischaemic stroke and venous thromboembolism.
Specifically, anticoagulant intervention with oral vitamin K
antagonists, including warfarin, is used to prevent recurrent
stroke. However, this drug is substantially underused owing to
concerns over the risk of bleeding (1). Among the different types of stroke,
ischemia stroke is the main type, which makes up 60–80% of all
stroke evens. Neuronal infarct and behavioral dysfunction caused by
ischemia induced brain injury are common in patients with stroke.
Tissue plasminogen activator (tPA) is the only approved
pharmacological treatment for ischemic stroke (2) In cases that result in a negative
outcome with tPA (3), the
identification of a neuroprotective therapy is required.
Inflammation may serve an important role in regulating tissue
damage and dysfunction (4).
Previous reports indicated that dysfunctional energy metabolism may
induce inflammation in cerebral ischemia injury (5). Therefore, pharmacological treatment
with an anti-inflammatory drug may have obvious effects in treating
stroke.
AMP-activated protein kinase (AMPK) responds to
changes in the AMP:ATP ratio and is considered an index of cellular
energy levels (6), and serves as a
sensor of energy balance and is activated in response to low energy
supply (7). AMPK expression is
abundant in the brain, and cerebral AMPK is rapidly activated in
response to cerebral ischemia (8).
The effects of AMPK on the nervous system under pathological
conditions are being examined (9).
Based on its sensitivity to AMP, AMPK may be activated by nutrient
deprivation-induced metabolic stresses, such as hypoxia or glucose
deprivation (10); ischemia may
induce phosphorylation of AMPK at Thr172 (11). Furthermore, previous studies have
indicated that activation of AMPK protected against global cerebral
ischemia and focal ischemia (8,12,13).
Dexmedetomidine (DEX), an α2 adrenergic receptor
(AR) agonist, has been reported to have protective effects against
I/R injury in different tissues (14,15),
particularly in the brain (16).
DEX has also been reported to serve an anti-inflammatory role in
ischemia injury in rats (17–21).
However, the protective effects and mechanisms of DEX on brain
ischemic injury have not been fully examined. For example, it is
unknown if the anti-inflammatory effects of DEX may be associated
with AMPK pathway. Therefore, the present study aimed to
investigate the effects and mechanism of DEX on cerebral
ischemia-induced inflammation.
Materials and methods
Materials
DEX was purchased from Jiangsu Singch Pharm. Co.,
Ltd. (Jiangsu, China) and the α2-AR antagonist yohimbine (YOH) was
purchased from Tocris Bioscience (Bristol, UK)
2,3,5-Triphenyltetrazolium chloride (TTC) and pentobarbital sodium
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Primary antibodies against AMPK and ELISA kits were purchased from
R&D Systems Europe, Ltd., (Abingdon, UK). Rabbit anti-goat,
goat anti-rabbit and goat anti-mouse secondary antibodies were
purchased from R&D Systems Europe, Ltd.
Animals
A total of 126 male Sprague-Dawley (SD) rats (8
weeks, 220–250 g) were purchased from the Experimental Animal
Center of Shanghai Jiaotong University (Shanghai, China) and housed
in a controlled environment with a 12-h light/dark cycle, 60±5%
humidity and 22±2°C with access to water and food ad
libitum. All procedures were conducted in accordance with The
National Institute of Health Guide for the Care and Use of
Laboratory Animals and this study was approved by the Renji
Hospital Laboratory Animal Ethics Committee (reference no.
20170723-006).
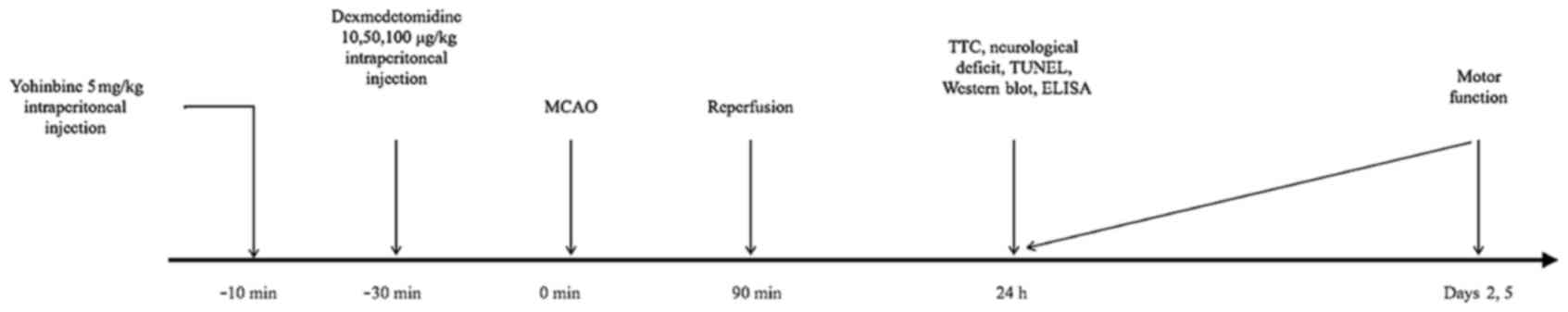
As illustrated in Fig.
1, SD rats were divided to seven experimental groups (18
rats/group): Sham surgery [treated with intraperitoneal (i.p.)
saline]; middle cerebral artery occlusion (MCAO) surgery (90 min);
DEX10 [10 µg/kg i.p. injection 30 min prior to MCAO]; DEX50 (50
µg/kg i.p. 30 min prior to MCAO); DEX100 (100 µg/kg i.p. 30 min
prior to MCAO); DEX50+Yohimbine [YOH; 5 mg/kg 10 min prior to DEX
(50 µg/kg i.p.) administration and MCAO] and YOH (5 mg/kg 40 min
prior to MCAO).
Establishment of middle cerebral
artery occlusion (MCAO) model rats
The MCAO model was established as previously
described (22). Briefly, rats
were anesthetized with an i.p injection of 3% pentobarbital sodium
(50 mg/kg; Sigma-Aldrich; Merck KGaA), and the middle cerebral
artery (MCA) was occluded by threading a monofilament sterile nylon
suture with a heat-rounded tip through the internal carotid artery,
which was advanced until it blocked the origin of the MCA. At 90
min following ischemia induction, reperfusion was initiated by
withdrawal of the monofilament. In the Sham surgery group, the MCA
was separated only and MCAO was not performed. During all surgical
procedures rats were maintained at 37°C using a heating blanket and
a heat lamp.
Drug treatments
DEX was dissolved in normal saline and administered
by i.p injection at three single doses (10, 50 100 µg/kg;
0.25 ml administration) 30 min prior to MCAO surgery. YOH (5 mg/kg;
0.25 ml) was administered by i.p 10 min prior to the
dexmedetomidine (50 µg/kg). The concentrations of DEX and YOH were
selected according to the previous reports (19,23–26).
The vehicle control was normal saline, which was administrated by
i.p injection in 0.25 ml 30 min prior to ischemia induction
(Fig. 1). SD rats were divided to
seven experimental groups (18 rats/group): Sham surgery; middle
cerebral artery occlusion (MCAO) surgery (90 min); DEX10 [10 µg/kg
i.p injection 30 min prior to MCAO]; DEX50 (50 µg/kg
i.p 30 min prior to MCAO); DEX100 (100 µg/kg i.p 30
min prior to MCAO); DEX50+Yohimbine [YOH; 5 mg/kg 10 min prior to
DEX (50 µg/kg i.p) administration and MCAO] and YOH (5 mg/kg
40 min prior to MCAO).
Evaluation of neurological
deficit
Neurological deficit scores were evaluated as
reported previously (26): 0, no
motor deficits (normal); 1, forelimb weakness and torso turning to
the ipsilateral side when held by tail (slight); 2, circling to the
contralateral side, with normal posture at rest (moderate); 3,
unable to bear weight on the affected side at rest (severe); 4, no
spontaneous locomotor activity or barrel rolling (serious).
Evaluation of infarct volume
Infarct volume was evaluated by TTC staining at 24 h
following I/R. A total of 6 rats/group were euthanized and the
brains were quickly removed. The brains were sliced into five
coronal sections (2 mm) and stained with 1% solution of TTC at 37°C
for 20 min, followed by fixation in 4% paraformaldehyde at room
temperature for 30 min. Images of TTC-stained sections were
captured by light microscopy (×1; DSC-HX9V, SONY Corporation,
Tokyo, Japan) and the digital images were analyzed using Image-Pro
Plus v6.0 (Media Cybernetics, Inc., Rockville, MD, USA) image
analysis software. Lesion volumes were calculated by multiplying
the area by the thickness of slices. The percentage of hemisphere
lesion volume (%HLV) was calculated by the following formula
(27): %HLV = {[total infarct
volume-(the volume of intact ipsilateral hemisphere-the volume of
intact contralateral hemisphere)]/contralateral hemisphere volume}
×100.
Detection of tumor necrosis factor
(TNF)-α and interleukin (IL)-6 expression in cortex
At 24 h post-I/R, 6 rats/group were sacrificed and
the brain cortex tissue were obtained from the infarcted cerebral
hemisphere and homogenized at 4°C in lysis buffer for ELISA. The
production levels of TNF-α and IL-1β were measured in brain tissue
homogenates using specific ELISA kits (TNF-α: cat. no. MTA00B;
IL-1β: cat. no. MLB00C; R&D Systems Europe, Ltd.), according to
the manufacturer's protocol; results were expressed as pg/ml.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
At 24 h following I/R, 6 rats/group were euthanized,
rats were anesthetized with 4% chloral hydrate and intracardially
perfused first with 250 ml saline and then fixed with 250 ml 4%
paraformaldehyde (at 4°C for 30 min). Then the fixed brains were
processed by embedding in paraffin blocks, and the brains (2-µm)
were sectioned. Apoptotic cells in the cortex were determined by
the terminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL) method. TUNEL staining was performed 6 times according to
the manufacturer's protocol (R&D Germany); TUNEL-positive cells
emitted a green fluorescent color and were quantified using
fluorescence microscopy (Olympus BX53; Olympus corporation) at
magnification, ×40, and 5 fields for each section were examined
from the ischemic cortex. The average percent of TUNEL-positive
cells out of the total number of cells were determined. Nuclei were
stained with DAPI.
Western blot analysis
At 24 h post-I/R, the cortex of 6 rats/group were
obtained and homogenized. Total protein extracted from cortex
homogenates with a ProteinExt™ Mammalian Total Protein
Extraction kit (TransGenBiotech, Beijing, China) was used to
analyze protein expression by western blotting. Following that,
protein concentrations were measured using a BCA Protein
Quantification kit (Yeasen, Shanghai, China) with a microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein
samples (40 µg/lane) were separated by 12% SDS-PAGE and transferred
to a polyvinylidene fluoride membrane (EMD Millipore, Billerica,
MA, USA). Membranes were placed in QuickBlock Blocking Buffer for
Western Blot (Beyotime Institute of Biotechnology, Beijing, China)
for 30 min to block non-specific binding sites at 4°C prior to
incubating with mouse primary antibodies (Cell Signaling
Technology, Inc., Danvers, MA, USA) against AMPKα (1:1,000; cat.
no. 2532), phosphorylated (p)-AMPK (1:1,000; cat. no. 4186) and
β-actin (1:2,000; Abcam; cat. no. ab8226) at 4°C overnight. The
membranes were subsequently washed with TBST (0.05% Tween-20),
incubated with the appropriate secondary antibodies (horseradish
peroxidase-conjugated anti-mouse IgG secondary antibody; R&D
Systems Europe, Ltd.; cat. no. HAF007, 1:2,000) at room temperature
for 2 h and washed with TBST. This experiment was repeated 6 times.
The protein bands were detected using a Bio-Rad Imaging System
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and quantified
using the Quantity One software package (version 2, Bio-Rad
Laboratories, Inc.). The expression of p-AMPK was normalized to
β-actin.
Motor function tests
A total of 6 rats/group were subjected to neuromotor
tests (screen clinging, horizontal bar and prehensile traction) at
days 1, 2 and 5 post-I/R, as previously described (28), with a score of 9 being the best
possible score. The rats were placed on a screen (29×30 cm), which
was rotated in the vertical plane. The time that the rat was able
to hold onto the vertical screen was recorded for a maximum of 15
seconds (screen clinging). In addition, the rats were placed at the
center of a horizontal wooden rod (diameter, 2.5 cm) and the time
that the rat was able to remain balanced on the rod was recorded
for a maximum of 30 sec (horizontal bar). Finally, the time that
the animal was able to cling to a horizontal rope was recorded for
a maximum of 5 sec (prehensile traction). The tests were performed
by a researcher who was blinded to animal group assignments.
Statistical analyses
Data were analyzed by one-way analysis of variance
followed by Tukey's post hoc test except total motor scores using
GraphPad Prism 5.0 (Graphpad, Inc., La Jolla, CA, USA). Neuromotor
scores using the Tukey's Multiple Comparison Test data were
expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference. Each
experiment was repeated six times.
Results
DEX suppresses I/R-induced neuronal
injury and apoptosis in the cortex of rats
Compared with rats in the MCAO group, the
neurological scores and infarct volumes were significantly
decreased in rats in each of the three DEX pre-treatment groups
(P<0.01; Fig. 2A and B). No
significant differences were identified for neurological scores and
infarct volume in the YOH group compared with the MCAO group.
Compared with the DEX10 group, the neurological scores and infarct
volumes were significantly decreased in DEX50 and DEX100 groups
(P<0.05); However, no significant differences were identified
between the DEX50 and the DEX100 group (P>0.05). The
neurological scores and infarct volumes were significantly
increased in rats in the DEX50+YOH group (P<0.001).
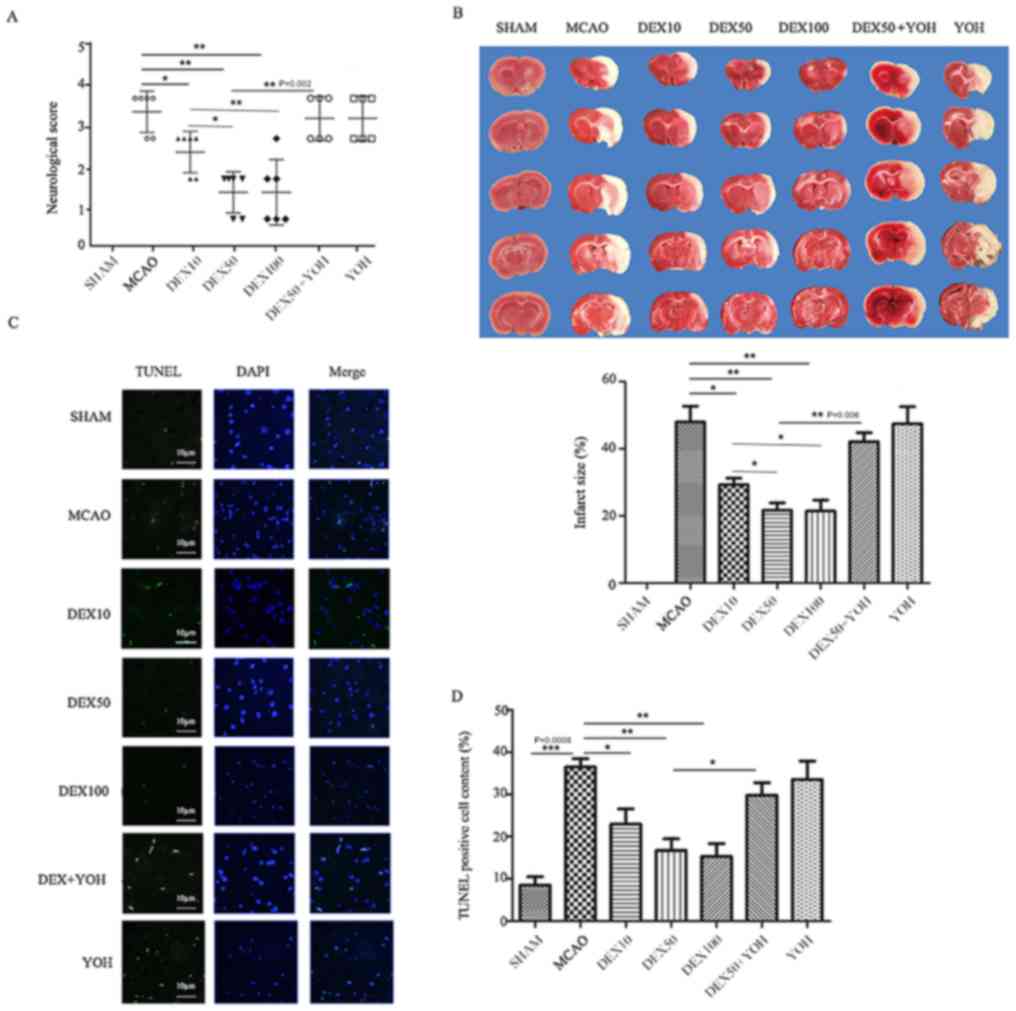 | Figure 2.DEX suppresses MACO-induced neuronal
death and apoptosis in rats. (A) Neurological score. Score of Sham
group was 0. (B) TTC staining of brain tissue was used to determine
ischemia-induced infarct size in the cortexes in rats in the
various treatment groups. Infarct size, the Sham group was scored
0. (C) Apoptotic neurons were detected using TUNEL staining
(green); nuclei were stained with DAPI (blue); scale bar, 10 µm.
(D) The percent of TUNEL-positive cells was determined for each
experimental group. Data are presented as the mean ± standard
deviation; *P<0.05, **P<0.01 and ***P<0.001. MCAO, middle
cerebral artery occlusion; TTC, 2,3,5-triphenyltetrazolium
chloride; TUNEL, terminal deoxynucleotidyl-transferase-mediated
dUTP nick end labeling. DEX10/50/100, dexmedetomidine pre-treatment
(10/50/100 µg/kg); YOH, yohimbine. |
Compared with the Sham group, the percent of
TUNEL-positive cells and infarct size in the cortex of ischemic
hemisphere in MCAO group increased (P<0.001; Fig. 2C and D). Compared with the MCAO
group, the percent of TUNEL-positive cells was significantly
decreased in the DEX treatment groups (P<0.05 or
P<0.01); however, no significant difference in apoptotic
cells was identified in the YOH group compared with the MCAO group.
Compared with the DEX10 group, the percent of TUNEL-positive cells
was significantly decreased in the DEX50 and DEX100 groups
(Fig. 2D; P<0.01). Compared
with the DEX50 group, there was a significant increase in apoptotic
cells in the DEX50+YOH group (P<0.01).
DEX alleviates I/R-induced
inflammation in rats via activation of AMPK
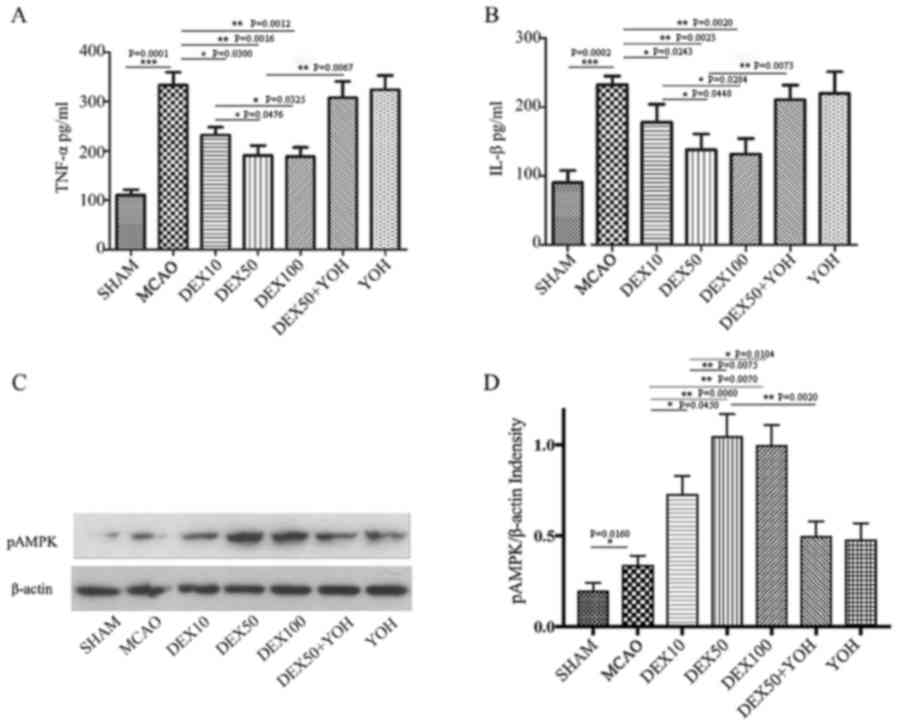
The production levels of TNF-α and IL-1β in the
brain tissues were examined to elucidate the effects of DEX on
MCAO-induced inflammation (Fig. 3A and
B, respectively). Compared with the Sham group, the levels of
TNF-α and IL-1β were significantly increased in the MCAO group
(P<0.001). Compared with the MCAO group, the levels of TNF-α and
IL-1β were significantly decreased in the three DEX treatment
groups (P<0.01). YOH had no observable effects on the production
of TNF-α and IL-1β in the cortex compared with the MCAO group.
Compared to DEX50 group, the levels of TNF-α and IL-1β were higher
in the DEX50+YOH-treatment group (P<0.01).
The expression levels of p-AMPK were also examined
in the ischemic brain cortex. The levels of p-AMPK were elevated in
ischemic cortex (Fig. 3C and D).
Compared with the MCAO group, pretreatment with DEX (10, 50 and 100
µg/kg) resulted in a significant increase in the expression levels
of p-AMPK (P<0.01). p-AMPK expression levels were
increased in DEX50 and DEX100 groups compared with the DEX10 group
(P<0.05). Compared with the DEX50 group, p-AMPK expression was
significantly lower in the DEX50+YOH group (P<0.01).
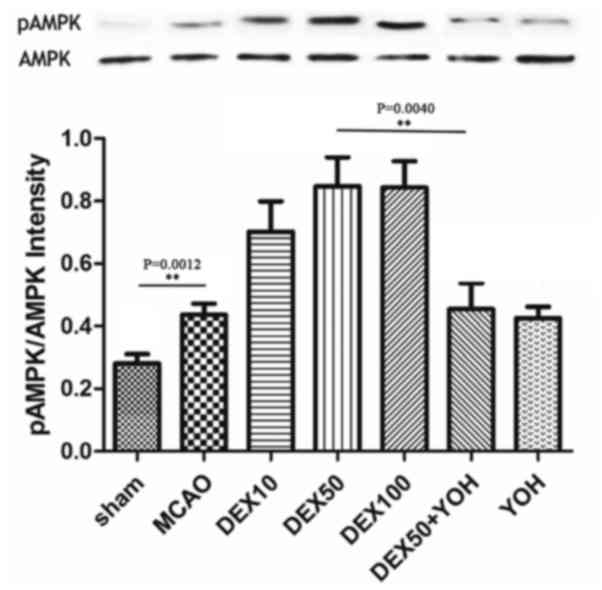
Furthermore, the ratio of p-AMPK/AMPK as illustrated in Fig. 4.
DEX improves motor functions following
forebrain ischemia
Focal cerebral ischemia resulted in reduced motor
function scores in MCAO rats compared with Sham group rats at day 1
following I/R (Fig. 5A), and the
motor functions remained low at days 2 and 5 (Fig. 5B and C, respectively). The decline
of motor function was partially improved by pre-treatment with DEX,
and the higher doses (50 and 100 µg/kg) improved the motor function
scores more significantly compared with the lower dose (10 µg/kg;
P<0.05). No significant differences in motor functions were
identified in rats in the YOH groups compared with the MCAO group;
However, compared with the DEX50 group, pretreatment of YOH
resulted in a significant reduction in the motor score
(P<0.01).
Discussion
Results from the present study demonstrated that
different doses of DEX (10, 50 100 µg/kg), an α2-AR agonist, were
able to effectively reduce the cortex area of brain injury, cell
apoptosis and inflammation; DEX pre-treatment also significantly
improved motor function scores. The protective effects of DEX on
I/R-induced cerebral brain cell injury may function through the
AMPK pathway. In addition, the α2-AR antagonist YOH was revealed to
inhibit the effects of DEX.
DEX functions include antianxiety, analgesia and
restraining the sympathetic nerve activity (29). Previous clinical and animal studies
have demonstrated that DEX attenuated cerebral injury and decreased
the occurrence of disordered cerebral function during the
perioperative period (30,31); in vivo experiments
demonstrated that DEX decreased cerebral infarction area and
improved the function of motor (32,33).
In order to investigate further the effects of DEX pretreatment on
MCAO and the related molecular mechanism. In the present study, DEX
was administered 30 min prior to ischemia induction, and the
results demonstrated that DEX pre-treatment effectively alleviate
MCAO-induced cerebral injury, reduce necrotic areas (TTC staining
area) of cerebral ischemia, improve the neurological score and
motor score. Post-I/R analysis of cortex cell apoptosis
demonstrated that DEX pre-treatment was able to reduce brain cell
apoptosis. The effects of DEX on both neurological and motor
revealed that the different concentrations DEX protected the brain
from ischemic injury, and the efficacy of high concentrations if
DEX (50 and 100 µg/kg) were more efficient compared with the lower
concentration of DEX (10 µg/kg).
The present study confirmed that inflammation may be
one of the important factors that induced I/R injury (34,35).
I/R-induced inflammation has been explored previously as a
treatment target. DEX has been widely used as a sedative drug
(36), many previous studies also
demonstrated the DEX inhibited infection or non-infection induced
by inflammation in vivo or in vitro (37–39).
Particularly, DEX was reported to serve an anti-inflammatory role
in ischemic injury in intestines, lungs and kidneys (20,40,41).
The present study explored the effects of DEX on inflammation
following cerebral ischemia in rats, and the results demonstrated
that DEX pre-treatment decreased the levels of TNF-α and IL-1β in
MCAO rats, which was similar to previous studies (16,19,42).
In addition, the effects of different concentrations of DEX on
inflammation demonstrated that the anti-inflammatory effects are
increased with high concentrations of DEX compared with the lower
concentration, as indicated by the reduced levels of TNF-α and
IL-1β expression in DEX50 and DEX100 groups compared with the DEX10
group. The present study explored the effects of DEX on MCAO and
focused on the changes in expression of inflammation-related
factors in brain tissue in MCAO rats. In the future, the changes of
inflammation factor expressions will be examined in plasma.
Although DEX was able to reduce I/R-induced
inflammation, the related mechanisms were not clear. Abnormal
cellular energy metabolism has been reported as an important factor
for inflammation reactions (43).
AMPK is considered a key enzyme in the process of energy metabolism
and the AMPK pathway is the energy sensitive protein kinase
(44). These factors serve a key
role in inflammatory disease development. Activated AMPK has been
reported to inhibit the inflammation reaction (45,46).
The present study demonstrated that DEX affected the expression of
P-AMPK in the cortex of MCAO rats in a dose-dependent manner, as
illustrated in Fig. 3C and D. A
previous study reported that increased cAMP inhibits AMPK through
the activated protein kinase A (PKA) pathway. The PKA inhibitor H89
was previously demonstrated to increase the activity of AMPK
(47). Other studies reported that
DEX suppresses the expression of cAMP via α2-AR and subsequently
affects the generation of PKA (48,49).
Therefore, it was speculated that DEX may activate the AMPK pathway
by inhibiting PKA and attenuate the levels of inflammatory factors,
thus inhibiting the inflammation reaction following MCAO. Local
cerebral I/R injury reduced the function of cerebral pallium, which
is the center of motor functions, and the changes of motor
functions were examined for 5 days post-MCAO surgery; the motor
function score was reduced at days 1, 2 and 5 in untreated rats.
However, motor function scores were significantly improved in rats
that received DEX pre-treatment. DEX exerts neuroprotective effects
via α2-AR, and this effect was demonstrated to be reversed by the
α2-AR antagonist YOH.
There are some limitations in the present study.
First, the dose of DEX should be studied further, including the
post-ischemia and intravenous dose. Furthermore, the effects of DEX
post-treatment on MCAO-induced injury and the associated molecular
mechanism require more investigation. Second, the activation
mechanism of DEX on AMPK is unclear or whether the upstream and
downstream signal pathways of AMPK were involved in regulating
MCAO-induced injury
In conclusion, DEX was demonstrated to inhibit
inflammation, alleviate cerebral injury and improve the motor
function score. DEX may exert protective effects by activating AMPK
and inhibiting the generation of inflammatory factors. Future
studies will continue to explore the mechanism of DEX on activating
AMPK.
Acknowledgements
The authors would like to thanks Jihua Xin for help
with Motor function tests.
Funding
The present study is supported by The National
Natural Science Foundation of China (grant no. NSFC 81300996).
Availability of data and materials
All data generated or materials during this study
are included in this published article.
Authors' contributions
ZH and ZW participated in research design; WZ, HD
and ZW conducted experiments; WZ and XM performed data analysis;
and WZ and ZW wrote or contributed to the writing of the
manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from the Renji Hospital
Laboratory Animal Ethics Committee (reference no.
20170723-006).
Patient consent for publication
Not applicable.
Competing interests
Not applicable.
References
|
1
|
Hankey GJ: Anticoagulant therapy for
patients with ischaemic stroke. Nat Rev Neurol. 8:319–328. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grossman AW and Broderick JP: Advances and
challenges in treatment and prevention of ischemic stroke. Ann
Neurol. 74:363–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng YD, Al-Khoury L and Zivin JA:
Neuroprotection for ischemic stroke: Two decades of success and
failure. NeuroRx. 1:36–45. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wong CH and Crack PJ: Modulation of
neuro-inflammation and vascular response by oxidative stress
following cerebral ischemia-reperfusion injury. Curr Med Chem.
15:1–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miao Y and Liao JK: Potential serum
biomarkers in the pathophysiological processes of stroke. Expert
Rev Neurother. 14:173–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spasic MR, Callaerts P and Norga KK:
AMP-activated protein kinase (AMPK) molecular crossroad for
metabolic control and survival of neurons. Neuroscientist.
15:309–316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hardie DG: Energy sensing by the
AMP-activated protein kinase and its effects on muscle metabolism.
Proc. Nutr. Soc. 70:92–99. 2011.
|
|
8
|
McCullough LD, Zeng Z, Li H, Landree LE,
McFadden J and Ronnett GV: Pharmacological inhibition of
AMP-activated protein kinase provides neuroprotection in stroke. J
Biol Chem. 280:20493–20502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Huang Y, Xu Y, Ruan W, Wang H,
Zhang Y, Saavedra JM, Zhang L, Huang Z and Pang T: A dual AMPK/Nrf2
activator reduces brain inflammation after stroke by enhancing
microglia M2 polarization. Antioxid Redox Signal. 28:141–163. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zang Y, Yu LF, Pang T, Fang LP, Feng X,
Wen TQ, Nan FJ, Feng LY and Li J: AICAR induces astroglial
differentiation of neural stem cells via activating the JAK/STAT3
pathway independently of AMP-activated protein kinase. J Biol Chem.
283:6201–6208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C,
Zhu CH and Miao JY: Apelin-13 protects against apoptosis by
activating AMP-activated protein kinase pathway in ischemia stroke.
Peptides. 75:96–100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ashabi G, Khodagholi F, Khalaj L,
Goudarzvand M and Nasiri M: Activation of AMP-activated protein
kinase by metformin protects against global cerebral ischemia in
male rats: Interference of AMPK/PGC-1α pathway. Metab Brain Dis.
29:47–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ashabi G, Khalaj L, Khodagholi F,
Goudarzvand M and Sarkaki A: Pre-treatment with metformin activates
Nrf2 antioxidant pathways and inhibits inflammatory responses
through induction of AMPK after transient global cerebral ischemia.
Metab Brain Dis. 30:747–754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gu J, Sun P, Zhao H, Watts HR, Sanders RD,
Terrando N, Xia P, Maze M and Ma D: Dexmedetomidine provides
renoprotection against ischemia-reperfusion injury in mice. Crit
Care. 15:R1532011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang XY, Liu ZM, Wen SH, Li YS, Li Y, Yao
X, Huang WQ and Liu KX: Dexmedetomidine administration before, but
not after, ischemia attenuates intestinal injury induced by
intestinal ischemia-reperfusion in rats. Anesthesiology.
116:1035–1046. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng X, Wang H, Xing X, Wang Q and Li W:
Dexmedetomidine protects against transient global cerebral
ischemia/reperfusion induced oxidative stress and inflammation in
diabetic rats. PLoS One. 11:e01516202016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Wang Z, Wang Y, Zhou G and Li H:
The effect of dexmedetomidine on inflammatory response of septic
rats. BMC Anesthesiol. 15:682015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Y, Zhang R, Li C, Yin X, Lv C, Wang Y,
Zhao W and Zhang X: Dexmedetomidine attenuates acute lung injury
induced by lipopolysaccharide in mouse through inhibition of MAPK
pathway. Fundam Clin Pharmacol. 29:462–471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren X, Ma H and Zuo Z: Dexmedetomidine
postconditioning reduces brain injury after brain hypoxia-ischemia
in neonatal rats. J Neuroimmune Pharmacol. 11:238–247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu G, Song H, Qiu L, He A, Tong F, Wan Q,
Wang X, Xia Y and Huang L: Dexmedetomidine preconditioning inhibits
the long term inflammation induced by renal ischemia/reperfusion
injury in rats. Acta Cir Bras. 31:8–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shou-Shi W, Ting-Ting S, Ji-Shun N and
Hai-Chen C: Preclinical efficacy of dexmedetomidine on spinal cord
injury provoked oxidative renal damage. Ren Fail. 37:1190–1197.
2015.PubMed/NCBI
|
|
22
|
Yu K, Wu Y, Hu Y, Zhang Q, Xie H, Liu G,
Chen Y, Guo Z and Jia J: Neuroprotective effects of prior exposure
to enriched environment on cerebral ischemia/reperfusion injury in
rats: The possible molecular mechanism. Brain Res. 1538:93–103.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sato K, Kimura T, Nishikawa T, Tobe Y and
Masaki Y: Neuroprotective effects of a combination of
dexmedetomidine and hypothermia after incomplete cerebral ischemia
in rats. Acta Anaesthesiol Scand. 54:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Laudenbach V, Mantz J, Lagercrantz H,
Desmonts JM, Evrard P and Gressens P: Effects of
alpha(2)-adrenoceptor agonists on perinatal excitotoxic brain
injury: Comparison of clonidine and dexmedetomidine.
Anesthesiology. 96:134–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuhmonen J, Pokorný J, Miettinen R,
Haapalinna A, Jolkkonen J, Riekkinen P Sr..Sivenius J:
Neuroprotective effects of dexmedetomidine in the gerbil
hippocampus after transient global ischemia. Anesthesiology.
87:371–377. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Cummins Reversible middle cerebral artery occlusion
without craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li MH, Ruan LY, Chen C, Xing YX, Hong W,
Du RH and Wang JS: Protective effects of polygonum multiflorum on
ischemic stroke rat model analysed by 1H NMR metabolic
profiling. J Pharm Biomed Anal. 155:91–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gionet TX, Thomas JD, Warner DS, Goodlett
CR, Wasserman EA and West JR: Forebrain ischemia induces selective
behavioral impairments associated with hippocampal injury in rats.
Stroke. 22:1040–1047. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Q, Wu D, Yang Y, Liu T and Liu H:
Dexmedetomidine alleviates hyperoxia-induced acute lung injury via
inhibiting NLRP3 inflammasome activation. Cell Physiol Biochem.
42:1907–1919. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schoeler M, Loetscher PD, Rossaint R,
Fahlenkamp AV, Eberhardt G, Rex S, Weis J and Coburn M:
Dexmedetomidine is neuroprotective in an in vitro model for
traumatic brain injury. BMC Neurol. 12:202012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ji F, Li Z, Nguyen H, Young N, Shi P,
Fleming N and Liu H: Perioperative dexmedetomidine improves
outcomes of cardiac surgery. Circulation. 127:1576–1584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuhmonen J, Haapalinna A and Sivenius J:
Effects of dexmedetomidine after transient and permanent occlusion
of the middle cerebral artery in the rat. J Neural Transm (Vienna).
108:261–271. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding XD, Zheng NN, Cao YY, Zhao GY and
Zhao P: Dexmedetomidine preconditioning attenuates global cerebral
ischemic injury following asphyxial cardiac arrest. Int J Neurosci.
126:249–256. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trendelenburg G: Molecular regulation of
cell fate in cerebral ischemia: Role of the inflammasome and
connected pathways. J Cereb Blood Flow Metab. 34:1857–1867. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang R, Wang ST, Wang YD, Wu G, Du Y, Qian
MQ, Liang XG, Elbatreek MH, Yang HY, Liu ZR, et al:
Stress-responsive heme oxygenase-1 isoenzyme participates in
Toll-like receptor 4-induced inflammation during brain ischemia.
Neuroreport. 27:445–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X, Zhang K, Wang W, Xie G and Fang X:
Dexmedetomidine sedation reduces atrial fibrillation after cardiac
surgery compared to propofol: A randomized controlled trial. Crit
Care. 20:1–8. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sezer A, Memis D, Usta U and Sut N: The
effect of dexmedetomidine on liver histopathology in a rat sepsis
model: An experimental pilot study. Ulus Travma Acil Cerrahi Derg.
16:108–112. 2010.PubMed/NCBI
|
|
38
|
Wu Y, Liu Y, Huang H, Zhu Y, Zhang Y, Lu F
and Zhou C, Huang L, Li X and Zhou C: Dexmedetomidine inhibits
inflammatory reaction in lung tissues of septic rats by suppressing
TLR4/NF-κB pathway. Mediators Inflamm. 2013:5621542013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yeh YC, Wu CY, Cheng YJ, Liu CM, Hsiao JK,
Chan WS, Wu ZG, Yu LC and Sun WZ: Effects of dexmedetomidine on
intestinal microcirculation and intestinal epithelial barrier in
endotoxemic rats. Anesthesiology. 125:355–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun Y, Gao Q, Wu N, Li SD, Yao JX and Fan
WJ: Protective effects of dexmedetomidine on intestinal
ischemia-reperfusion injury. Exp Ther Med. 10:647–652. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gao S, Wang Y, Zhao J and Su A: Effects of
dexmedetomidine pretreatment on heme oxygenase-1 expression and
oxidative stress during one-lung ventilation. Int J Clin Exp
Pathol. 8:3144–3149. 2015.PubMed/NCBI
|
|
42
|
Sifringer M, von Haefen C, Krain M,
Paeschke N, Bendix I, Bührer C, Spies CD and Endesfelder S:
Neuroprotective effect of dexmedetomidine on hyperoxia-induced
toxicity in the neonatal rat brain. Oxid Med Cell Longev.
2015:5303712015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Falkowska A, Gutowska I, Goschorska M,
Nowacki P, Chlubek D and Baranowska-Bosiacka I: Energy metabolism
of the brain, including the cooperation between astrocytes and
neurons, especially in the context of glycogen metabolism. Int J
Mol Sci. 16:25959–25981. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li XD, Wang LL, Zhou XE, Ke J, de Waal PW,
Gu X, Eileen Tan MH, Wang D, Wu D, Eric Xu H and Melcher K:
Erratum: Structural basis of AMPK regulation by adenine nucleotides
and glycogen. Cell Res. 25:3982015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yerra VG and Kumar A: Adenosine
monophosphate-activated protein kinase abates
hyperglycaemia-induced neuronal injury in experimental models of
diabetic neuropathy: Effects on mitochondrial biogenesis, autophagy
and neuroinflammation. Mol Neurobiol. 54:2301–2312. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng YF, Young GH, Lin JT, Jang HH, Chen
CC, Nong JY, Chen PK, Kuo CY, Kao SH, Liang YJ and Chen HM:
Activation of AMP-activated protein kinase by adenine alleviates
TNF-Alpha-induced inflammation in human umbilical vein endothelial
cells. PLoS One. 10:e01422832015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hurtado de Llera A, Martin-Hidalgo D, Gil
MC, Garcia-Marin LJ and Bragado MJ: The calcium/CaMKKalpha/beta and
the cAMP/PKA pathways are essential upstream regulators of AMPK
activity in boar spermatozoa. Biol Reprod. 90:292014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gu XY, Liu BL, Zang KK, Yang L, Xu H, Pan
HL, Zhao ZQ and Zhang YQ: Dexmedetomidine inhibits
Tetrodotoxin-resistant Nav1.8 sodium channel activity through
Gi/o-dependent pathway in rat dorsal root ganglion neurons. Mol
Brain. 8:152015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tanabe K, Matsushima-Nishiwaki R, Kozawa O
and Iida H: Dexmedetomidine suppresses interleukin-1β-induced
interleukin-6 synthesis in rat glial cells. Int J Mol Med.
34:1032–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|