Introduction
Hepatic fibrosis is a common consequence of chronic
liver diseases of various etiologies, such as hepatitis virus,
alcoholic liver disease (ALD), non-alcoholic steatohepatitis
(NASH), and others (1). The
development of liver fibrosis encompasses activation of hepatic
stellate cells (HSCs) and their crosstalk with liver parenchymal
cells, such as hepatocytes and biliary epithelial cells (2). HSCs become activated and
differentiate into myofibroblasts, thus playing a critical role in
the production of collagen and excessive accumulation of
extracellular matrix (ECM), which are two main features of hepatic
fibrogenesis (3). Liver fibrosis
is a form of chronic liver injury that is strongly associated with
hypoxia. Moreover, exposure of HSCs to hypoxia affects development
of hepatic fibrosis (4,5).
During the past decades, increasing evidence has
suggested that overexpression of hypoxia-inducible factor 1-α
(HIF1-α) is closely associated with liver fibrogenesis (6–8).
Indeed, overexpression of HIF1-α in vitro results in HSC
activation and upregulation of genes associated with matrix
deposition (9). HIF1-α and
collagen 1A1 (Col1A1) expression increase in HSCs under hypoxia
(4,10). Transfection with short interfering
RNA against HIF1-α (siHIF1-α) prevents human HSC migration
(7), while upregulation of HIF1-α
is associated with viral hepatitis-derived fibrosis, HSC
activation, and mitogen activated protein kinase (MAPK) activity
(11). Additionally, expression of
fibrogenic mediators is decreased in bile duct ligated
HIF1-α-knockout mice compared to controls (6). Altogether, this strong evidence
indicates that HIF1-α is highly involved in hepatic fibrosis;
however, the underlying regulatory mechanism is still not fully
understood.
Previously, it was reported that the
Rho/Rho-associated coiled-coil-forming kinase 1 (ROCK1) pathway is
involved in HSC activation (12).
Furthermore, ROCK inhibition with Y-27632 was demonstrated to
suppress HSC proliferation and type I collagen production (13). Several studies have clearly
indicated that there is crosstalk between ROCK1 and HIF1-α
(14–16). However, the relationship between
HIF1-α and ROCK1 in HSCs remains unexplored.
In the present study, we measured changes in HIF1-α
and ROCK1 expression in HSCs under hypoxia at different time
points. In order to functionally address the role of HIF1-α in HSC
proliferation and collagen synthesis, and the interplay between
HIF1-α and ROCK1 signaling, we performed knockdown experiments
using siRNA transfection. Finally, we investigated whether ROCK1
inhibition mediates HIF1-α-derived HSC activation, thus providing
an attractive target for the treatment of hepatic fibrosis.
Materials and methods
Cell culture
The HSC-T6 cell line was maintained in room air or
in hypoxic conditions (1% O2) in an incubator at 37°C.
HSC-T6 cells were cultured in Dulbecco's modified Eagle's medium
(DMEM) containing 10% fetal bovine serum (FBS; both Gibco; Thermo
Fisher Scientific, Inc. Waltham, MA, USA), 100 U/ml penicillin, and
100 µg/ml streptomycin. Culture medium was changed every 24 h, and
the ROCK1 inhibitor, Y-27632 (100 µM) (Merck KGaA, Darmstadt,
Germany), was added.
siRNA transfection
One day prior to transfection, HSC-T6 cells were
seeded in 6-well plates at a density of 2×105 cells per
well or in 96-well plates at a density of 1×104 cells
per well. Cells were transfected with 100 nM siHIF1-α or mock siRNA
(as a control) (Shanghai GenePharma Co., Ltd., Shanghai, China)
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Briefly,
Lipofectamine-siRNA was incubated for 20 min before being added to
cells. The transfection medium was replaced with fresh medium after
6 h and cells were collected after another 48 h for the follow-up
tests.
Enzyme linked immunosorbent assay
(ELISA)
We measured secreted collagen 3A1 (Col3AI) using a
commercially available ELISA kit (Uscn Life Sciences, Inc., Wuhan,
China). After cells were transfected with siRNA or treated with the
ROCK1 inhibitor, supernatants were collected at 48 h. Samples were
added into wells according to the manufacturers' instructions. The
optical density (OD) values were read at 450 nm with an ELISA
reader. The Col3A1 concentration was measured using a standard
curve diagram.
Immunofluorescence detection
HSC-T6 cells were cultured in hypoxic conditions at
37°C as described above, and then collected at 0, 12 and 48 h. The
cells were fixed in 4% paraformaldehyde for 15 min, permeabilized
with 0.1% Triton X-100 for 15 min and blocked with 4% bovine serum
albumin (BSA) for 1 h. Cells were incubated with a mouse α-smooth
muscle actin (α-SMA) antibody diluted 1:100 in phosphate buffered
saline (PBS) for 1 h and then washed with PBS three times. Next,
cells were incubated with Fluorescein Isothiocyanate (FITC)
conjugated anti-mouse secondary antibody diluted 1:200 for 1 h and
washed with PBS. After mounting with anti-fade mounting medium,
cells were observed and fluorescent images were taken using a
fluorescence microscope.
Western blot analysis
All protein concentrations were quantified using a
BCA protein assay kit (Beyotime Institute of Biotechnology, Haimen,
China). 30 µg protein was loaded into 8–10% gradient SDS-PAGE gels,
and transferred to a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
skimmed milk for 1 h and then incubated with primary antibodies
(β-actin, HIF1-α, ROCK1, α-SMA, Col1A1, and Col3A1) at 4°C
overnight. Goat anti-mouse IgG or anti-rabbit IgG (Jackson, USA)
conjugated to horseradish peroxidase was then added for 1 h at
37°C. Finally, antibody binding was developed using an Odyssey
infrared scanner and Western Bright ECL reagents (Thermo Fisher
Scientific, Inc.). Samples were washed three times with 1X Tris
buffered saline and 0.1% Tween-20 (TBS/T).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted with TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), and cDNA synthesis
was performed using random hexamers with 1.0 mg RNA. RT-qPCR was
performed using SYBR-Green I (Toyobo Life Science, Osaka, Japan).
The reaction mixture volume was 20 µl. Primer sequences were as
follows:
β-actin (131) forward,
5′-CGTAAAGACCTCTATGCCAACA-3′ and reverse,
5′-GGAGGAGCAATGATCTTGATCT-3′; HIF-1α (36) forward,
5′-GTCGGACAGCCTCACCAAACAGAGC-3′ and reverse,
5′-GTTAACTTGATCCAAAGCTCTGAG-3′; Col1A1 (95) forward,
5′-GTACATCAGCCCAAACCCCAAG-3′ and reverse,
5′-CGGAACCTTCGCTTCCATACTC-3′; Col3A1 (88) forward,
5′-GACTGCCCCAACCCAGAGATC-3′ and reverse,
5′-TACCATCAGGAATGACAGGAGCAG-3′; α-SMA (77) forward,
5′-GTGCTGTCCCTCTATGCCTCTGG-3′ and reverse,
5′-GGCACGTTGTGAGTCACACCATC-3′; ROCK1 (123) forward,
5′-CGGTATCTCTACATGGTGATGGAG-3′ and reverse,
5′-ATCCAATGCAAGAACTACTTCTGC-3′
All PCR reactions were normalized to β-actin.
Relative quantification of gene expression is shown using the
2−∆∆Cq method.
MTT assay
Cells were cultured at a density of
1.0×104 cells per well in 96-well plates (each group
with 6 duplicates) for 24 h. Then, cells were either transfected
with siRNA or treated with the ROCK1 inhibitor, Y-27632, and
incubated for 48 h. Thereafter, 50 µl of MTT (5 mg/ml) (Nanjing
KeyGen Biotech Co., Ltd., Nanjing, China) was added to each well
and cells were incubated at 37°C in an incubator for another 4 h.
Finally, the medium was discarded and replaced with 150 µl
dimethyl-sulfoxide (DMSO). Once the violet crystal was dissolved,
the OD of each well was measured using a microplate reader at a
wavelength of 570 nm.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 20.0; SPSS Inc., Chicago, IL, USA). Data are
shown as mean ± standard deviation, and statistical analysis was
performed using one-way analysis of variance with Tukey's post hoc
test for multiple comparisons. For comparison of two groups,
t-tests were used. P<0.05 was considered to indicate a
statistically significant difference. Each experiment was repeated
in triplicate.
Results
Hypoxia influences collagen synthesis
and activation of HSCs and expression of HIF1-α and ROCK1
It is well known that hypoxia can promote a stem
cell-like phenotype in multiple myeloma cells (17,18).
To investigate the effect of hypoxia on collagen synthesis and
activation of HSCs, western blotting was used to detect the level
of Col1A1, Col3A1 and α-SMA protein. Cells were cultured under
normoxia or hypoxia condition for 48 h. Compared with normoxia
group, the expression of Col1A1, Col3A1 and α-SMA was increased in
hypoxia group. We also found the different expression of HIF1-α and
ROCK1 between normoxia and hypoxia conditions. Cells were both
cultured for 48 h, but the levels of HIF1-α and ROCK1 were
upregulated under hypoxia condition. These results indicated that
hypoxia could increase collagen synthesis, activated the HSCs, and
induced the expression of HIF1-α and ROCK1 (Fig. 1).
 | Figure 1.Effect of 1% O2 on Col1A1,
Col3A1, α-SMA, HIF1-α and ROCK1 expression in HSC-T6 cells. HSC-T6
cells were incubated under normoxic or hypoxic conditions (1%
O2) and collected 48 h later. Col1A1, Col3A1, α-SMA,
HIF1-α and ROCK1 protein expression were then measured by western
blotting. Col1/3 A1, collagen type 1/3 α1 chain; α-SMA, α-smooth
muscle actin; HIF1, hypoxia inducible factor 1; ROCK,
Rho-associated coiled-coil-forming kinase 1. |
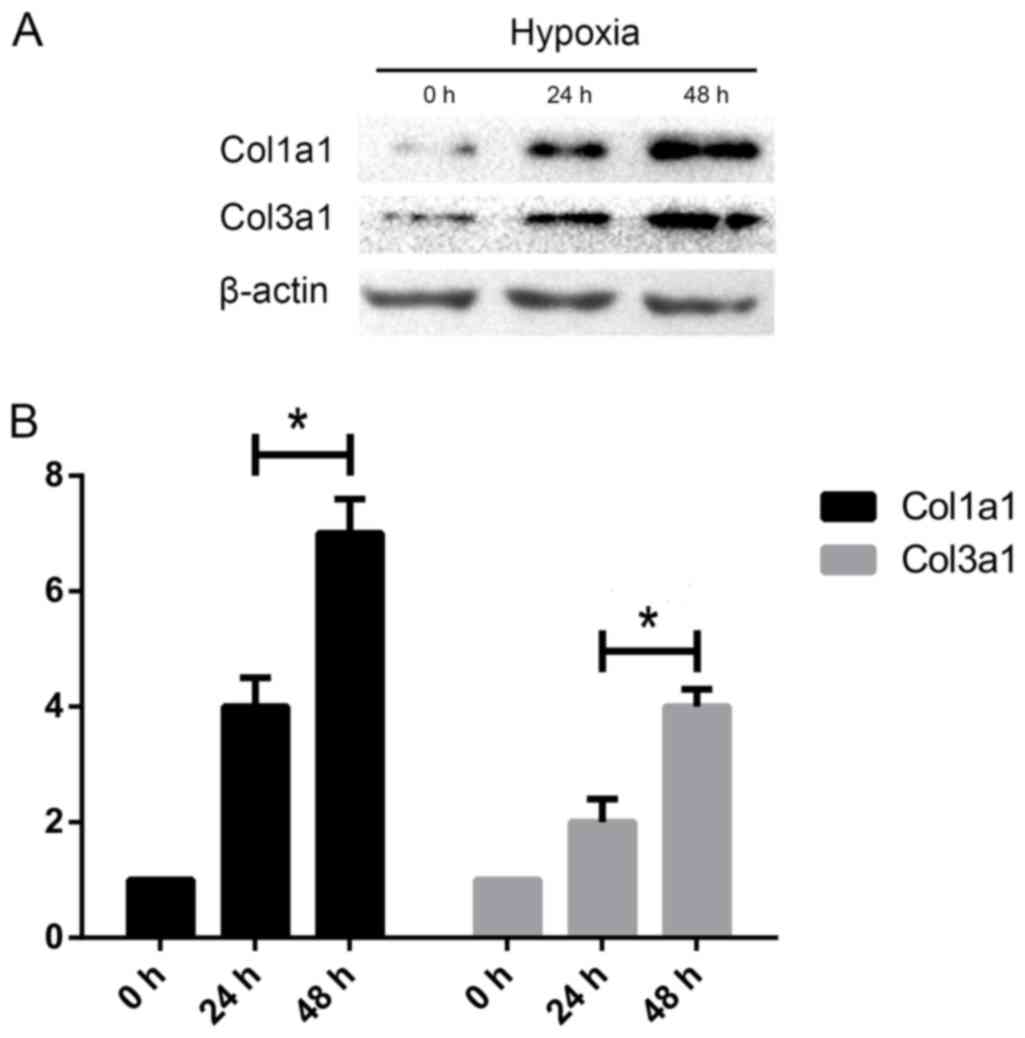
The length of hypoxia influences
collagen synthesis in HSCs
To analyze whether the length of hypoxia can
phenotypically alter HSCs, we investigated the effects of different
hypoxia time on HSC collagen synthesis in the HSC-T6 rodent cell
line. Compared with the ‘12 h’ group, Col1A1 and Col3A1 protein
expression were elevated in ‘24 h’ group when cells were exposed to
hypoxia, suggesting that hypoxia increases expression of makers
associated with matrix deposition with time (Fig. 2).
The length of hypoxia influences HSC
activation
Next, we aimed to investigate whether length of
hypoxia had an effect on the activation of HSCs. The cells were
incubated under hypoxia for 0, 24 and 48 h. We measured levels of
α-SMA as a marker of HSC activation. We found an increase in α-SMA
protein expression using western blot and immunofluorescence
analyses as the duration increased (Fig. 3). These data indicate that HSC are
activated in response to hypoxia time.
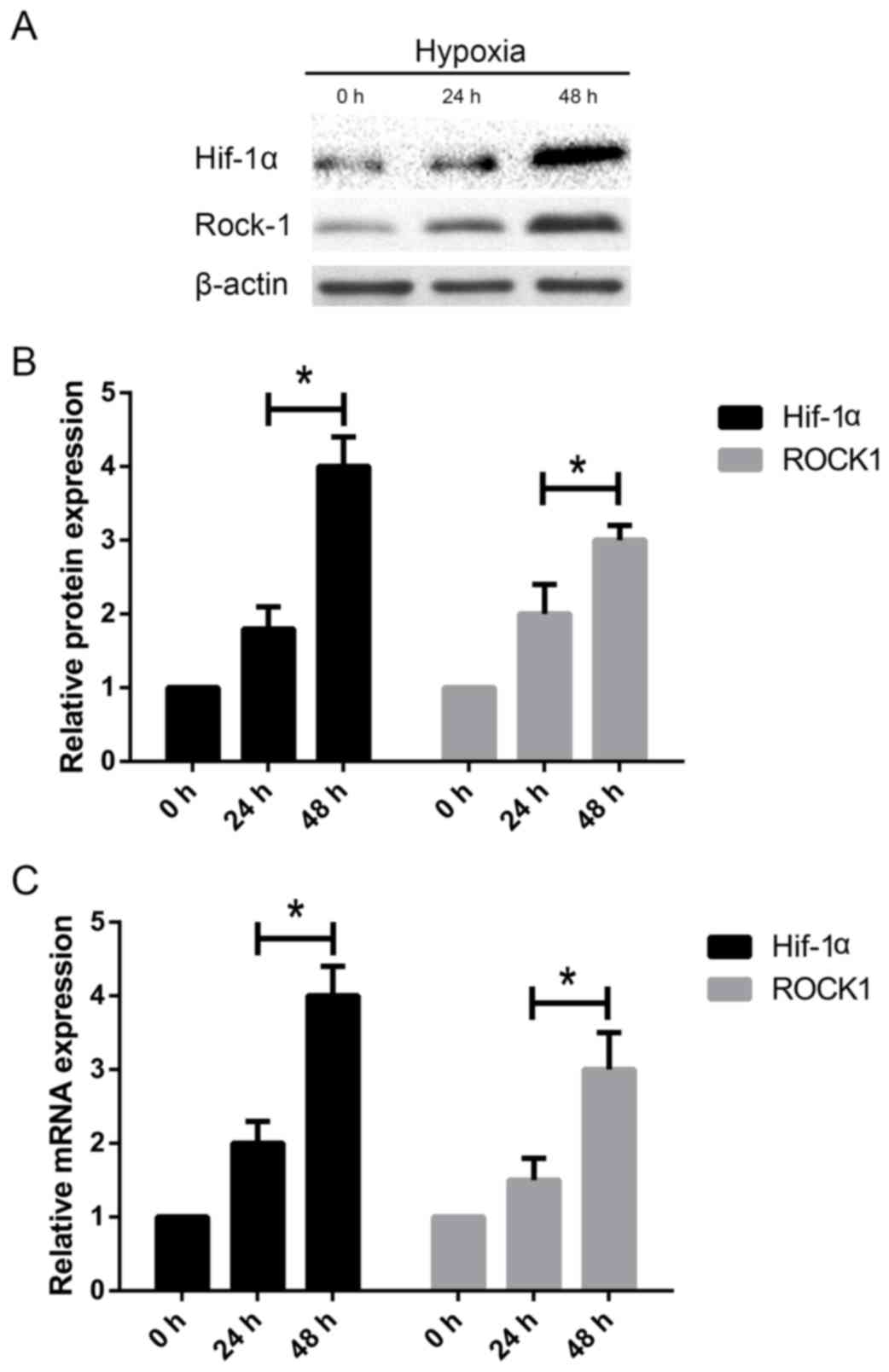
The length of hypoxia influences
HIF1-α and ROCK1 upregulation in HSCs
Since HIF1-α and ROCK1 are associated with HSC
activation and matrix deposition, we further investigated if there
were changes in the mRNA and protein expression of HIF1-α and ROCK1
in HSC-T6 cells under different hypoxic conditions. Western blot
analysis showed that HIF1-α and ROCK1 protein levels were increased
in HSC-T6 cells with the increase of time (Fig. 4A-B). Accordingly, the RT-PCR
analysis showed that HIF1-α and ROCK1 mRNA expression increased
3-fold and 2-fold, respectively, compared to untreated HSC-T6 cells
at 0 h (Fig. 4C). These data
suggest that HIF1-α and ROCK1 may play an important role in HSC
activation.
Knockdown of HIF1-α inhibits ROCK1
expression and HSC activation under hypoxia
Next, we investigated if HIF1-α alters
hypoxia-induced activation of HSC-T6 cells. HSC-T6 cells were
cultured under hypoxic conditions. siHIF1-α was transfected into
cells to silence HIF1-α expression. HSC-T6 cells were transfected
with siHIF1-α for 48 h and α-SMA expression was measured. Our
results showed that knockdown of HIF1-α decreased α-SMA protein and
mRNA expression (Fig. 5A-B).
Accordingly, knockdown of HIF1-α also attenuated expression of
Col1A1, a typical marker of activated HSCs (Fig. 5). Col3A1 secretion was decreased
when cells were transfected with siHIF1-α under hypoxia (Fig. 5D). Additionally, we measured ROCK1
expression in siHIF1-α-transfected HSC-T6 cells. We found that
ROCK1 protein expression and mRNA levels were markedly
downregulated following HIF1-α knockdown (Fig. 5A-C). Taken together, these findings
indicate that decreasing HIF1-α expression attenuates ROCK1
expression and HSC activation in response to hypoxia.
 | Figure 5.Effect of HIF1-α knockdown on ROCK1
expression and HSC activation under hypoxia. HSC-T6 cells were
transfected with siHIF1-α and non-specific siRNA. (A) ROCK1, α-SMA
and Col1A1 protein levels were measured by western blotting. (B)
ROCK1, α-SMA and Col1A1 mRNA levels were measured by reverse
transcription-quantitative polymerase chain reaction. (C) Relative
protein expression (n=3). (D) Col3A1 secretion was measured by
ELISA. Data are presented as the mean ± standard deviation.
*P<0.05, as indicated. HIF1, hypoxia inducible factor 1; ROCK,
Rho-associated coiled-coil-forming kinase 1; HSC, hepatic stellate
cell; si, small interfering; Col1/3 A1, collagen type 1/3 α1 chain;
α-SMA, α-smooth muscle actin; NS, non-specific; ELISA, enzyme
linked immunosorbent assay. |
Inhibition of ROCK1 downregulates
HIF1-α expression and HSC activation under hypoxia
Next, we evaluated whether ROCK1 is required for
hypoxia-induced activation of HSC-T6 cells. We inhibited ROCK1
activity using Y-27632, a specific ROCK1 inhibitor. We observed
that ROCK1 inhibition decreased α-SMA protein expression and mRNA
levels in HSCs under hypoxic conditions (Fig. 6A-C). Similarly, western blot and
RT-PCR analyses showed that Col1A1 was downregulated in response to
ROCK1 inhibition (Fig. 6A-C). When
cells were treated with the inhibitor under hypoxia conditions,
Col3A1 secretion was concomitantly decreased, as shown by ELISA
(Fig. 6D). Interestingly, HIF1-α
protein expression and mRNA transcript levels were decreased in
hypoxia-exposed HSC-T6 cells compared to cells incubated under
normoxic conditions (Fig. 6A-C).
Our data collectively demonstrate that HIF1-α activation in HSCs
cultured under hypoxic conditions is ROCK1-dependent.
 | Figure 6.Effect of ROCK-1 inhibition on HIF1-α
expression and HSC activation under hypoxia. HSC-T6 cells were
challenged with Y-27632, a specific ROCK1 inhibitor. (A) HIF1-α,
α-SMA, Col1A1 and Col3A1 protein levels were measured by western
blotting. (B) HIF1-α, α-SMA and Col1A1 mRNA levels were measured by
reverse transcription-quantitative polymerase chain reaction. (C)
Relative protein expression (n=3). (D) Col3A1 secretion was
measured by ELISA. Data are presented as the mean ± standard
deviation. *P<0.05, as indicated. HIF1, hypoxia inducible factor
1; ROCK, Rho-associated coiled-coil-forming kinase 1; HSC, hepatic
stellate cell; Col1/3 A1, collagen type 1/3 α1 chain; SMA, smooth
muscle actin; ELISA, enzyme linked immunosorbent assay. |
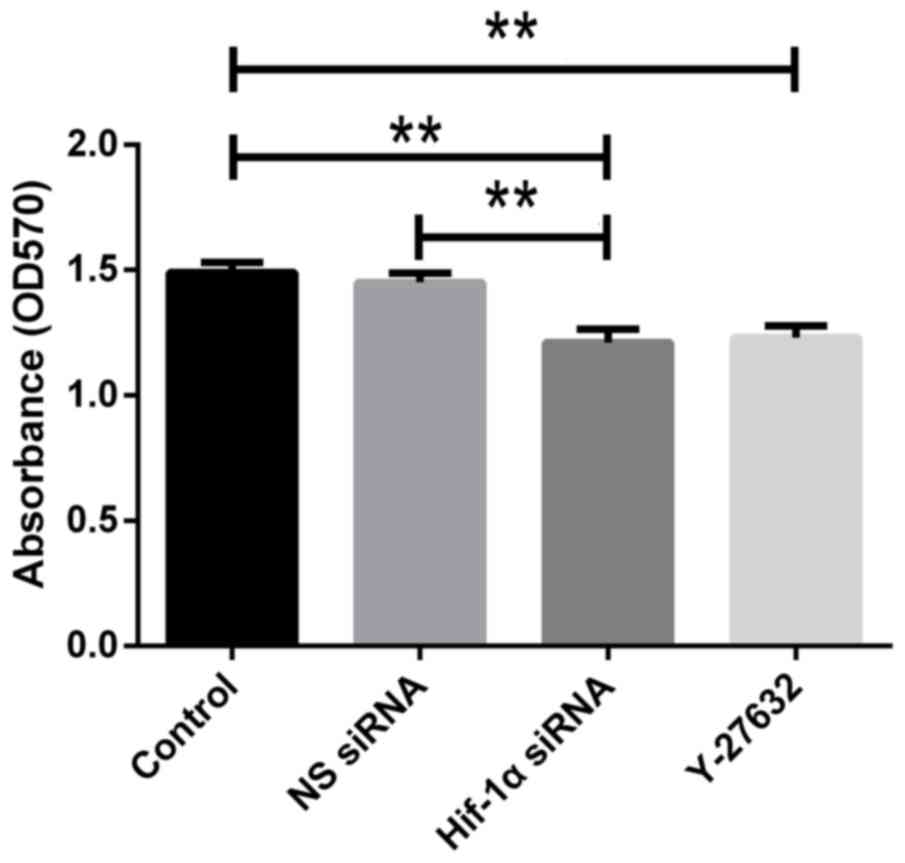
Effect of HIF1-α and ROCK1 on HSC
proliferation under hypoxia
HIF1-α and ROCK1 are essential in cancer cell
proliferation. Here, we determined if HIF1-α and ROCK1 are required
for proliferation of hypoxia-exposed HSC-T6 cells using the MTT
assay. We observed that the proliferation of cells was reduced
after transfection with HIF1-α siRNA compared with control one. The
proliferation was also suppressed in cells added Y-27632 compared
with control group (Fig. 7). Our
results collectively indicate that knockdown of HIF1-α or
inhibition of ROCK1 significantly decreases HSC proliferation.
Discussion
Liver fibrosis is a reversible inflammatory response
characterized by HSC activation and, consequently, excessive ECM
accumulation. Constant hepatic fibrogenesis can result in major
clinical outcomes including portal hypertension, ascites,
esophageal varices, and cirrhosis. Understanding the etiology of
fibrogenesis is critical for reversing hepatic fibrosis (3,19).
Numerous molecular pathways have been associated with liver
fibrogenesis. Hence, it is crucial to characterize the pathogenesis
of hepatic fibrosis in order to identify novel therapeutic
strategies.
Hypoxia is an early event in liver injury (6). Indeed, several studies have shown
that hypoxia is associated with the initiation and progression of
hepatic fibrosis (5,6). Hypoxia induces HIF1-α activation,
which has important effects on malignant cancers (20,21).
Moreover, HIF1-α plays determinant roles in cardiac development,
pulmonary hypertension, diabetes and cancer, such as colorectal,
pancreatic and ovarian cancers (22–26).
Recently, the role of HIF1-α in hepatic fibrosis has gained
considerable attention (27).
Moreover, the RhoA/ROCK1 cascade is related to HSC activation and
ECM deposition (28,29). However, the crosstalk between
HIF1-α and ROCK1 in HSC regulation remains unexplored.
In the present study, we investigated whether HIF1-α
expression influences cell proliferation and collagen synthesis in
HSCs in response to hypoxia. Our experiments clearly show that
HIF1-α expression is upregulated in activated HSCs under hypoxia.
The expression was elevated with hypoxia time increasing.
Additionally, transfection of HSCs with siHIF1-α decreased HSC
activation, suggesting that HIF1-α is directly related to HSC
activation, consistent with previous studies (7).
Recently, Kutscher et al (30) found that HIF1-α is required for
endothelial progenitor cell functions, including proliferation,
invasion and cell survival. Hu et al (31) suggested that HIF1-α might be a new
therapeutic target for rheumatoid arthritis as it prevents
interactions of synovial fibroblasts with T and B cells. In another
study, Marhold et al (32)
revealed that HIF1-α is critical for maintaining quiescence in
cancer stem cells. According to these data, HIF1-α may have
additional roles in different systems. HSC activation has been
linked to collagen deposition (1).
In the present study, we detected increased levels of collagen
under hypoxia. When cells were exposed to hypoxia longer, the
collagen expression was induced more. Importantly, silencing HIF1-α
expression decreased collagen deposition and secretion; however,
matrix deposition was not completely suppressed, suggesting that
other factors apart from HIF1-α affect HSC activation.
Therefore, we further explored the potential
interplay between HIF1-α and ROCK1 during HSC activation under
hypoxia. Wojciak-Stothard et al (33) showed that activation of RhoA/ROCK1
is enhanced under hypoxia in human pulmonary artery endothelial and
smooth muscle cells. Due to the involvement of the RhoA/ROCK1
signaling pathway in numerous fibrotic processes, Knipe et
al (34) suggested that
further studies are necessary to shed light on the function of this
pathway in fibrogenesis. In our study, upregulation of ROCK1 was
observed in response to hypoxia in HSCs. Due to the increasing
hypoxia time, ROCK1 expression was increased. Furthermore, ROCK1
inhibition attenuated HSC proliferation and collagen synthesis.
Interestingly, specific ROCK1 inhibitors were effective in
relieving inflammatory pain (35)
and improving spinal cord injury (36).
Our results demonstrate that HIF1-α expression is
also reduced in HSCs challenged with a ROCK1 inhibitor, suggesting
that upregulation of HIF1-α is partly ROCK1-dependent. Conversely,
silencing HIF1-α suppressed ROCK1 activation in HSCs. These results
strongly support crosstalk between HIF1-α and ROCK1 in response to
hypoxia. In view of the functions mentioned above, the interplay
between HIF1-α and ROCK1 is associated with HSC activation.
Recently, Gilkes et al (14) indicated that hypoxia might induce
HIF1-α activation via RhoA-ROCK1 expression, resulting in increased
breast cancer cell motility. In human ovarian cancer cells, Ohta
et al (16) showed that
silencing RhoA, ROCK1 or ROCK2 decreased HIF1-α expression.
Moreover, Yin et al (37)
concluded that the role of HIF1-α in hypoxia-induced apoptosis was
RhoA-dependent in a neuroblastoma cell line.
In conclusion, our data clearly indicate that there
is crosstalk between HIF1-α and ROCK1 in HSCs under hypoxia.
Moreover, our results suggest that this crosstalk is essential for
regulating HSC activation, providing novel insights into the
underlying mechanisms of hepatic fibrogenesis and potential
therapeutic targets for treatment of liver fibrosis.
Acknowledgements
The authors would like to thank Medjaden Bioscience
Ltd. (Hong Kong, China) for assisting in the preparation of this
manuscript.
Funding
The present study was supported by The Natural
Science Foundation of Zhejiang Province (grant no.
LY14H030010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH and RF designed the study, and YH, DH, HY and WX
performed the data analysis. YH and DH drafted and wrote the
manuscript. RF revised the manuscript critically for intellectual
content. All authors gave intellectual input to the study and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HSCs
|
hepatic stellate cells
|
|
α-SMA
|
α-smooth muscle actin
|
|
COL1A1
|
collagen type 1 α1 chain
|
|
COL3A1
|
collagen type 3 α1 chain
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
References
|
1
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rockey DC: Translating an understanding of
the pathogenesis of hepatic fibrosis to novel therapies. Clin
Gastroenterol Hepatol. 11:224–231.e1-5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corpechot C, Barbu V, Wendum D, Kinnman N,
Rey C, Poupon R, Housset C and Rosmorduc O: Hypoxia-induced VEGF
and collagen I expressions are associated with angiogenesis and
fibrogenesis in experimental cirrhosis. Hepatology. 35:1010–1021.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Poli G: Pathogenesis of liver fibrosis:
Role of oxidative stress. Mol Aspects Med. 21:49–98. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon JO, Welch TP, Gonzalez FJ and Copple
BL: Reduced liver fibrosis in hypoxia-inducible
factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver
Physiol. 296:G582–G592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Novo E, Povero D, Busletta C, Paternostro
C, di Bonzo LV, Cannito S, Compagnone A, Bandino A, Marra F,
Colombatto S, et al: The biphasic nature of hypoxia-induced
directional migration of activated human hepatic stellate cells. J
Pathol. 226:588–597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:pp. 5510–5514. 1995; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Copple BL, Bai S, Burgoon LD and Moon JO:
Hypoxia-inducible factor-1α regulates the expression of genes in
hypoxic hepatic stellate cells important for collagen deposition
and angiogenesis. Liver Int. 31:230–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Novo E, Cannito S, Zamara E, Valfrè di
Bonzo L, Caligiuri A, Cravanzola C, Compagnone A, Colombatto S,
Marra F, Pinzani M and Parola M: Proangiogenic cytokines as
hypoxia-dependent factors stimulating migration of human hepatic
stellate cells. Am J Pathol. 170:1942–1953. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Huang Y, Guan F, Xiao Y, Deng J,
Chen H, Chen X, Li J, Huang H and Shi C: Hypoxia-inducible
factor-1alpha and MAPK co-regulate activation of hepatic stellate
cells upon hypoxia stimulation. PLoS One. 8:e740512013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kato M, Iwamoto H, Higashi N, Sugimoto R,
Uchimura K, Tada S, Sakai H, Nakamuta M and Nawata H: Role of Rho
small GTP binding protein in the regulation of actin cytoskeleton
in hepatic stellate cells. J Hepatol. 31:91–99. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwamoto H, Nakamuta M, Tada S, Sugimoto R,
Enjoji M and Nawata H: A p160ROCK-specific inhibitor, Y-27632,
attenuates rat hepatic stellate cell growth. J Hepatol. 32:762–770.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gilkes DM, Xiang L, Lee SJ, Chaturvedi P,
Hubbi ME, Wirtz D and Semenza GL: Hypoxia-inducible factors mediate
coordinated RhoA-ROCK1 expression and signaling in breast cancer
cells. Proc Natl Acad Sci USA. 111:pp. E384–E393. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matoba K, Kawanami D, Okada R, Tsukamoto
M, Kinoshita J, Ito T, Ishizawa S, Kanazawa Y, Yokota T, Murai N,
et al: Rho-kinase inhibition prevents the progression of diabetic
nephropathy by downregulating hypoxia-inducible factor 1α. Kidney
Int. 84:545–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohta T, Takahashi T, Shibuya T, Amita M,
Henmi N, Takahashi K and Kurachi H: Inhibition of the Rho/ROCK
pathway enhances the efficacy of cisplatin through the blockage of
hypoxia-inducible factor-1α in human ovarian cancer cell. Cancer
Biol Ther. 13:25–33. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van den Beucken T, Koch E, Chu K,
Rupaimoole R, Prickaerts P, Adriaens M, Voncken JW, Harris AL,
Buffa FM, Haider S, et al: Hypoxia promotes stem cell phenotypes
and poor prognosis through epigenetic regulation of DICER. Nat
Commun. 5:52032014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barsoum IB, Smallwood CA, Siemens DR and
Graham CH: A mechanism of hypoxia-mediated escape from adaptive
immunity in cancer cells. Cancer Res. 74:665–674. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ji RC: Hypoxia and lymphangiogenesis in
tumor microenvironment and metastasis. Cancer Lett. 346:6–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wigerup C, Påhlman S and Bexell D:
Therapeutic targeting of hypoxia and hypoxia-inducible factors in
cancer. Pharmacol Ther. 164:152–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krishnamachary B, Berg-Dixon S, Kelly B,
Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi P
and Semenza GL: Regulation of colon carcinoma cell invasion by
hypoxia-inducible factor 1. Cancer Res. 63:1138–1143.
2003.PubMed/NCBI
|
|
24
|
Ball MK, Waypa GB, Mungai PT, Nielsen JM,
Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK,
Steinhorn RH, et al: Regulation of hypoxia-induced pulmonary
hypertension by vascular smooth muscle hypoxia-inducible factor-1α.
Am J Respir Crit Care Med. 189:314–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He G, Jiang Y, Zhang B and Wu G: The
effect of HIF-1α on glucose metabolism, growth and apoptosis of
pancreatic cancerous cells. Asia Pac J Clin Nutr. 23:174–180.
2014.PubMed/NCBI
|
|
26
|
Birner P, Schindl M, Obermair A,
Breitenecker G and Oberhuber G: Expression of hypoxia-inducible
factor 1alpha in epithelial ovarian tumors: Its impact on prognosis
and on response to chemotherapy. Clin Cancer Res. 7:1661–1668.
2001.PubMed/NCBI
|
|
27
|
Zhan L, Huang C, Meng XM, Song Y, Wu XQ,
Yang Y and Li J: Hypoxia-inducible factor-1alpha in hepatic
fibrosis: A promising therapeutic target. Biochimie. 108:1–7. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Murata T, Arii S, Nakamura T, Mori A,
Kaido T, Furuyama H, Furumoto K, Nakao T, Isobe N and Imamura M:
Inhibitory effect of Y-27632, a ROCK inhibitor, on progression of
rat liver fibrosis in association with inactivation of hepatic
stellate cells. J Hepatol. 35:474–481. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tada S, Iwamoto H, Nakamuta M, Sugimoto R,
Enjoji M, Nakashima Y and Nawata H: A selective ROCK inhibitor,
Y27632, prevents dimethylnitrosamine-induced hepatic fibrosis in
rats. J Hepatol. 34:529–536. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kutscher C, Lampert FM, Kunze M,
Markfeld-Erol F, Stark GB and Finkenzeller G: Overexpression of
hypoxia-inducible factor-1 alpha improves vasculogenesis-related
functions of endothelial progenitor cells. Microvasc Res.
105:85–92. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu F, Liu H, Xu L, Li Y, Liu X, Shi L, Su
Y, Qiu X, Zhang X, Yang Y, et al: Hypoxia-inducible factor-1α
perpetuates synovial fibroblast interactions with T cells and B
cells in rheumatoid arthritis. Eur J Immunol. 46:742–751. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marhold M, Tomasich E, El-Gazzar A, Heller
G, Spittler A, Horvat R, Krainer M and Horak P: HIF1α Regulates
mTOR signaling and viability of prostate cancer stem cells. Mol
Cancer Res. 13:556–564. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wojciak-Stothard B, Zhao L, Oliver E,
Dubois O, Wu Y, Kardassis D, Vasilaki E, Huang M, Mitchell JA,
Harrington LS, et al: Role of RhoB in the regulation of pulmonary
endothelial and smooth muscle cell responses to hypoxia. Circ Res.
110:1423–1434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Knipe RS, Tager AM and Liao JK: The Rho
kinases: Critical mediators of multiple profibrotic processes and
rational targets for new therapies for pulmonary fibrosis.
Pharmacol Rev. 67:103–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang C, Song S, Zhang Y, Ge Y, Fang X,
Huang T, Du J and Gao J: Inhibition of the Rho/Rho kinase pathway
prevents lipopolysaccharide-induced hyperalgesia and the release of
TNF-α and IL-1β in the mouse spinal cord. Sci Rep. 5:145532015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dyck SM, Alizadeh A, Santhosh KT, Proulx
EH, Wu CL and Karimi-Abdolrezaee S: Chondroitin sulfate
proteoglycans negatively modulate spinal cord neural precursor
cells by signaling through LAR and RPTPσ and modulation of the
Rho/ROCK pathway. Stem Cells. 33:2550–2563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yin CP, Guan SH, Zhang B, Wang XX and Yue
SW: Upregulation of HIF-1α protects neuroblastoma cells from
hypoxia-induced apoptosis in a RhoA-dependent manner. Mol Med Rep.
12:7123–7131. 2015. View Article : Google Scholar : PubMed/NCBI
|