Introduction
Tyrosine kinase inhibitors (TKIs), such as gefitinib
and erlotinib, target epidermal growth factor receptors (EGFRs) and
have been demonstrated to be efficacious in treating non-small cell
lung cancers (NSCLCs) (1–3). The dysregulation of EGFR signaling is
closely associated with cell proliferation and EGFR TKI resistance
(4,5), which highlights the importance of
complete EGFR signaling inhibition in lung cancer treatment.
Geranylgeranylation is an important biochemical
modification for cellular localization and the function of small G
proteins (6,7). Previous studies have reported that
geranylgeranylation is critical for cancer cell proliferation and
invasion (8,9), which indicates the potential
anticancer function of inhibiting geranylgeranylation. The
geranylgeranyl transferase 1 (GGT1) inhibitor (GGTI)-298 inhibits
protein geranylgeranylation. Recent studies in vitro and in
animal models have revealed that GGTI-298 inhibited cell
proliferation and induced apoptosis when applied as a single agent
or in combination with chemotherapeutics (10–12).
In addition, GGTIs inhibited EGF-mediated EGFR tyrosine
phosphorylation, indicating that the GGT1 substrate may serve a
vital role in the process of EGFR phosphorylation (13). However, the molecular mechanism
underlying the GGTI-induced inhibition of EGFR phosphorylation has
not been well studied.
Ras homolog family member A (RhoA), a widely studied
protein in the Rho family of guanosine triphosphate (GTP)-ases,
participates in various physiological processes, including
cytoskeleton organization, cell cycle progression, cell survival
and cell migration (14). A
growing number of studies have revealed that RhoA interacts with
various signaling molecules to exert effects on cell proliferation
and apoptosis (15–17). Thus, a deeper understanding of
RhoA-associated signaling regulation will support the
identification of novel strategies and modalities in cancer
treatment.
In the present study, a potentially favorable
interaction between GGTI-298 and the EGFR TKI gefitinib was
identified. The two combined stimulated cytotoxicity and induced
apoptosis in lung cancer cells. The synergistic mechanism was also
studied.
Materials and methods
Cell culture and agents
The NSCLC cell lines (HCC827, A549, H1975, SPCA1 and
H1299) were purchased from American Type Culture Collection
(Manassas, VA, USA). All cell lines were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with or without 10% heat inactivated
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
depending on the assay. Cell cultures were maintained at 37°C in a
humidified (5% CO2, 95% air) incubator. GGTI-298 and
gefitinib were purchased from Selleck Chemicals (Houston, TX, USA),
and dimethylsulfoxide (DMSO) and EGF were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Immunoblotting analysis
A549 cells were treated with 5 µM GGTI-298 in
serum-free medium for 48 h. Prior to protein extraction, the
cultures were exposed to 30 ng EGF for 30 min. Cells (HCC827, A549,
H1975, SPCA1 and H1299) were lysed with ice-cold
Radioimmunoprecipitation Assay buffer [150 mM NaCl, 1% NP-40, 50 mM
Tris-HCl (pH 7.4), 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml
leupeptin, 1 mM deoxycholic acid and 1 mM EDTA] containing a
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The
protein concentration was detected using the BCA assay (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). In total, ~20 µg total
protein was loaded on to a 12% SDS-PAGE gel, separated
electrophoretically and then transferred to nitrocellulose
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
blocked with 5% nonfat milk at room temperature for 30 min and were
washed three times with phosphate buffer saline (PBS). The
membranes were incubated with the following primary antibodies:
Phosphorylated (p)-EGFR (cat. no. 3777), cyclin D1 (cat. no. 2922),
phosphatase and tensin homolog (PTEN; cat. no. 9552), GAPDH (cat.
no. 97166), glutathione S-transferase (GST; all Cell Signaling
Technology, MA, USA), p-extracellular signal-regulated kinase (ERK;
cat. no. EP197Y), ERK (cat. no. ab 17942), p-protein kinase B (AKT;
cat. no. EP2109Y), AKT (cat. no. EPR16798; all Abcam, Cambridge,
UK), EGFR (cat. no. sc-03-G), 3-hydroxy-3-methylglutaryl-co-enzyme
A reductase (HMGCR; cat. no. sc-271595) and RhoA (cat. no. sc-418;
all Santa Cruz Biotechnology, Inc., Dallas, TX, USA). All of the
primary antibodies were added at a 1:1,000 dilution overnight at
4°C. Subsequently, the membranes were incubated with the
horseradish peroxidase conjugated anti-mouse (cat. no. HA1022) or
anti-rabbit (cat. no. HA1001) secondary antibodies with dilution of
1:5,000 (Huabio, Hangzhou, China) at room temperature for 3 h and
were washed with PBS. Protein expression was detected using the
ODYSSEY Infrared Imaging System (LI-COR Biosciences, Lincoln, NE,
USA) and quantified using Image J 1.48 (National institute of
Health, Bethesda, MA, USA).
Cell proliferation assay
HCC827 and A549 cells were seeded (~5×103
cells per well in 100 µl DMEM with 10% FBS) in a 96-well
flat-bottomed plate (Corning Incorporated, Corning, NY, USA) at 24
h prior to treatment, namely, HCC827 and A549 cells were treated
with gefitinib (HCC827, 10 nM; A549, 5 µM) and/or GGTI-298
(concentration indicated) for 48 h at 37°C in a humidified (5%
CO2, 95% air) incubator. Following incubation at 37°C,
cell growth was measured using a Cell Counting Kit-8 (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan). Synergistic
activity between gefitinib and GGTI-298 was assessed using
combination index (CI) analysis, where a CI <1 indicated
synergism, CI=1 indicated additive effects and CI >1 indicated
antagonism (18).
Cell apoptosis assay
HCC827 and A549 cells were seeded (~5×103
cells per well in 100 µl medium) in a 6-well flat-bottomed plate 24
h prior to treatment, namely, HCC827 and A549 cells were treated
with DMSO, gefitinib (HCC827, 10 nM; A549, 5 µM), GGTI-298 (HCC827,
5 µM; A549, 7.5 µM) or a combination of gefitinib and GGTI-298 for
48 h at 37°C in a humidified (5% CO2, 95% air)
incubator. Apoptotic cells were detected using Hoechst 33342
(Sigma-Aldrich; Merck KGaA) DNA staining (room temperature for 10
min), according to the kit instructions. Apoptotic cells were
characterized by nuclear condensation and DNA fragmentation, which
were observed by fluorescence microscopy (Nikon Corporation, Tokyo,
Japan).
Wound healing assay
HCC827 and A549 cells (~5×105) were
seeded in a 6-well culture plate for 24 h at 37°C. Wounding was
performed by scraping through the cell monolayer with a 200 µl
pipette tip. Medium and nonadherent cells were removed and replaced
with 2 ml of fresh serum-free medium containing the indicated
reagents (DMSO, gefitinib, GGTI-298 or a combination of gefitinib
and GGTI-298). Cells were permitted to migrate for 48 h at 37°C.
Wound healing was photographed microscopically (light) at 0, 24 and
48 h. Three fields of view for each wound were randomly selected,
and the average wound width was calculated. The relative wound
width was calculated as follows: wound width at the indicated time
(0, 24 or 48 h)/wound width at 0 h.
Pull-down assays
Briefly, to detect the levels of active RhoA, equal
amounts (~8×106 cells) of A549 cell lysates [20 mM
Hepes, 100 mM NaCl, 0.5% NP-40, 10% glycerol, 0.2% deoxycholic
acid, and 10 mM MgCl2, (pH 7.5)] were incubated with
GST-Rho binding domain proteins immobilized on
glutathione-Sepharose beads at 4°C overnight. The beads were then
washed three times with ice-cold phosphate buffered solution. Bound
proteins were eluted from the beads with SDS-PAGE sample buffer.
RhoA protein was then analyzed by immunoblotting with anti-RhoA
antibody, which was performed as previously described.
Transfection
Small interfering (si)-RNAs against RhoA and the
control were synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). The sequence of the RhoA siRNA was
5′-GACAUGCUUGCUCAUAGUCTT-3′, and the sequence of the control siRNA
was 5′-AAUUCUCCGAACGUGUCACGUUU-3′. A549 cells were transfected with
50 nM control siRNA and RhoA siRNA for 72 h using Lipofectamine
2000™ (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions.
Statistical analysis
All statistical analyses were performed with SPSS
14.0 for Windows software (SPSS, Inc., Chicago, IL, USA). The
results were analyzed using the Student's t-test when two groups
were compared, or by one-way analysis of variance with Tukey's post
hoc test for multiple comparisons. P<0.05 was considered to
indicated a statistically significant difference. Values are
presented as the mean ± standard error of the mean.
Results
Synergic antiproliferation effect of
gefitinib and GGTI-298 in NSCLC cell lines A549 and HCC827
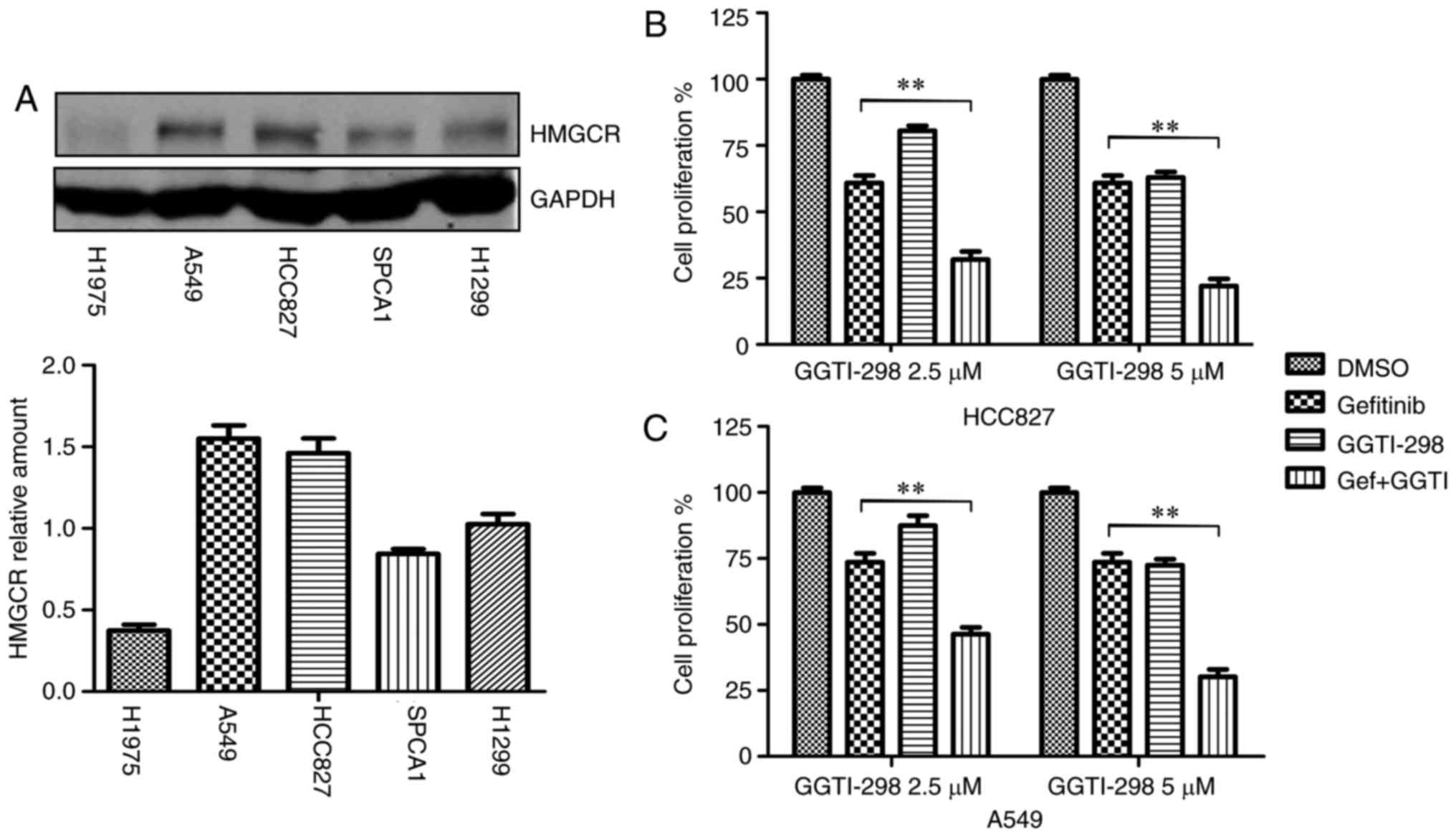
As GGTI-298 is an inhibitor of a branch of the
mevalonate pathway (19), the
present study first detected the relative protein expression of
HMGCR (the rate-limiting enzyme of mevalonate pathway) in five cell
lines (H1975, A549, HCC827, SPCA1 and H1299) and then the two cell
lines with a relatively high expression of HMGCR, HCC827 and A549,
were selected for further experimentation (Fig. 1A). To determine the effect of
GGTI-298 on gefitinib sensitivity, lung cancer cells were treated
with gefitinib, GGTI-298 and combination of gefitinib and GGTI-298.
The results revealed that the combined treatment of gefitinib and
GGTI-298 inhibited cell proliferation more thoroughly than either
of the single drug treatments, in a dose-dependent manner (Fig. 1B and C). The CI values indicated
synergy, with values of 0.48 and 0.58 for HCC827 and A549 cells,
respectively.
Enhanced effects of gefitinib and
GGTI-298 on the apoptosis and migration of lung cancer cells
To investigate the effect of GGTI-298 on
gefitinib-induced apoptosis, Hoechst assays were conducted. The
results demonstrated that a combination of GGTI-298 and gefitinib
markedly promoted cell apoptosis when compared with each
monotherapy group (Fig. 2A and B).
Addition of GGTI-298 increased apoptosis by >25% when compared
with gefitinib alone (percentage of apoptotic cells: HCC827,
gefitinib vs. gefitinib+GGTI-298=18.3 vs. 48.3%, P<0.01; A549,
gefitinib vs. gefitinib+GGTI-298=12 vs. 39.7%, P<0.01; Fig. 2B). Then, wound healing assays were
conducted to investigate the influence of GGTI-298 and gefitinib on
cell migration. The results revealed that migration ability was
significantly inhibited with a combination of gefitinib and
GGTI-298 when compared with either single drug treatment following
48 h (Fig. 2C and D). The relative
wound widths (wound width at the indicated time/wound width at 0 h)
at 48 h of gefitinib and gefitinib+GGTI-298 were 57.5 and 89.0% in
HCC827 cells, and 53.5 and 83.6% in A549 cells, respectively
(P<0.05; Fig. 2D).
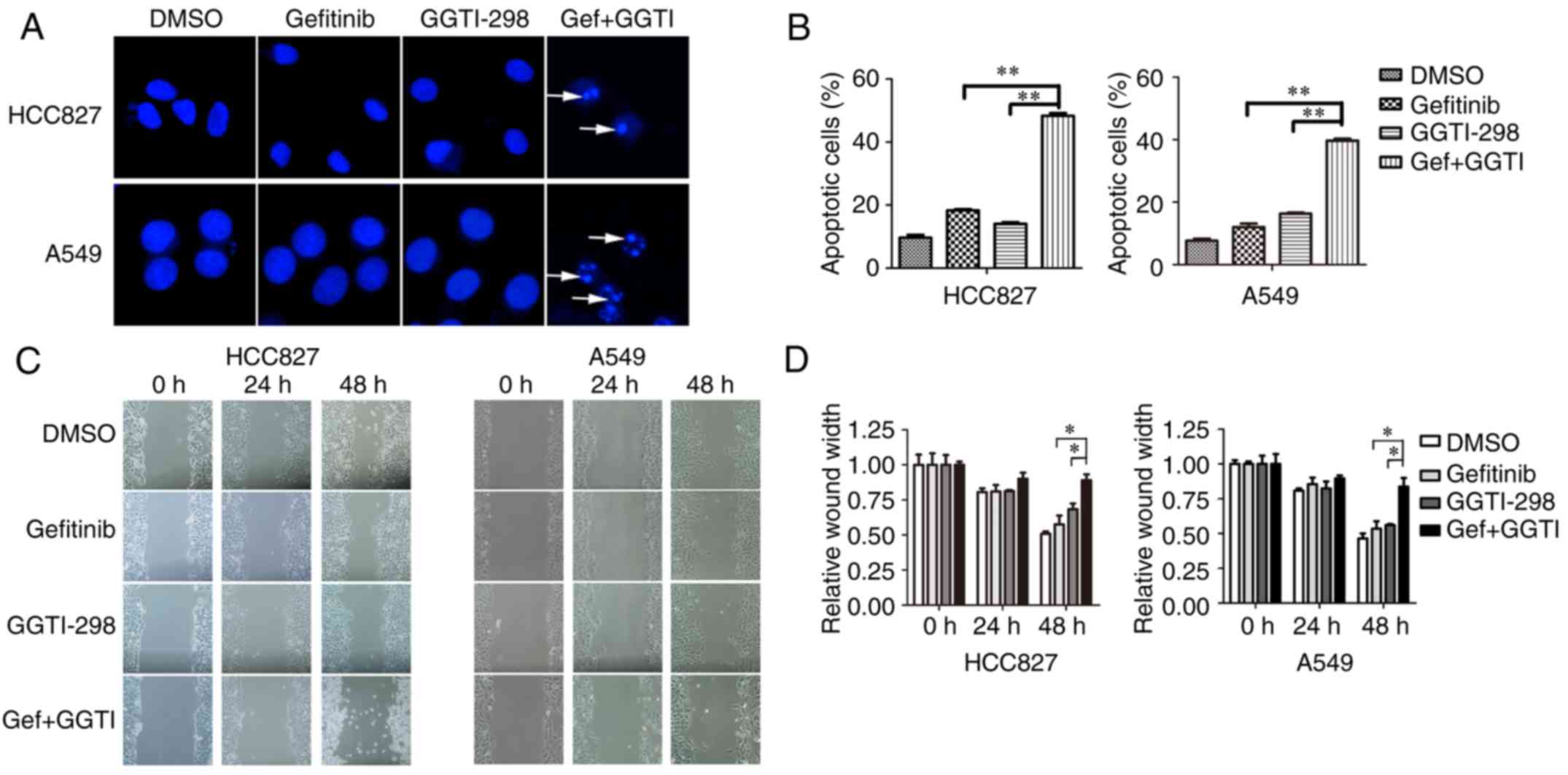 | Figure 2.Synergistic effects of gefitinib and
GGTI-298 on apoptosis and migration. (A) Cell apoptosis was
measured by Hoechst staining and (B) the number of apoptotic cells
was recorded. HCC827 and A549 cells were treated with DMSO,
gefitinib (HCC827, 10 nM; A549, 5 µM), GGTI-298 (HCC827, 5 µM;
A549, 7.5 µM) or a combination of gefitinib and GGTI-298 for 48 h.
Representative images of immunofluorescence are presented.
Apoptotic cells are indicated by white arrows (magnification,
×400). At least 300 cells were scored in every replicate. (C) Cell
migration was detected using wound healing assays for HCC827 and
A549 cells treated with DMSO, gefitinib (HCC827, 10 nM; A549, 5
µM), 5 µM GGTI-298 or a combination of gefitinib and GGTI-298 for
48 h. (D) The ratio of the relative wound width was calculated as
follows: wound width at the indicated time (0, 24 or 48 h)/wound
width at 0 h. Representative images from the wound healing assays
are shown. The results were presented as the mean ± standard error
of the mean (n=3 biological replicates). *P<0.05 and
**P<0.01, as indicated. GGTI/GGTI-298, geranylgeranyl
transferase 1 inhibitor; Gef, gefitinib; DMSO,
dimethylsulfoxide. |
Effects of gefitinib and GGTI-298 on
the EGFR signaling pathway and cyclin D1
To gain a greater understanding of the mechanism
underlying the combined effect of GGTI-298 and gefitinib, the
present study evaluated protein expression by performing
immunoblotting. Protein expression was examined following 48 h of
treatment with gefitinib and/or GGTI-298. As shown in Fig. 3, GGTI-298 treatment effectively
inhibited the expression of pEGFR and pAKT. Furthermore, inhibition
of the EGFR-AKT signaling pathway was substantially amplified when
GGTI-298 and gefitinib were combined (Fig. 3). However, the present study did
not observe marked changes in pERK expression with GGTI-298
treatment alone (Fig. 3). The
protein expression of cyclin D1 was also analyzed, which is a
proliferation-associated protein and a downstream target of AKT
(20). Consistent with the pEGFR
and pAKT results, treatment with gefitinib and GGTI-298 induced
additional cyclin D1 downregulation when compared with either
monotherapy (Fig. 3).
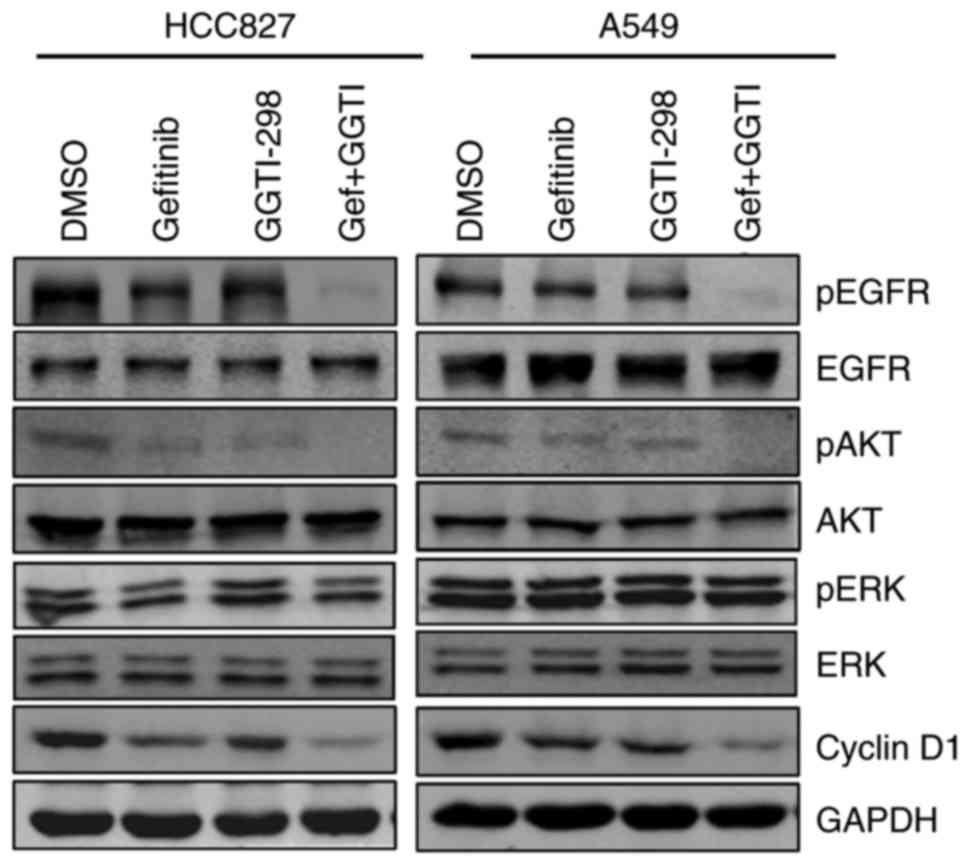 | Figure 3.Effects of gefitinib and GGTI-298 on
the EGFR signaling pathway and cyclin D1. Protein levels for the
EGFR signaling pathway and cyclin D1 were measured by
immunoblotting analysis. HCC827 and A549 cells were treated with
DMSO, gefitinib (HCC827, 20 nM; A549, 5 µM), 5 µM GGTI-298 or a
combination of gefitinib and GGTI-298 for 48 h. Representative
results are shown (n=3 biological replicates). GGTI/GGTI-298,
geranylgeranyl transferase 1 inhibitor; Gef, gefitinib; DMSO,
dimethylsulfoxide; p-, phosphorylated; EGFR, epidermal growth
factor receptor; AKT, protein kinase B; ERK, extracellular
signal-regulated kinase. |
GGTI-298 inhibits the EGFR-AKT
signaling pathway through RhoA
The present study then examined the specific
mechanism underlying pAKT downregulation. Immunoblotting was
employed to analyze the expression of pEGFR (which is expressed
upstream of pAKT), PTEN (a negative regulator of pAKT) and pAKT.
The results revealed that pEGFR and pAKT expression increased
markedly following treatment with EGF, while GGTI-298 treatment
reversed the activation of pEGFR and pAKT signaling induced by EGF
(Fig. 4A). However, PTEN
expression remained essentially unchanged (Fig. 4B). These results revealed that
GGTI-298 may have inhibited pAKT signaling primarily through
regulating pEGFR expression. Further studies were then conducted to
elucidate how GGTI-298 mediates the downregulation of pEGFR.
GGTI-298, a GGT1 inhibitor, prevents the geranylgeranylation of
small G proteins such as RhoA (19), one of the most intensely studied
proteins within the Rho family of GTPases, which is involved in
various signaling regulations. Thus, it was hypothesized that
GGTI-298 may have inhibited pEGFR through RhoA. Firstly, the
present study detected the expression of RhoA. As shown in Fig. 4C, total RhoA increased in A549
cells following treatment with 5 µM GGTI-298 when compared with the
untreated (DMSO) group. Then, pull-down assays were conducted to
detect active RhoA, which is responsible for the bioactivity of
RhoA (21). Compared with control
cells, GGTI-298 markedly impaired RhoA activation (Fig. 4C). These results indicated that
GGTI-298 inhibited RhoA activation in A549 cells. To detect the
involvement of the RhoA protein in EGFR signaling, cells were
transfected with siRNAs for RNA interference and the effects were
analyzed. As detected by immunoblotting, the transfection of
RhoA-specific siRNA resulted in the marked inhibition of total and
active RhoA expression, and the downregulation of pEGFR (Fig. 4D). Taken together, these results
validated the critical role of RhoA in the regulation of pEGFR and
suggested that GGTI-298 may inhibit pEGFR signaling primarily
through RhoA in A549 cells.
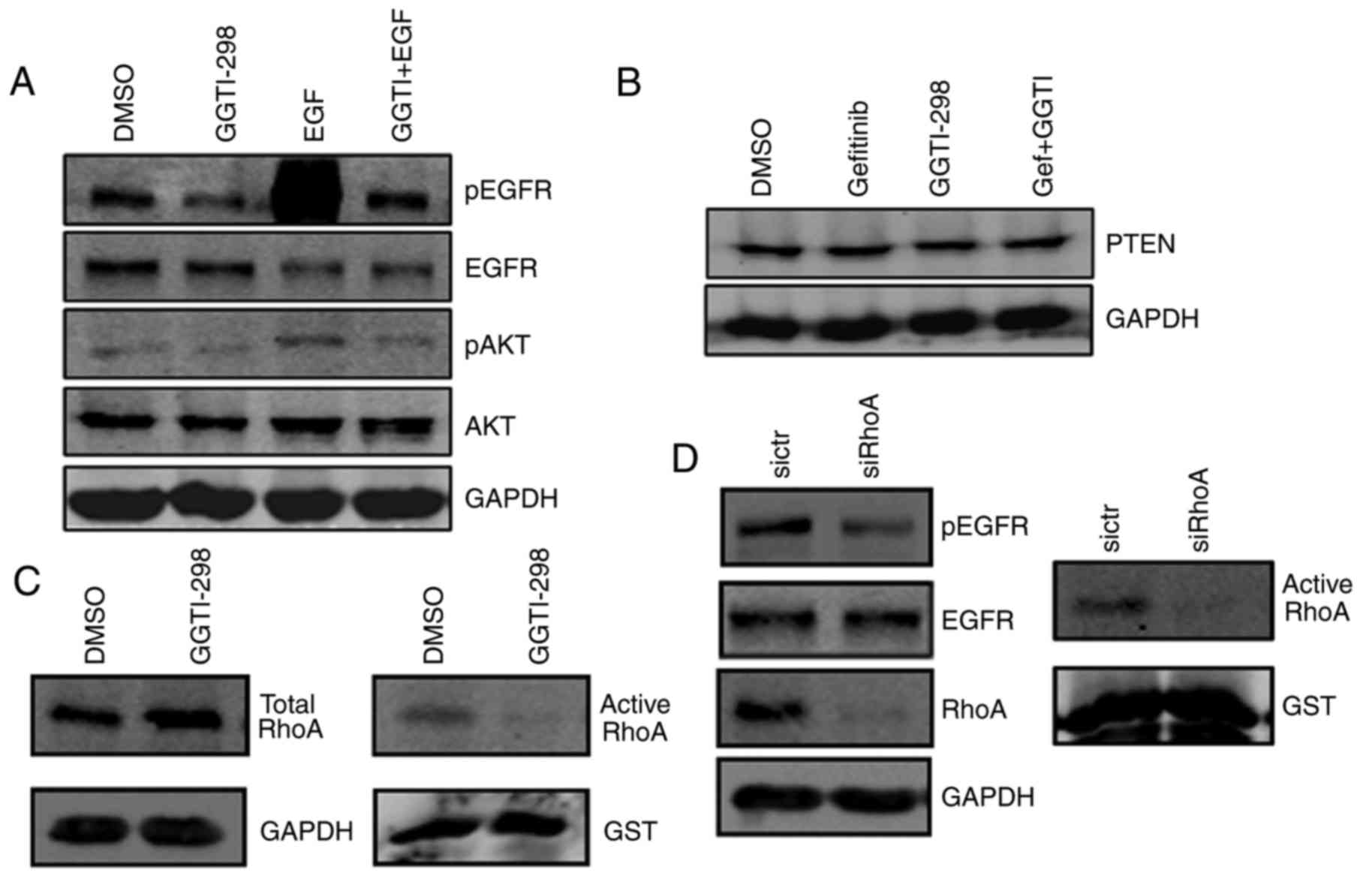 | Figure 4.GGTI-298 inhibits the EGFR-AKT
signaling pathway through RhoA. (A) GGTI-298 inhibited the
phosphorylation of AKT and EGFR induced by EGF. A549 cells were
treated with 5 µM GGTI-298 in serum-free medium for 48 h. Prior to
protein extraction, cultures were exposed to 30 ng EGF for 30 min,
as indicated in the figure. (B) PTEN levels were measured by
immunoblotting analysis. A549 cells were treated with DMSO, 5 µM
gefitinib, 5 µM GGTI-298 or a combination of gefitinib and GGTI-298
for 48 h. GAPDH was used as an internal control. (C) Activity of
RhoA was measured in A549 cells treated with DMSO and GGTI-298 for
48 h. (D) A549 cells were transfected with control siRNA or siRhoA,
and the activity of RhoA and the phosphorylation of EGFR were
measured 72 h following transfection. GST was used as a control in
the pull-down assays. Representative results are shown (n=3
biological replicates). GGTI/GGTI-298, geranylgeranyl transferase 1
inhibitor; Gef, gefitinib; DMSO, dimethylsulfoxide; p-,
phosphorylated; EGFR, epidermal growth factor receptor; AKT,
protein kinase B; PTEN, phosphatase and tensin homolog; RhoA, Ras
homolog family member A; si-/siRNA, small interfering RNA; GST,
glutathione S-transferase. |
Discussion
Treatment of advanced NSCLC was revolutionized by
EGFR TKIs such as gefitinib and erlotinib. Inadequate inhibition of
EGFR signaling is considered to significantly contribute to TKI
resistance and disease progression (22). Thus, effective inhibition of EGFR
function through multiple mechanisms may augment EGFR TKI activity
and overcome TKI resistance. The combination therapy consisting of
afatinib and cetuximab previously presented promising clinical
efficacy (22), indicating that
the co-inhibition of EGFR may a potential method for enhancing the
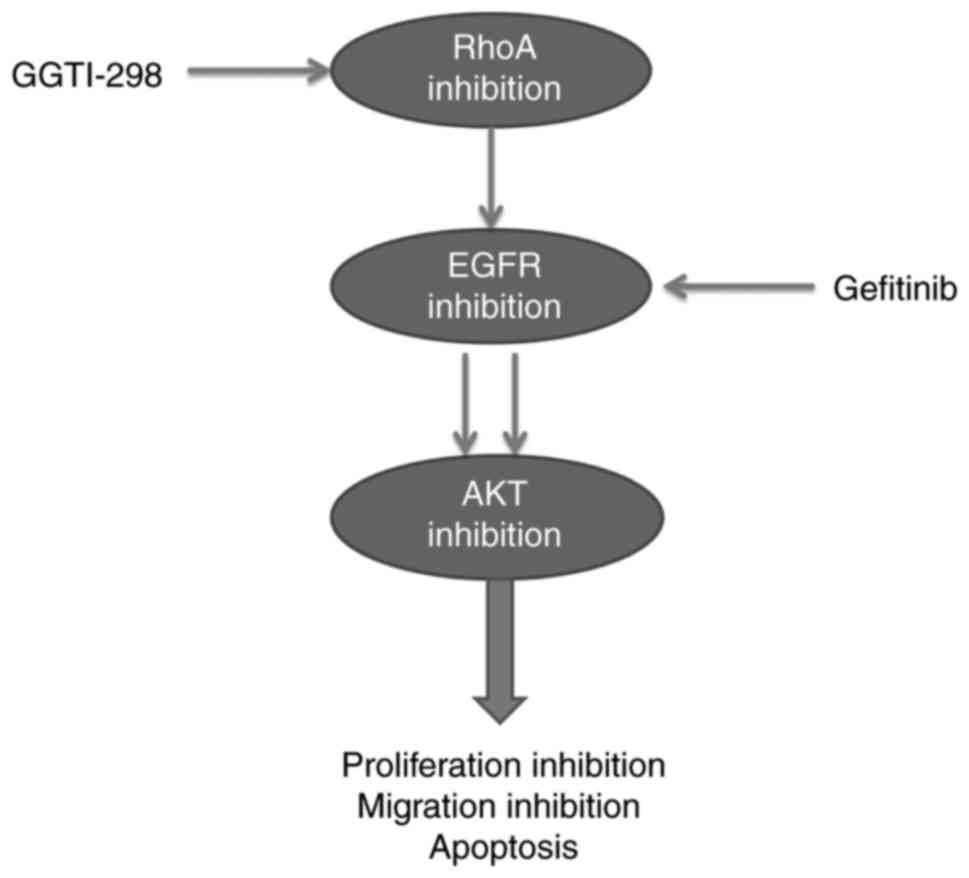
antitumor response. The present study revealed that GGTI-298, a
GGT1 inhibitor, inhibited the EGFR signaling pathway mainly through
RhoA, and a combined treatment of GGTI-298 and gefitinib impaired
the phosphorylation of EGFR more thoroughly than either compound
alone, significantly inhibiting cell proliferation and migration,
and promoting apoptosis.
EGFR signaling is regulated by multiple protein
molecules (23–26). In the present study, the EGFR-AKT
signaling pathway was inhibited by the GGT1 inhibitor GGTI-298.
Further experiments demonstrated that there was a slight
upregulation of total RhoA expression following GGTI-298 treatment.
However, the expression of active RhoA was markedly inhibited by
GGTI-298. These results indicated that GGTI-298 inhibited RhoA
activation. Crosstalk involving RhoA and other proteins has been
widely reported (14,27,28).
In the present study, transfection experiments with siRNA against
RhoA decreased the level of pEGFR. A study has previously
demonstrated that actin organization served an important role in
EGFR localization and activation (29), and the bioactivity of RhoA was
critical for actin organization (30), which further supports the present
study's results regarding the effect of RhoA on EGFR signaling.
Taken together, these results indicated that GGTI-298 may inhibit
the EGFR-AKT signaling pathway mainly through RhoA in A549
cells.
AKT and ERK are two main downstream signaling
proteins of the EGFR signaling pathway (31). In the present study, the expression
of pAKT was markedly decreased following GGTI-298 treatment.
Mechanistic studies further revealed that GGTI-298 inhibited the
EGFR activation induced by EGF; however, it had little effect on
PTEN expression, which negatively regulates pAKT expression. These
results suggested that GGTI-298 inhibited pAKT through EGFR, though
not PTEN. However, the expression of pERK was essentially unchanged
following treatment with GGTI-298. It was hypothesized that Ras,
which is upstream of pERK, may mediate activity mainly through
farnesylation, but not geranylgeranylation, which is consistent
with the results of Sebti et al (32).
Recent studies have demonstrated the potential for
mevalonate metabolite depletion to affect EGFR activity (33,34).
Targeting the mevalonate pathway inhibited the function of EGFR
(35). Combinations of statins,
inhibitors of the mevalonate pathway, and an EGFR inhibitor, such
as gefitinib and cetuximab, induced a potent synthetical
cytotoxicity (31,36). However, it should be noted that
statins inhibit the geranylgeranylation of RhoA as well as the
synthesis of many other types of bioactive substances such as
cholesterol and ubiquinone (19).
Cholesterol is a critical component for the structure of animal
cell membranes (37), and
ubiquinone participates in aerobic cellular respiration (38). Thus, the use of statins may cause
multiple side effects associated with these bioactive substances.
In the present study, the results revealed that the inhibition of
RhoA geranylgeranylation by GGTI-298 also suppressed the EGFR-AKT
signaling pathway, and it was not involved in the synthesis of
cholesterol and ubiquinone (19).
Upon combination treatment with GGTI-298 and gefitinib, the
synthetical loss of EGFR-AKT activation in A549 and HCC827 cells
resulted in a marked increase in the inhibition of cell
proliferation and migration inhibition, and increased levels of
apoptosis (Fig. 5). The effect of
the GGTI alone was not satisfactory (39), which indicated that a
combination-based therapy with a GGTI and other agents may be a
potential option for augmenting the antitumor efficiency of GGTIs.
Thus, these results were suggestive of the potential of combining
these two drugs targeting EGFR, through distinct mechanisms; they
therefore represent a novel, promising therapeutic strategy in lung
cancer.
In conclusion, the results of the present study
suggested that the antitumor efficacy could be markedly increased
by combining gefitinib with GGTI-298, a specific inhibitor of
protein geranylgeranylation. However, further investigation is
required to evaluate the clinical efficacy of this combinatorial
treatment in lung cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81621003), the
National Key R&D Program of China (grant nos. 2016YFC1303200
and 2016YFC1303203) and the National Natural Science Foundation of
China (grant no. 81572731).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BSL and XYD designed and performed experiments,
analyzed data and prepared the manuscript. HWX, HJX and QLT
conducted the experiments associated with Figs. 1 and 2. QYG and YZN analyzed the data and
contributed to the manuscript. FB supervised these experiments,
analyzed data, and prepared the manuscript. All authors reviewed
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37 Suppl 4:S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yokoyama K, Goodwin GW, Ghomashchi F,
Glomset JA and Gelb MH: A protein geranylgeranyltransferase from
bovine brain: Implications for protein prenylation specificity.
Proc Natl Acad Sci USA. 88:pp. 5302–5306. 1991; View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reid TS, Terry KL, Casey PJ and Beese LS:
Crystallographic analysis of CaaX prenyltransferases complexed with
substrates defines rules of protein substrate selectivity. J Mol
Biol. 343:417–433. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kusama T, Mukai M, Tatsuta M, Matsumoto Y,
Nakamura H and Inoue M: Selective inhibition of cancer cell
invasion by a geranylgeranyltransferase-I inhibitor. Clin Exp
Metastasis. 20:561–567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun J, Qian Y, Chen Z, Marfurt J, Hamilton
AD and Sebti SM: The geranylgeranyltransferase I inhibitor GGTI-298
induces hypophosphorylation of retinoblastoma and partner switching
of cyclin-dependent kinase inhibitors. A potential mechanism for
GGTI-298 antitumor activity. J Biol Chem. 274:6930–6934. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dan HC, Jiang K, Coppola D, Hamilton A,
Nicosia SV, Sebti SM and Cheng JQ: Phosphatidylinositol-3-OH
kinase/AKT and survivin pathways as critical targets for
geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene.
23:706–715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sonnemann J, Bumbul B and Beck JF:
Synergistic activity of the histone deacetylase inhibitor
suberoylanilide hydroxamic acid and the bisphosphonate zoledronic
acid against prostate cancer cells in vitro. Mol Cancer Ther.
6:2976–2984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berndt N, Hamilton AD and Sebti SM:
Targeting protein prenylation for cancer therapy. Nat Rev Cancer.
11:775–791. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
McGuire TF, Qian Y, Vogt A, Hamilton AD
and Sebti SM: Platelet-derived growth factor receptor tyrosine
phosphorylation requires protein geranylgeranylation but not
farnesylation. J Biol Chem. 271:27402–27407. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li F, Jiang Q, Shi KJ, Luo H, Yang Y and
Xu CM: RhoA modulates functional and physical interaction between
ROCK1 and Erk1/2 in selenite-induced apoptosis of leukaemia cells.
Cell Death Dis. 4:e7082013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Del Re DP, Miyamoto S and Brown JH: Focal
adhesion kinase as a RhoA-activable signaling scaffold mediating
Akt activation and cardiomyocyte protection. J Biol Chem.
283:35622–35629. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Basile JR, Gavard J and Gutkind JS:
Plexin-B1 utilizes RhoA and Rho kinase to promote the
integrin-dependent activation of Akt and ERK and endothelial cell
motility. J Biol Chem. 282:34888–34895. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hebert M, Potin S, Sebbagh M, Bertoglio J,
Breard J and Hamelin J: Rho-ROCK-dependent ezrin-radixin-moesin
phosphorylation regulates Fas-mediated apoptosis in Jurkat cells. J
Immunol. 181:5963–5973. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chou TC, Motzer RJ, Tong Y and Bosl GJ:
Computerized quantitation of synergism and antagonism of taxol,
topotecan, and cisplatin against human teratocarcinoma cell growth:
A rational approach to clinical protocol design. J Natl Cancer
Inst. 86:1517–1524. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sorrentino G, Ruggeri N, Specchia V,
Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio
R, Piazza S, et al: Metabolic control of YAP and TAZ by the
mevalonate pathway. Nat Cell Biol. 16:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li P, Wei J and Gao X: Insulin promotes
the proliferation of human umbilical cord matrix-derived
mesenchymal stem cells by activating the Akt-Cyclin D1 axis. Stem
Cells Int. 2017:73716152017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng W, Gu L, Li X, Zheng J, Zhang Y, Duan
B, Cui J, Dong J and Du J: CD24 associates with EGFR and supports
EGF/EGFR signaling via RhoA in gastric cancer cells. J Transl Med.
14:322016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Janjigian YY, Smit EF, Groen HJ, Horn L,
Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, et al:
Dual inhibition of EGFR with afatinib and cetuximab in kinase
inhibitor-resistant EGFR-mutant lung cancer with and without T790M
mutations. Cancer Discov. 4:1036–1045. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fischer OM, Hart S, Gschwind A and Ullrich
A: EGFR signal transactivation in cancer cells. Biochem Soc Trans.
31:1203–1208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song S, Honjo S, Jin J, Chang SS, Scott
AW, Chen Q, Kalhor N, Correa AM, Hofstetter WL, Albarracin CT, et
al: The Hippo coactivator YAP1 mediates EGFR overexpression and
confers chemoresistance in esophageal cancer. Clin Cancer Res.
21:2580–2590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He C, Mao D, Hua G, Lv X, Chen X,
Angeletti PC, Dong J, Remmenga SW, Rodabaugh KJ, Zhou J, et al: The
Hippo/YAP pathway interacts with EGFR signaling and HPV
oncoproteins to regulate cervical cancer progression. EMBO Mol Med.
7:1426–1449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chrysogelos SA and Dickson RB: EGF
receptor expression, regulation, and function in breast cancer.
Breast Cancer Res Treat. 29:29–40. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Xia H, Ge X, Chen Q, Yuan D, Chen
Q, Leng W, Chen L, Tang Q and Bi F: CD44 acts through RhoA to
regulate YAP signaling. Cell Signal. 26:2504–2513. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H, Ung CY, Ma XH, Li BW, Low BC, Cao ZW
and Chen YZ: Simulation of crosstalk between small GTPase RhoA and
EGFR-ERK signaling pathway via MEKK1. Bioinformatics. 25:358–364.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lunn JA, Wong H, Rozengurt E and Walsh JH:
Requirement of cortical actin organization for bombesin,
endothelin, and EGF receptor internalization. Am J Physiol Cell
Physiol. 279:C2019–C2027. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Asanuma K, Yanagida-Asanuma E, Faul C,
Tomino Y, Kim K and Mundel P: Synaptopodin orchestrates actin
organization and cell motility via regulation of RhoA signalling.
Nat Cell Biol. 8:485–491. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee J, Lee I, Han B, Park JO, Jang J, Park
C and Kang WK: Effect of simvastatin on cetuximab resistance in
human colorectal cancer with KRAS mutations. J Natl Cancer Inst.
103:674–688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sebti SM and Hamilton AD:
Farnesyltransferase and geranylgeranyltransferase I inhibitors and
cancer therapy: Lessons from mechanism and bench-to-bedside
translational studies. Oncogene. 19:6584–6593. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mantha AJ, Hanson JE, Goss G, Lagarde AE,
Lorimer IA and Dimitroulakos J: Targeting the mevalonate pathway
inhibits the function of the epidermal growth factor receptor. Clin
Cancer Res. 11:2398–2407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Niknejad N, Morley M and Dimitroulakos J:
Activation of the integrated stress response regulates
lovastatin-induced apoptosis. J Biol Chem. 282:29748–29756. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dimitroulakos J, Lorimer IA and Goss G:
Strategies to enhance epidermal growth factor inhibition: Targeting
the mevalonate pathway. Clin Cancer Res. 12:4426s–4431s. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hwang KE, Kwon SJ, Kim YS, Park DS, Kim
BR, Yoon KH, Jeong ET and Kim HR: Effect of simvastatin on the
resistance to EGFR tyrosine kinase inhibitors in a non-small cell
lung cancer with the T790M mutation of EGFR. Exp Cell Res.
323:288–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Simons K and Sampaio JL: Membrane
organization and lipid rafts. Cold Spring Harb Perspect Biol.
3:a0046972011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ernster L and Dallner G: Biochemical,
physiological and medical aspects of ubiquinone function. Biochim
Biophys Acta. 1271:195–204. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lobell RB, Liu D, Buser CA, Davide JP,
DePuy E, Hamilton K, Koblan KS, Lee Y, Mosser S, Motzel SL, et al:
Preclinical and clinical pharmacodynamic assessment of L-778,123, a
dual inhibitor of farnesyl:protein transferase and
geranylgeranyl:protein transferase type-I. Mol Cancer Ther.
1:747–758. 2002.PubMed/NCBI
|