Introduction
Allergic rhinitis (AR) is a common chronic
inflammatory disease of the nasal mucosa which is characterized by
sneezing, itching, nasal obstruction, and rhinorrhea (1). Symptoms of AR are frequently
bothersome, impair the quality of life and increase their
socioeconomic burden. Despite numerous experimental therapies that
have been made to improve the functional outcome of patients with
AR, advances in therapy for this condition is still unsatisfactory
(2). Thus, the development of an
effective therapy for this disease is essential.
Emerging evidence indicates that allergic diseases
including AR are often caused by numerous inflammatory mediators
such as histamine, chemokines, and cytokines from immune cells
(3,4). Over the past several years, IL-4 and
IL-13 produced by T helper type 2 (TH2) cells, have been found to
contribute to the pathogenesis of AR (5). For example, IL-13 has been found to
promote mucus production and the secretion of inflammatory
cytokines in airway epithelial cells (NEC) (6). In addition, IL-1β, IL-6, IL-8
inflammatory molecules produced by mast cell contributed to the
influx of immune cells into nasal mucosa tissue of AR, and
accelerated inflammatory reactions (7). Recently, Wang et al (8), found that platycodin D (PLD)
inhibited IL-13 induced inflammatory response in NECs by blocking
the activation of NF-κB and MAPK signaling pathways. Therefore,
reducing the production of inflammatory molecules may be an
effective way to treat AR (9).
MicroRNAs (miRNAs) are a group of small,
evolutionarily conserved, noncoding RNAs of 18–25 nucleotides in
length that regulate gene expression through mRNA cleavage and/or
translation inhibition by binding to the 3′UTR of target genes
(10). Recent studies found that a
large number of abnormal miRNAs expression was closely associated
with multiple allergic inflammatory diseases, such as asthma
(11,12), AR (13) and atopic dermatitis (14,15).
For example, Case et al (16) showed that miR-21 inhibited
toll-like receptor 2 (TLR2) agonist-induced lung inflammation in an
animal model of asthma, which indicate the potential therapeutic
effects of miR-21 on allergic diseases. Collison et al
(17), demonstrated that
inhibition of miR-145 significantly attenuated allergic
inflammation of the nasal mucosa in mite-induced allergic airways
disease. However, little attention has been paid on the evaluation
of miRNAs function in AR.
In the present study, we found that miR-16 screened
by microarray was downregulated in nasal mucosal samples from AR
patients and overexpression of miR-16 inhibited inflammatory
cytokines and mucus production in IL-13-induced cell model of AR.
Furthermore we identified IKKβ, a key catalytic subunit of IKK
complex as a direct target of miR-16 and investigated the roles of
miR-16 on the activation of NF-κB pathway in IL-13-stimulated nasal
epithelial cells.
Materials and methods
Preparation of nasal mucosal
specimens
Nasal mucosal samples were obtained from inferior
turbinate sections from 25 patients with perennial AR and 25
patients with nonallergic rhinitis (NAR control group) according to
the method of Ruocco et al (18). Diagnosed based on the case history,
nasal endoscopy, allergen skin-prick tests, and specific IgE
assays. No patient had received oral steroids or other medications
for 4 weeks prior to study recruitment. The presemt study was
approved by the Institutional Review Board at Hangzhou First
People's Hospital, Nanjing Medical University. All participants
provided written informed consent.
microRNA expression profiling
Total RNA was extracted from nasal mucus from AR
patients and NAR controls using the miRNeasy mini kit (Qiagen, West
Sussex, UK) according to manufacturer's instructions. Samples were
labeled with Hy3 using the miRCURY™ Array Labelling kit (Exiqon,
Inc., Woburn, MA, USA) and hybridized to the miRCURY LNA™ Array
(v.16.0; Agilent Technologies, Inc., Santa Clara, CA, USA). After
washing, the chips were scanned with the Axon GenePix 4000B
Microarray Scanner (Molecular Devices, LLC, Sunnyvale, CA, USA).
The procedure and images process method as described previously
(19). The miRNA expressions of
all differentially expressed samples were clearly displayed by a
hierarchical clustering heat map.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from nasal mucosal specimens
and nasal epithelial cells using the miRNeasy mini kit (Qiagen) and
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's instructions. The
concentration and purity of RNA were determined
spectrophotometrically. Then the RNA was reverse transcribed into
cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo
Fisher Scientific, Inc.). qPCR was performed using the Step One
Plus Real-Time PCR System (Thermo Fisher Scientific, Inc.). Primer
sequences: miR-16, sense 5′-TAGCAGCACGTAAATATTGGCG-3′, antisense
5′-TGCGTGTCGTGGAGTC-3′; U6 sense 5′-TGCGGGTGCTCGCTTCGCAGC-3′; U6
antisense 5′-CCAGTGCAGGGTCCGAGGT-3′; mucin 5AC (MUC5AC) sense
5′-CGACAACTACTTCTGCGGTGC-3′, antisense 5′-GCACTCATCCTTCCTGTCGTT-3′;
IKKβ, sense 5′-TGTCAGTGGAAGCCCGGATAG-3′, and antisense was
3′-AGGTTATGTGCTTCAGCCACCAG-5′; GAPDH, sense
5′-CAAGCTCATTTCCTGGTATGAC-3′, antisense,
5′-CAGTGAGGGTCTCTCTCTTCCT-3′. The relative quantification of gene
expression was analyzed with 2−ΔΔCT method, normalized
with U6 or GAPDH expression.
Cell cultures and treatment
Human nasal epithelial cell (JME/CF15) and human
293T cell lines were obtained from Academy of Military Medical
Science, Beijing, China. JME/CF15 cells were routinely cultured in
RPMI 1640 (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), 2 mM glutamine, and 1 mM pyruvate and
incubated at 37°C with 5% CO2 and 95% humidity. 293T
cells were maintained in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc.) containing 10% FBS and
1% streptomycin-penicillin mix and incubated at 37°C with 5%
CO2. The JME/CF15 cells were either unstimulated or
stimulated with IL-13 (10 ng/ml) for 24 h or 30 min as previously
described (8).
Cell transfection
MiR-16 mimics, miR-16 inhibitor and the
corresponding negative control (mimics NC and inhibitor NC) were
purchased from RiboBio (Guangzhou RiboBio Co., Ltd., Guangzhou,
China). For enforced expression of IKKβ in JME/CF15 cells, open
reading frame region of human IKKβ gene was amplified and inserted
into the pcDNA3.1 eukaryotic expression vector (Invitrogen; Thermo
Fisher Scientific, Inc.). The JME/CF15 cells were seeded at 6-well
plate and transfection was performed with miR-16 mimics, mimics NC,
miR-16 inhibitor and inhibitor NC (100 nmol) or 2 µg/ml IKKβ
plasmid using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) on the following day according to the
manufacturer's instructions.
Enzyme-linked immunosorbent assay
(ELISA)
After treatment, the concentrations of
granulocyte-macrophage colony-stimulating factor (GM-CSF), Eotaxin,
IL-1β, IL-6 and IL-8 in the cell culture supernatants were measured
using ELISA kit in accordance with the manufacturers' instructions.
ELISA kits were purchased from R&D Systems, Inc., (Minneapolis,
MN, USA). All assays were performed in three independent times.
Luciferase reporter assay
To detect IKKβ as the direct binding target of
miR-16, a luciferase reporter assay was performed. 293T cells were
cultured in 24-well plate at a density of 1.5×106 cells
for 24 h, and then miR-16 mimics, miR-16 inhibitor, and IKKβ 3′-UTR
wild type (wt) or mutation (mut) of the putative miR-16 target
region were co-transfected into this cells using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.). Forty-eight hours after
transfection, the cells were lysed and their luciferase activity
was assayed using dual-luciferase reporter assay system (Promega
Corporation, Madison, WI, USA). Luciferase activity was normalized
to corresponding Renilla luciferase activity. All experiments were
performed in three independent times.
Western blot
Proteins were obtained from cells the using RIPA
lysis buffer (Beyotime Institute of Biotechnology, Haimen, China).
Histone protein was extracted from cells using NE-PER™ Nuclear and
Cytoplasmic Extraction Reagents from Thermo Fisher Scientific,
Inc., according with the manufacturers' instructions. Then the
protein concentration was determined using BCA Protein Assay kit
(Pierce; Thermo Fisher Scientific, Inc.). Proteins (50 µg) were
separated by 8% SDS-PAGE gel and transferred to polyvinylidene
difluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA).
Western blotting of MUCA5C, IKKβ, IκB-α, nuclear p-p65, histone 3
and β-actin was performed using a primary rabbit monoclonal
anti-MUCA5C antibody, anti-IKKβ (1:500, Abcam, Cambridge, UK),
anti-IκB-α (1:1,000), anti-p-IκB-α (1:1,000), anti-nuclear p-p65
(1:1,000), anti-histone 3 (1:1,000; all from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and anti-β-actin antibody
(1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA)
overnight at 4°C and followed by incubating with secondary antibody
(goat anti-rabbit IgG conjugated with the horseradish peroxidase,
1:4,000 dilution; Cell Signaling Technology, Inc.) for 1 h at room
temperature. The protein bands were visualized by ECL detection
reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA).
Relative band intensities were determined by densitometry using
Scion image software (v.4.0).
Statistical analysis
Statistical analyses were performed with SPSS v.13.0
software (SPSS, Inc., Chicago, IL, USA). All data were recorded as
means ± SD. For numerical variables, the results were evaluated by
the Student's t-test (comparison between 2 groups) or one way ANOVA
to make multiple-group comparisons followed by the post hoc Tukey's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-16 was downregulated in nasal
mucosal from AR patients
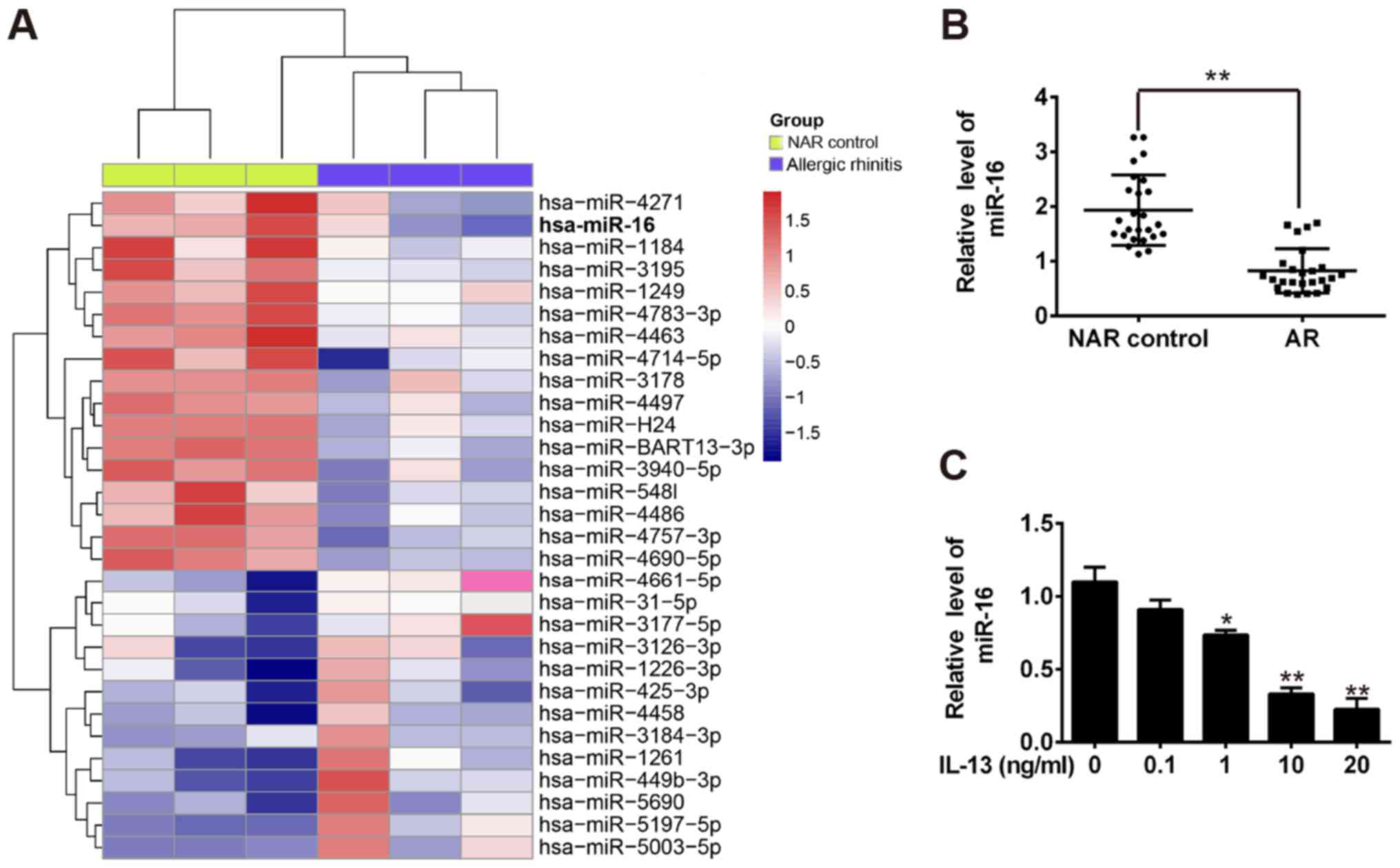
To identify the miRNAs that differed in expression
between AR patients and NAR control group. Microarray analysis
revealed a significant downregulation of 17 miRNAs and upregulation
of 13 miRNAs in AR group compared with NAR control group (Fig. 1A). Among the aberrantly expressed
miRNAs, miR-16 was one of the top downregulated miRNAs in nasal
mucosal tissues and altered expression of miR-16 has been found to
play a critical role in inflammation in several types of diseases
(20,21). We also measured its expression in
25 pairs of the nasal mucosal tissues among AR patients and NAR
control group by qRT-PCR and observed that miR-16 was significantly
decreased in AR group compared with NAR control group (Fig. 1B). Thus, miR-16 was chose as the
candidate for further study.
Recently, the IL-13-induced cell model of AR has
been found to have similar pathologic changes reminiscent of AR and
can be used in preliminary profiling therapies (22–24).
Hence, we evaluated the expression of miR-16 in the cell model of
IL-13-induced AR. As shown in Fig.
1C, IL-13 decreased the expression of miR-16 in this cell lines
in a dose-dependent manner. These results suggest that miR-16 might
be associated with the development of AR.
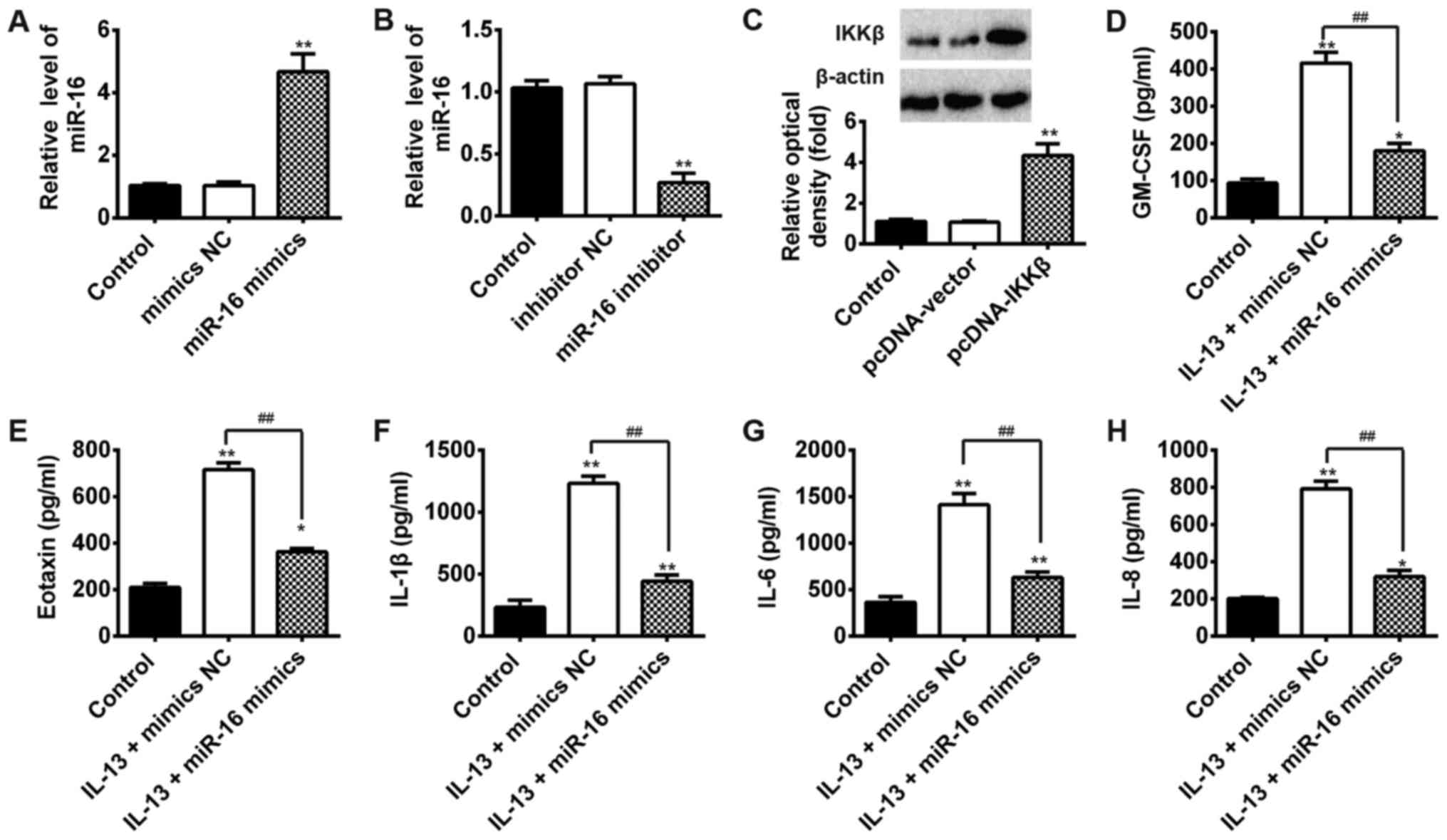
Validation of cell transfection
efficiency
qPCR was performed to assess the transfection
efficiency of miR-16 mimics or NC and miR-16 inhibitor or inhibitor
NC and pcDNA-IKKβ or pcDNA-vector. Compared with the mimics NC
group, the expression levels of miR-16 were markedly increased in
cells transfected with miR-16 mimics (Fig. 2A). Similarly, compared with the
inhibitor NC group, miR-16 expression levels were significantly
decreased following transfection with miR-16 inhibitor (Fig. 2B). In addition, compared with empty
vector group, the expression levels of IKKβ were obviously
increased in cells transfected with pcDNA-IKKβ (Fig. 2C).
Overexpression of miR-16 inhibits the
levels of inflammatory cytokines in IL-13-induced JME/CF15
cells
To further determine the protective effect of miR-16
in AR, the expression levels of inflammatory cytokines (TNF-α, IL-6
and IL-1β) were examined using ELISA. The results showed that IL-13
significantly increased the protein levels of GM-CSF, Eotaxin,
IL-1β, IL-6 and IL-8 that are common markers for activated NF-κB
pathway (7), whereas
overexpression of miR-16 inhibited these protein expression levels
in IL-13-stimulated JME/CF15 cells (Fig. 2D-H). All these results showed that
overexpression of miR-16 could inhibit inflammatory responses by
inhibiting these pro-inflammatory cytokines in AR.
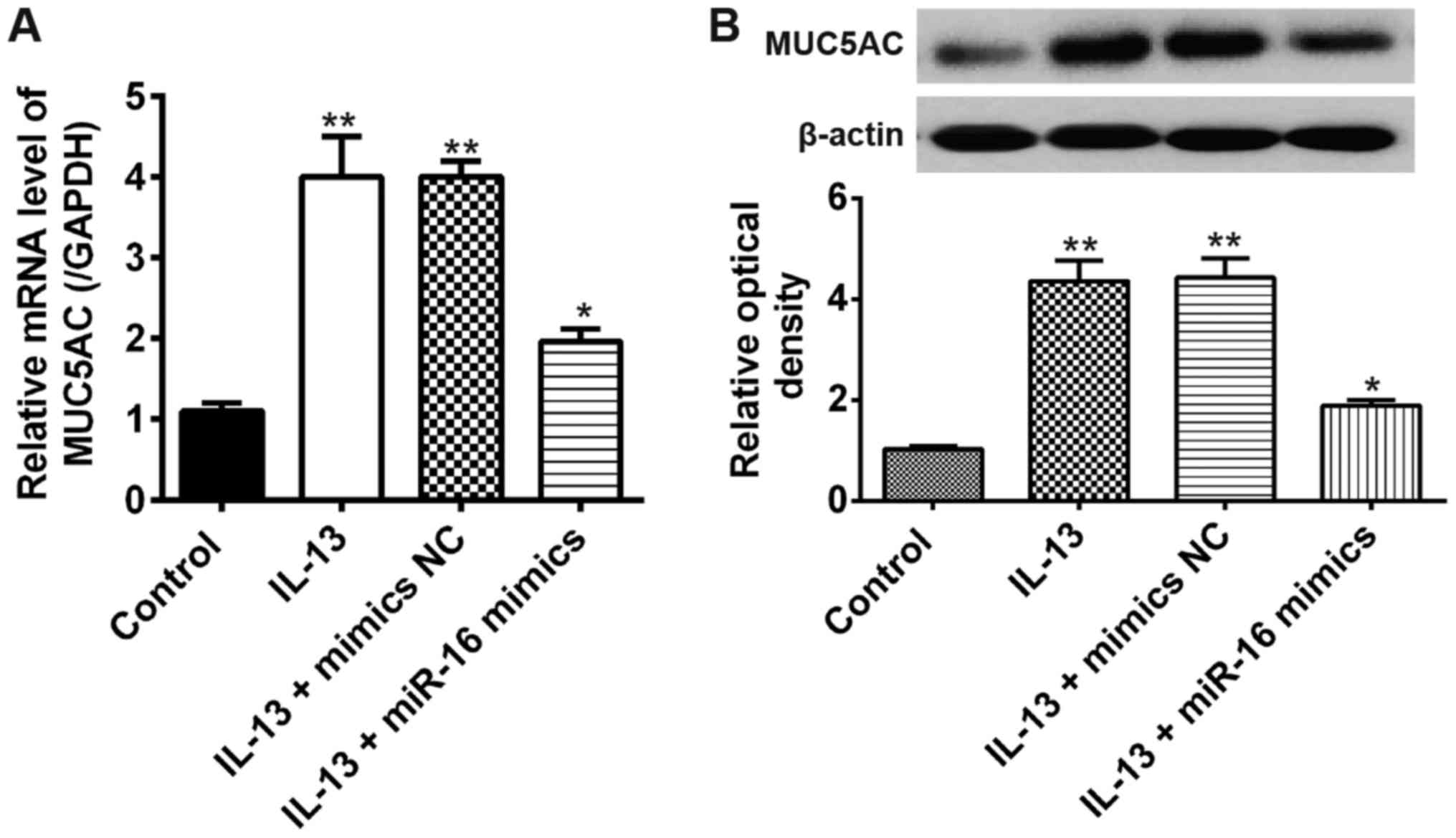
Overexpression of miR-16 suppresses
IL-13-induced mucus production in JME/CF15 cells
It has been demonstrated that mucus hypersecretion
(particularly MUC5AC) is a common feature of allergic airway
disorders including AR (25,26).
To evaluate the effects of miR-16 on mucus hypersecretion, we
measured the mRNA expression of MUC5AC in miR-16 mimics transfected
JME/CF15 cells following IL-13 stimulation. Compared with the
control group, the mRNA expression levels of MUC5AC were obviously
increased by IL-13 stimulation, whereas this promoting effect was
suppressed after miR-16 mimics transfection (Fig. 3A). Similarly, the result of Western
blot showed that overexpression of miR-16 inhibited IL-13-induced
MUC5AC protein expression in JME/CF15 cells (Fig. 3B). All these results showed that
overexpression of miR-16 exert its anti-AR activity by inhibiting
mucus production in JME/CF15 cells.
IKKβ was a direct target of
miR-16
To investigate the molecular mechanism by which
miR-16 functions in inflammatory responses in AR, two
bioinformatics algorithms (TargetScan and miRanda) were used to
search for potential targets of miR-16. According to the results of
these analyses, we focused on IKKβ for experimental verification.
In addition, IKKβ was reported to be a key catalytic subunit of IKK
complex and associated with the activation of NF-κB pathway
(27–29). As suggested in Fig. 4A, the complementary sequence of
miR-16 was found in the 3′-UTR of IKKβ mRNA. Subsequently, JME/CF15
cells were transfected with miR-16 mimics, miR-16 inhibitor or
controls and the mRNA of IKKβ was determined by qRT-PCR. The
results showed that overexpression of miR-16 significantly reduced,
whereas inhibition of miR-16 promoted the mRNA expression of IKKβ
(Fig. 4B). To further test whether
that miR-16 could directly target 3′-UTR of IKKβ, a luciferase
reporter assay was conducted. The results showed that miR-16 mimics
significantly decreased, whereas miR-16 inhibitor increased the
luciferase activity of IKKβ with wt 3′-UTR (Fig. 4C). The luciferase activity of
mutant IKKβ was not affected by miR-16. (Fig. 4C). In addition, we also examined
whether miR-16 could modulate the protein expression of IKKβ in
JME/CF15 cells. The results of Western blot showed that
overexpression of miR-16 significantly reduced, whereas inhibition
of miR-16 promoted the protein expressions of IKKβ (Fig. 4D). These results indicate that IKKβ
is a main functional target of miR-16 in the development of AR.
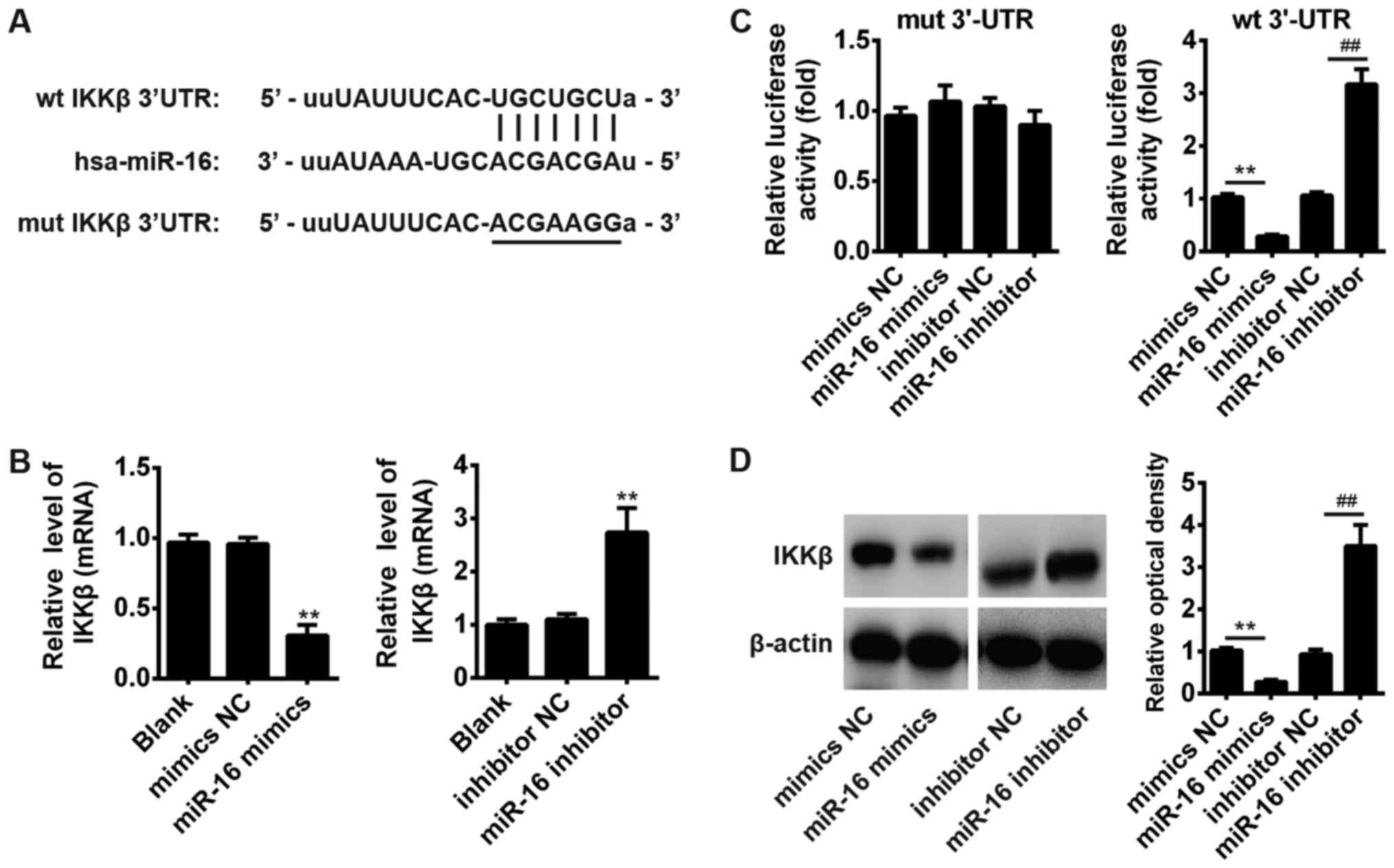 | Figure 4.IKKβ was a direct target of miR-16.
(A) The predicted miR-16 binding sites on IKKβ. (B) miR-16 mimics,
miR-16 inhibitor and controls were transfected into JME/CF15 cells,
then the mRNA levels of IKKβ were detected by RT-qPCR. **P<0.01
vs. mimics NC, inhibitor NC or Blank group, one-way ANOVA, Tukey's
post hoc test. (C) Luciferase activity in 293T cells co-transfected
with miR-16 mimics, miR-16 inhibitor and luciferase reporters
containing IKKβ wild type or mutant type (MUT) 3′-UTR. Histogram
indicates the values of luciferase measured 48 h after
transfection. **P<0.01 vs. mimics NC group;
##P<0.01 vs. inhibitor NC group, one-way ANOVA,
Tukey's post hoc test. (D) miR-16 mimics, miR-16 inhibitor and
controls were transfected into JME/CF15 cells, then the protein
levels of IKKβ were detected by western blot assays. Data represent
the mean ± SD of three independent experiments. **P<0.01 vs.
mimics NC group; ##P<0.01 vs. inhibitor NC group,
one-way ANOVA, Tukey's post hoc test. IKKβ, IκB kinase β; NC,
negative control. |
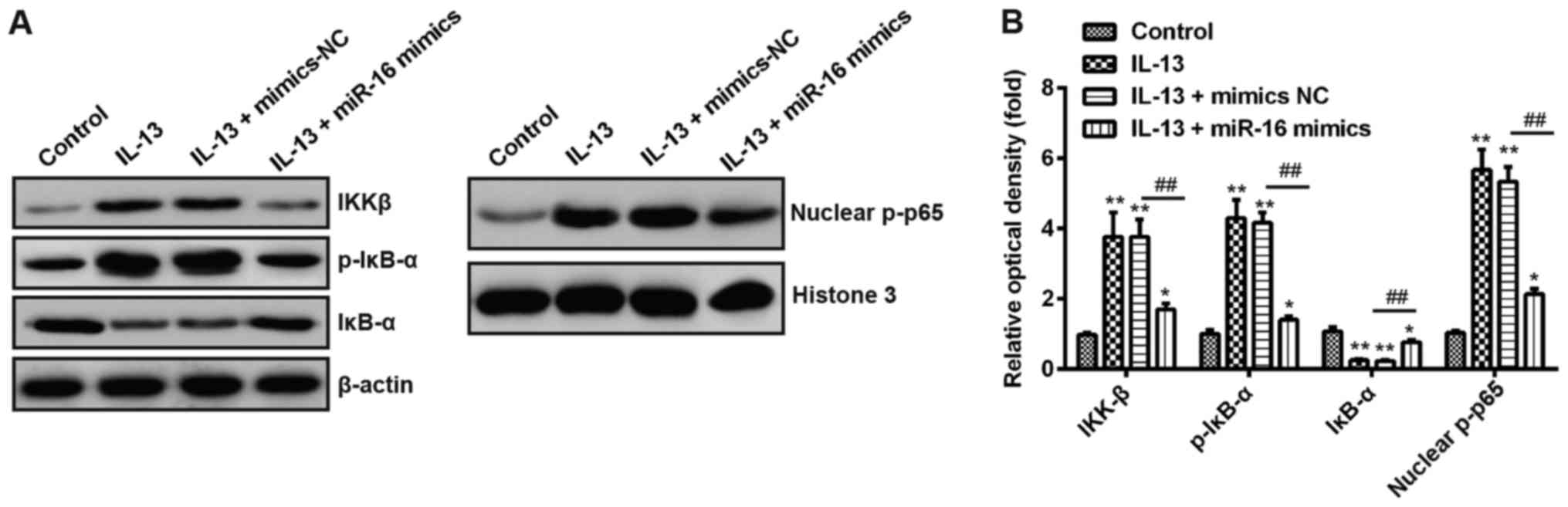
Effects of miR-16 on NF-κB signaling
pathway in IL-13-induced JME/CF15 cells
Recent studies have been shown that miR-16
negatively modulated the activation of the NF-κB pathway in
glioblastoma multiforme (GBM) (30) and in the diabetic retinopathy
(31). It is well-known that NF-κB
signaling pathway plays important roles in allergic diseases
including AR (32–35). Thus, we sought to determine whether
NF-κB pathway involves in the protective effect of miR-16 in AR. As
shown in Fig. 5A and B, IL-13
treatment significantly increased the protein expression level of
IKKβ, p-p65 and p-IκB-α, decreased the expression levels of IκB-α,
while overexpression of miR-16 reduced the expression of IKKβ,
nuclear p-p65, p-IκB-α, and promoted IκB-α expression in
IL-13-treated JME/CF15 cells. These results indicate that miR-16
inhibits inflammatory response via blocking the activation of NF-κB
signaling pathway.
miR-16 inhibits IL-13-induced
inflammation through IKKβ
IKKβ has been shown to play an essential role in the
activation of NF-κB signaling pathway (27–29).
Thus, we then explored whether IKKβ could rescue the inhibition of
NF-κB and pro-inflammatory cytokines mediated by miR-16. We found
that overexpression of IKKβ along with miR-16 followed by IL-13
stimulation significantly rescued the inhibition of nuclear p-p65
and MUC5AC expression levels (Fig. 6A
and B). Moreover, the levels of the pro-inflammatory cytokines
including GM-CSF, Eotaxin, IL-1β, IL-6 and IL-8 were also rescued
(Fig. 6C-G). Taken together, these
data suggest that IL-13 inhibits miR-16 expression which is
involved in the negative regulation of IL-13-stimulated NF-κB
activation and pro-inflammatory cytokines production through
targeting IKKβ.
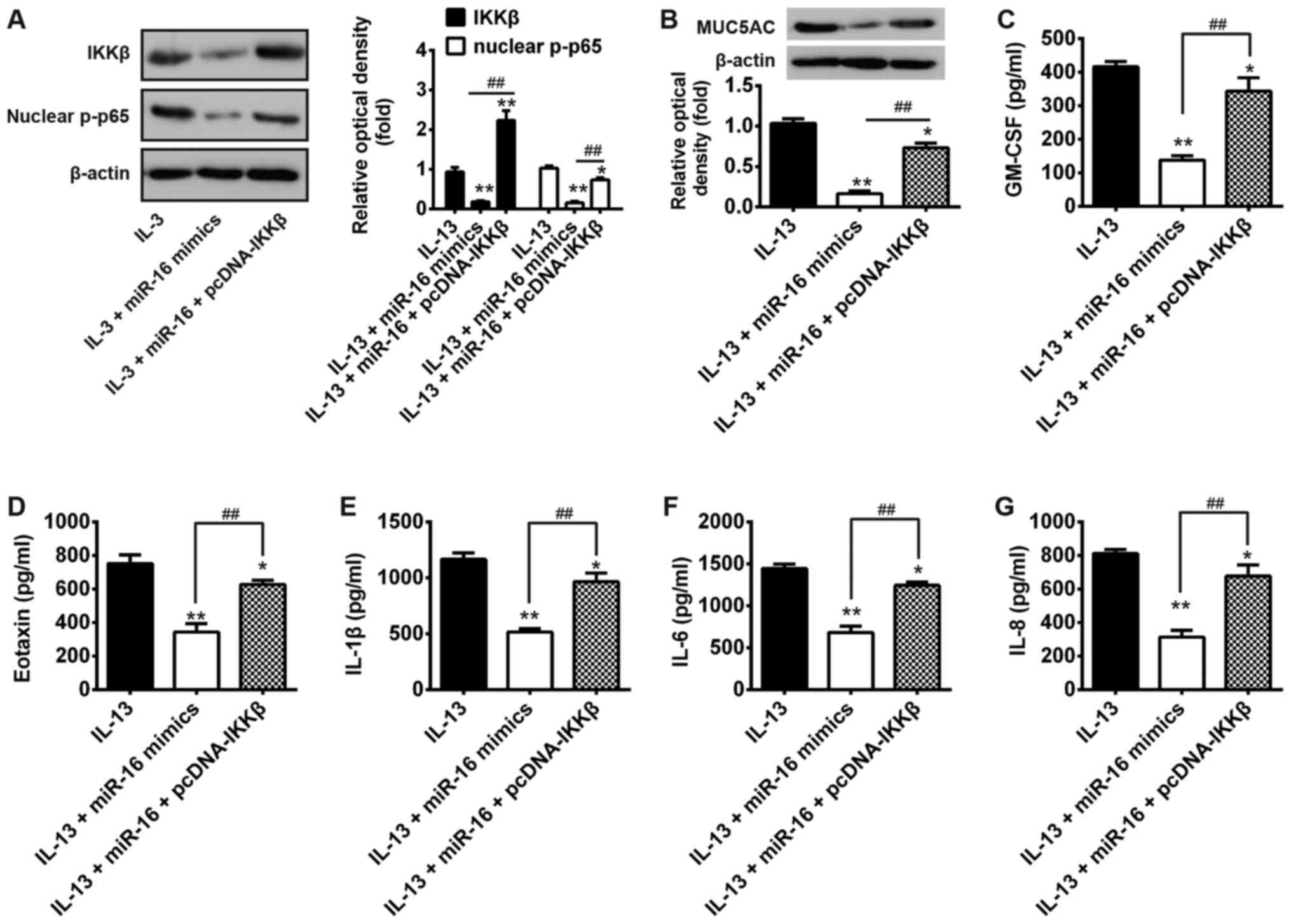 | Figure 6.miR-16 inhibits IL-13-induced mucus
production and inflammation through IKKβ. Cells were co-transfected
with miR-16 mimics and pcDNA-IKKβ for 24 h, then treated with 10
ng/ml IL-13 for another 24 h. (A and B) Protein levels of IKKβ,
nuclear p-p65 and MUCA5C were measured by western Blot. (C-G) The
levels of cytokines including GM-CSF, eotaxin, IL-1β, IL-6 and IL-8
were determined by ELISA. *P<0.05, **P<0.01 vs. IL-13 alone
group; ##P<0.01 vs. IL-13 + miR-16 mimics group,
one-way ANOVA, Tukey's post hoc test. IKKβ, IκB kinase β; GM-CSF,
granulocyte-macrophage colony-stimulating factor. |
Discussion
In the present study, miR-16 was found to be
downregulated in the nasal mucosal tissues from AR patients and
IL-13-induced cell model of AR. Moreover, we demonstrated that
miR-16 exerted its anti-inflammation activity through blocking
IL-13-stimulated NF-κB activation in JME/CF15 cells. Our findings
suggest that miR-16 may be a potential target for the prevention
and treatment of AR.
Previous studies have indicated that miRNAs have key
roles in allergic diseases. For example, miR-146a was increased in
keratinocytes and chronic lesional skin of patients with atopic
dermatitis (AD) and functioned as an important regulator in chronic
inflammation (36). Malmhäll et
al (37), found the
involvement of miR-155 in the regulation of allergic airway
inflammation by modulating TH2 responses. However, to date, little
attention has been paid to the role of miRNAs in the development of
AR. A recent report from Yu et al (13), analyzed the differentially
expressed miRNAs in AR using miRNA microarrays, and listed a
catalogue of miRNAs potentially involved in the development of AR.
Another study from Teng et al (38), found that miR-143 inhibited
IL-13-induced inflammatory response in NECs from AR patients by
targeting IL13Rα1. Thus, miR-143 may represent a promising
therapeutic target for allergic inflammation. These data promote us
to determine whether other miRNAs involved in the inflammation
response in AR. In this study, we identified a large number of
miRNAs that were significantly deregulated in nasal mucosal tissues
from AR patients using miRNA microarray. Among them, miR-16 was one
of the most downregulated miRNAs and also reported to play
different roles in inflammatory diseases (20,21).
Thus, we chose miR-16 for further study. Subsequently, we found
miR-16 was significantly decreased in AR patients and IL-13 treated
JME/CF15 cells. These results indicate that miR-16 may be related
to the pathogenesis of AR.
The pathophysiological mechanisms of AR are
complicated. During this period, allergic inflammation plays an
important role in the process of AR, which can be orchestrated by a
variety of cell mediators, such as IL-4 and IL-13 (39,40).
Among the key drivers, IL-13 pretreatment could cause severe sepsis
and inflammation (41).
Proinflammatory cytokines, such as GM-CSF, eotaxin and IL-1β were
obviously increased in the NECs following IL-13 stimulation
(38). In addition, IL-13-induced
cell model of AR has been widely used to simulate the major events
of AR. In this model, IL-13 treatment can lead to mucus
hypersecretion and inflammatory factors release in airway
epithelial cells (22,23). In our experiment, we used this
model to explore the roles of miR-16 in inflammatory responses in
AR. Consistent with above studies, we found that the levels of
MUC5AC and the pro-inflammatory cytokines including GM-CSF,
Eotaxin, IL-1β, IL-6 and IL-8 were increased in IL-13-treated
JME/CF15 cells, whereas overexpression of miR-16 reversed the
promoting effects of IL-13, suggesting the potential protective
effect of miR-16 in AR. Although the evidence highlighted the
important roles of miR-16 in AR, the underlying mechanism through
which miR-16 inhibits inflammatory responses in AR remains
unclear.
Recently, many new insights into the core signaling
pathways in allergic inflammatory diseases have been made,
especially NF-κB signaling pathways (8,34,42).
These pathways are often constitutively activated in subsets of
human nasal epithelial cells in AR. NF-κB is a pleiotropic
transcription factor that plays an important role in the
inflammatory process. For example, by regulation NF-κB signaling
pathway, PLD attenuated airway inflammation in a mouse model of
allergic asthma (36). In allergic
asthma, arsenictrioxide, a potent inhibitor of NF-κB, abrogated
allergen induced inflammatory response (43). Therefore, targeting NF-κB signaling
pathway could have benefit in the inhibition of inflammatory
reaction in allergic diseases. Shin et al (44), found that miR-16 was directly
regulated by NF-κB in gastric cancer. Thus, we hypothesis that
miR-16 may exert a significant therapeutic effect on AR through
inhibiting the activation of NF-κB pathway. In a previous study, it
was found that IKK, by phosphorylating the IκB, played an important
role in the activation of NF-κB signaling pathway (29). In our study, we found miR-16 could
directly target the 3′-UTR of IKKβ and negatively regulated the
expression of IKKβ. Furthermore, we found that overexpression of
miR-16 could reverse IL-13-induced the protein expression of NF-κB
p65, IKKβ and IκB-α, indicate that miR-16 is involved in the
negative regulation of IL-13-stimulated NF-κB activation. More
importantly, we found that IKKβ could rescue the inhibition of
NF-κB and pro-inflammatory cytokines mediated by miR-16. Based on
the above results, we felt safe to conclude that miR-16 inhibited
IL-13-induced inflammatory response in AR by inhibiting the
activation of IKKβ/NF-κB signaling pathway.
In summary, our results showed that miR-16 was
downregulated in AR and IL-13 treated JME/CF15 cells, and
overexpression of miR-16 inhibited inflammatory response by
blocking the activation of IKKβ/NF-κB signaling pathways. On this
basis, it is proposed that the miR-16/IKKβ/NF-κB axis might
represent a beneficial way of regulating nasal inflammation and
preventing the development of AR.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YQG performed the experiments, contributed to data
analysis and wrote the paper. ZZY analyzed the data. YQG
conceptualized the study design, contributed to data analysis and
experimental materials. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All individuals provided informed consent for the
use of human specimens for clinical research. The present study was
approved by the Ethics Committee of the Hangzhou First People's
Hospital, Nanjing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bousquet J, Khaltaev N, Cruz AA, Denburg
J, Fokkens WJ, Togias A, Zuberbier T, Baena-Cagnani CE, Canonica
GW, van Weel C, et al: Allergic rhinitis and its impact on asthma
(ARIA) 2008 update (in collaboration with the World Health
Organization, GA(2)LEN and AllerGen). Allergy. 63 Suppl 86:S8–S160.
2008. View Article : Google Scholar
|
|
2
|
Steelant B, Farré R, Wawrzyniak P, Belmans
J, Dekimpe E, Vanheel H, Van Gerven L, Kortekaas Krohn I, Bullens
DMA, Ceuppens JL, et al: Impaired barrier function in patients with
house dust mite-induced allergic rhinitis is accompanied by
decreased occludin and zonula occludens-1 expression. J Allergy
Clin Immunol. 137:1043–1053.e5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bradding P, Feather IH, Wilson S, Bardin
PG, Heusser CH, Holgate ST and Howarth PH: Immunolocalization of
cytokines in the nasal mucosa of normal and perennial rhinitic
subjects. The mast cell as a source of IL-4, IL-5, and IL-6 in
human allergic mucosal inflammation. J Immunol. 151:3853–3865.
1993.PubMed/NCBI
|
|
4
|
Minty A, Chalon P, Derocq JM, Dumont X,
Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B,
et al: Interleukin-13 is a new human lymphokine regulating
inflammatory and immune responses. Nature. 362:248–250. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wills-Karp M, Luyimbazi J, Xu X, Schofield
B, Neben TY, Karp CL and Donaldson DD: Interleukin-13: Central
mediator of allergic asthma. Science. 282:2258–2261. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuperman DA, Huang X, Koth LL, Chang GH,
Dolganov GM, Zhu Z, Elias JA, Sheppard D and Erle DJ: Direct
effects of interleukin-13 on epithelial cells cause airway
hyperreactivity and mucus overproduction in asthma. Nat Med.
8:885–889. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galli SJ and Tsai M: IgE and mast cells in
allergic disease. Nat Med. 18:693–704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang B, Gao Y, Zheng G, Ren X, Sun B, Zhu
K, Luo H, Wang Z and Xu M: Platycodin D inhibits
interleukin-13-induced the expression of inflammatory cytokines and
mucus in nasal epithelial cells. Biomed Pharmacother. 84:1108–1112.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang T, Finn DF, Barlow JW and Walsh JJ:
Mast cell stabilisers. Biomed Pharmacother. 778:158–168. 2016.
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu TX, Munitz A and Rothenberg ME:
MicroRNA-21 is up-regulated in allergic airway inflammation and
regulates IL-12p35 expression. J Immunol. 182:4994–5002. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roush S and Slack FJ: The let-7 family of
microRNAs. Trends Cell Biol. 18:505–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu S, Zhang R, Liu G, Yan Z, Hu H, Yu S,
Zhang J, Yu S, Zhang R, et al: Microarray analysis of
differentially expressed microRNAs in allergic rhinitis. Am J
Rhinol Allergy. 25:e242–e246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sonkoly E, Wei T, Janson PC, Sääf A,
Lundeberg L, Tengvall-Linder M, Norstedt G, Alenius H, Homey B,
Scheynius A, et al: MicroRNAs: Novel regulators involved in the
pathogenesis of psoriasis? PLoS One. 2:e6102007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vennegaard MT, Bonefeld CM, Hagedorn PH,
Bangsgaard N, Løvendorf MB, Odum N, Woetmann A, Geisler C and Skov
L: Allergic contact dermatitis induces upregulation of identical
microRNAs in humans and mice. Contact Dermatitis. 67:298–305. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Case SR, Martin RJ, Jiang D, Minor MN and
Chu HW: MicroRNA-21 inhibits toll-like receptor 2 agonist-induced
lung inflammation in mice. Exp Lung Res. 37:500–508. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Collison A, Mattes J, Plank M and Foster
PS: Inhibition of house dust mite-induced allergic airways disease
by antagonism of microRNA-145 is comparable to glucocorticoid
treatment. J Allergy Clin Immunol. 128:160–167.e164e. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ruocco L, Fattori B, Romanelli A,
Martelloni M, Casani A, Samolewska M and Rezzonico R: A new
collection method for the evaluation of nasal mucus proteins. Clin
Exp Allergy. 28:881–888. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu H, Wu Y, Li L, Yuan W, Zhang D, Yan Q,
Guo Z and Huang W: MiR-344b-1-3p targets TLR2 and negatively
regulates TLR2 signaling pathway. Int J Chron Obstruct Pulmon Dis.
12:627–638. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen W, Guo S and Wang S: MicroRNA-16
alleviates inflammatory pain by targeting ras-related protein 23
(RAB23) and inhibiting p38 MAPK activation. Med Sci Monit.
22:3894–3901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang X, Xu Z, Yuan M, Zhang Y, Zhao B,
Wang J, Zhang A and Li C: MicroRNA-16 suppresses the activation of
inflammatory macrophages in atherosclerosis by targeting PDCD4. Int
J Mol Med. 37:967–975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsukura S, Stellato C, Georas SN,
Casolaro V, Plitt JR, Miura K, Kurosawa S, Schindler U and
Schleimer RP: Interleukin-13 upregulates eotaxin expression in
airway epithelial cells by a STAT6-dependent mechanism. Am J Respir
Cell Mol Biol. 24:755–761. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wills-Karp M: Interleukin-13 in asthma
pathogenesis. Curr Allergy Asthma Rep. 4:123–131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corren J: Role of interleukin-13 in
asthma. Curr Allergy Asthma Rep. 13:415–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Voynow JA, Gendler SJ and Rose MC:
Regulation of mucin genes in chronic inflammatory airway diseases.
Am J Respir Cell Mol Biol. 34:661–665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thai P, Loukoianov A, Wachi S and Wu R:
Regulation of airway mucin gene expression. Annu Rev Physiol.
70:405–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Menssen A, Häupl T, Sittinger M, Delorme
B, Charbord P and Ringe J: Differential gene expression profiling
of human bone marrow-derived mesenchymal stem cells during
adipogenic development. BMC Genomics. 12:4612011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Häcker H and Karin M: Regulation and
function of IKK and IKK-related kinases. Sci STKE. 2006:re132006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miao Z, Mao F, Liang J, Szyf M, Wang Y and
Sun ZS: Anxiety-related behaviours associated with microRNA-206-3p
and BDNF expression in pregnant female mice following psychological
social stress. Mol Neurobiol. 55:1097–1111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ye EA, Liu L, Jiang Y, Jan J, Gaddipati S,
Suvas S and Steinle JJ: miR-15a/16 reduces retinal leukostasis
through decreased pro-inflammatory signaling. J Neuroinflammation.
13:3052016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y, Garvin LM, Nickola TJ, Watson AM,
Colberg-Poley AM and Rose MC: IL-1β induction of MUC5AC gene
expression is mediated by CREB and NF-κB and repressed by
dexamethasone. Am J Physiol Lung Cell Mol Physiol. 306:L797–L807.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim HY, Nam SY, Hwang SY, Kim HM and Jeong
HJ: Atractylone, an active constituent of KMP6, attenuates allergic
inflammation on allergic rhinitis in vitro and in vivo models. Mol
Immunol. 78:121–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shakoory B, Fitzgerald SM, Lee SA, Chi DS
and Krishnaswamy G: The role of human mast cell-derived cytokines
in eosinophil biology. J Interferon Cytokine Res. 24:271–281. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nam SY, Chung CK, Seo JH, Rah SY, Kim HM
and Jeong HJ: The therapeutic efficacy of α-pinene in an
experimental mouse model of allergic rhinitis. Int Immunopharmacol.
23:273–282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rebane A, Runnel T, Aab A, Maslovskaja J,
Rückert B, Zimmermann M, Plaas M, Kärner J, Treis A, Pihlap M, et
al: MicroRNA-146a alleviates chronic skin inflammation in atopic
dermatitis through suppression of innate immune responses in
keratinocytes. J Allergy Clin Immunol. 134:836–847.e11. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Malmhäll C, Alawieh S, Lu Y, Sjöstrand M,
Bossios A, Eldh M and Rådinger M: MicroRNA-155 is essential for
T(H)2-mediated allergen-induced eosinophilic inflammation in the
lung. J Allergy Clin Immunol. 133:1429–1438, 1438.e1-7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Teng Y, Zhang R, Liu C, Zhou L, Wang H,
Zhuang W, Huang Y and Hong Z: miR-143 inhibits
interleukin-13-induced inflammatory cytokine and mucus production
in nasal epithelial cells from allergic rhinitis patients by
targeting IL13Rα1. Biochem Biophys Res Commun. 457:58–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Eifan AO and Durham SR: Pathogenesis of
rhinitis. Clin Exp Allergy. 46:1139–1151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Howarth PH, Salagean M and Dokic D:
Allergic rhinitis: Not purely a histamine-related disease. Allergy.
55 Suppl 64:S7–S16. 2000. View Article : Google Scholar
|
|
41
|
Li L, Xia Y, Nguyen A, Lai YH, Feng L,
Mosmann TR and Lo D: Effects of Th2 cytokines on chemokine
expression in the lung: IL-13 potently induces eotaxin expression
by airway epithelial cells. J Immunol. 162:2477–2487.
1999.PubMed/NCBI
|
|
42
|
Han D, Zhou B, Cheng L, Oh Y and Li H: P38
MAP-kinase pathway is involved in the production of CLC-3 in nasal
epithelial cells with allergic rhinitis induced by interleukin-4.
Laryngoscope. 116:1973–1977. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou LF, Zhu Y, Cui XF, Xie WP, Hu AH and
Yin KS: Arsenic trioxide, a potent inhibitor of NF-kappaB,
abrogates allergen-induced airway hyperresponsiveness and
inflammation. Respir Res. 7:1462006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shin VY, Jin H, Ng EK, Cheng AS, Chong WW,
Wong CY, Leung WK, Sung JJ and Chu KM: NF-κB targets miR-16 and
miR-21 in gastric cancer: Involvement of prostaglandin E receptors.
Carcinogenesis. 32:240–245. 2011. View Article : Google Scholar : PubMed/NCBI
|