Introduction
Acute lung injury/acute respiratory distress
syndrome (ALI/ARDS) is an acute progressing hypoxemic respiratory
failure characterized by acute diffuse alveolar damage, influx of
neutrophils and protein-rich exudates in alveolar spaces (1). Each year in the United States there
are 190,600 cases of acute lung injury, which are associated with
74,500 mortalities and 3.6 million hospital days (2). The underlying immunological
mechanisms are still not well defined. The uncontrolled excessive
lung inflammation may account for the high mortality rate. Patients
usually receive supportive treatments with mechanical ventilation
and immunosuppressants, but the therapeutics do not prolong the
survival rate in patients and animal models (3,4).
Altered alveolar macrophage activation and cell death are
considered a major player in the progression of the uncontrolled
acute lung inflammation among patients with ALI/ARDS (5–7).
Macrophages are heterogeneous cell components and
serve distinct roles in the different stages of diseases (8,9).
Following exposure to pathogens, macrophages are activated
releasing a variety of cytokines and other mediators. At later
stages of the disease, the activated macrophages undergo
noninflammatory apoptosis and are finally cleared by monocytes, an
important pathological process in inflammation resolution and
tissue remodeling (5). However, it
is clinically reported that a high percentage of patients with
severe sepsis and ALI/ARDS fail to recover from the acute lung
inflammation, accompanied by persistent existence of the
uncontrolled lung edema and excessive host response to pathogens
(10,11). According to previous reports it was
hypothesized that the uncontrolled lung inflammation may be caused
by both insufficient clearance of dead cells and release of
proinflammatory cytokines during immune cell death (12,13).
A previous study indicated that macrophages can undergo
inflammatory programmed cell death, called pyroptosis, under
certain pathological circumstances (12). The pyroptotic cells are identified
as caspase-1 and propidium iodide (PI)-double positive cells and
are characterized by their release of a large amount of hallmark
proteins such as active interleukin (IL)-1β and IL-18 (14). Prior to pyroptosis, the
nucleotide-binding domain, leucine-rich-containing family, pyrin
domain-containing-3 (NLRP3) protein is upregulated and forms a
protein complex known as the NLRP3 inflammasome, with
apoptosis-associated speck-like protein containing a caspase
recruitment domain (ASC) and pro-caspase-1. Active caspase-1 can
stimulate IL-1β and IL-18, causing the release of these active
cytokines from pyroptotic cells (15). ASC is an important component of the
NLRP3 inflammasome and participates in NLRP3 inflammasome
activation (16). It was reported
that ASC deficiency caused a defect in NLRP3 inflammasome assembly
and protected against liver ischemia/reperfusion damage in animal
models through suppression of the caspase-1/IL-1β signaling pathway
(17). Other studies also
demonstrated that the NLRP3 inflammasome can be activated by
neutrophil-derived extracellular histones and complement component
5a, ultimately causing excessive tissue inflammation (18,19).
A body of evidence confirmed that p38
mitogen-activated protein kinase (MAPK) signaling pathway
participates in the progression of ALI/ARDS (20,21).
p38 MAPK regulates cell growth, proliferation, differentiation,
migration, apoptosis and inflammation (22,23).
In animals with lipopolysaccharide (LPS)-induced acute lung injury,
p38 MAPK expression is upregulated (21). However, there is limited
information about the role of p38 signaling pathway in macrophage
pyroptosis and uncontrolled lung inflammation in the ALI/ARDS mouse
model. In the present study, two macrophage cell lines and an
ALI/ARDS mouse model were treated with p38 MAPK inhibitor SB203580
to block p38 MAPK signaling pathway prior to LPS treatment. The
present results revealed that blockage of p38 MAPK signaling
pathway with SB203580 suppressed macrophage pyroptosis and
LPS-induced acute lung injury through negative regulation of NLRP3
inflammasome activation.
Materials and methods
Cell culture and treatments
The murine macrophage cell lines, RAW264.7 and
NR8383 cells (American Type Culture Collection, Manassas, VA, USA),
were cultured in Roswell Park Memorial Institute (RPMI)-1640 and
F-12K medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
respectively, and supplied with 10% fetal bovine serum, 2 µM
glutamine (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin. NR8383 cells were treated
with 1 µg/ml LPS (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 4 h at 37°C, with or without pretreatment of 10 mM MAPK p38
inhibitor SB203580 (Selleck Chemicals, Houston, TX, USA) for 1 h at
37°C. Conditioned media and cells were collected for measurement of
protein expression levels by enzyme-linked immunosorbent assay
(ELISA), reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and western blot analysis.
Animal procedure
A total of 48 10-week-old male wild-type C57BL/6
mice were purchased from Shanghai Model Organisms Center, Inc.
(Shanghai, China). All mice were housed at 22°C with 12-h
light/dark cycle and free access to food and water. The animal
protocol was approved by the Laboratory Animal Care and Use
Committee at the Medical College of Fudan University, Zhongshan
Hospital. Mice were intratracheally treated with 5 mg/kg LPS with
or without 1 h intraperitoneal (i.p.) pretreatment with 20 mg/kg
p38 MAPK inhibitor SB203580 (LPS group and SB203580/LPS group) at
room temperature. Mice received PBS, and the same doses of SB203580
were used as controls (PBS and SB203580 groups). Following 24 h
LPS/PBS treatment, collection of bronchoalveolar lavage fluid (BAL)
was performed by intratracheal injection of 0.5 ml of PBS followed
by gentle aspiration. The lavage was repeated three times and the
recovery rate was >90%. The collected BAL fluid was centrifuged
at 400 × g for 5 min at 4°C and cell pellets were suspended in 1 ml
of PBS and cell count was manually measured with a hemocytometer by
light microscopy. The supernatant of BAL fluid was used for
cytokine assay and the lung tissues were collected for further
analysis.
Preparation of SB203580
SB203580 was purchased from Selleck Chemicals,
dissolved with DMSO (Sigma-Aldrich, Merck KGaA) according to the
manufacturer's protocol and diluted with PBS as a working solution.
A final concentration of 2% DMSO was introduced in the cell culture
media for each group, and 10% DMSO in solution was used for
administration into the mouse model. There were no signs of cell
toxicity and mouse mortality with the concentrations used in
vitro and in vivo (data not shown).
Wet/dry weight ratio
The wet and dry weights (W) of the left lung tissues
were measured prior to and following drying at 65°C for 48 h. Water
content was obtained by calculating the W/D weight ratio.
Lung histology
Lung tissue was fixed in 4% paraformaldehyde and
sections of 5 µm thickness were placed onto glass slides and
stained with hematoxylin and eosin (Beyotime Institute of
Biotechnology, Shanghai, China), according to the manufacturer's
protocol, for histopathological examination. The lung histology was
viewed under a light photomicroscope and evaluated for pathological
changes using a double-blind method. The severity of lung injury
was evaluated using a semiquantitative histological index,
including alveolar edema, hemorrhage, alveolar septal thickening
and infiltration of polymorphonuclear leukocytes. Each item was
divided into four grades from 0 to 3 (0, normal; 1, mild; 2,
moderate; 3, severe) and then calculated for a total acute lung
injury score (24).
Western blot analysis
The collected cells (1×106 cells/ml) were
incubated with 100 µl radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology) supplied with proteinase
inhibitor phenylmethylsulphonyl fluoride (Beyotime Institute of
Biotechnology) for 30 min on ice. The proteins of the lung tissues
were extracted using a Nuclear and Cytoplasmic Protein Extraction
kit (Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Protein concentration was determined by
bicinchoninic assay protein assay kit (Beyotime Institute of
Biotechnology). Each 20 µg protein sample was resolved on a 10%
SDS-PAGE gel. The protein was transferred onto polyvinylidene
fluoride membrane blots (Merck Millipore; Merck KGaA), and the
blots were incubated with blocking buffer (Shanghai Beyotime
Institute of Biotechnology) for 1 h at room temperature, followed
by incubation with the indicated primary antibodies against NLRP3
(1:1,000; NBP2-12446; Novus Biologicals, LLC, Littleton, CO, USA),
caspase-1 (1:1,000; Ab1872; Abcam, Cambridge, MA, USA), caspase-3
(1:1,000; 9665s; Cell Signaling Technology, Inc., Danvers, MA, USA)
cleaved-caspase-3 (1:1,000; 9664s; Cell Signaling Technology,
Inc.), Toll-like receptor (TLR)2 (1:1,000; ab108998; Abcam), TLR4
(1:1,000; sc-293072; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), nuclear factor (NF)-κB p65 (1:1,000; 3033p; Cell Signaling
Technology, Inc.), p-p38 (1:1,000; 4511S; Cell Signaling
Technology, Inc.) and MAPK p38 (1:1,000; 4511S; Cell Signaling
Technology, Inc) at 4°C overnight. The antibody anti-mouse β-actin
(1:1,000; BM0627; Boster Biological technology, Pleasanton, CA,
USA) was used for internal loading control and blots were incubated
with stripping buffer at 37°C for 15 min. The membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:1,000; anti-rabbit:A0208; anti-mouse A0216; Beyotime
Institute of Biotechnology) for 1 h at room temperature. Immune
reactivity was visualized using an enhanced chemiluminescent
reagent (Beyotime Institute of Biotechnology). Band intensity was
quantitatively analyzed by densitometric analysis on ImageJ
software version 1.37 (National Institutes of Health, Bethesda, MD,
USA).
RT-qPCR
cDNA was synthesized from 1 µg total RNA with a
ReverTra Ace qPCR RT Master Mix kit (Toyobo Life Science, Osaka,
Japan). RT-qPCR was performed using SYBR-Green PCR Master Mix-Plus
(Toyobo Life Science, Osaka, Japan). All the primers were
synthesized by Shanghai BioSune Biotechnology Co. Ltd. Primer
sequences are listed in Table I.
qPCR reaction was performed on a 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) under the condition of
95°C 1 min followed by 40 cycles of 95°C 15 sec, 60°C 60 sec, 72°C
45 sec. The expression level was quantified using the
2−ΔΔCq method (25),
relative to the internal control β-actin.
 | Table I.Primers used for detection of gene
expression. |
Table I.
Primers used for detection of gene
expression.
| Genes | Primer sequences
(5′-3′) |
|---|
| Mouse-IL-1β | Forward
AGAGCTTCAGGCAGGCAGTA |
|
| Reverse
AGGTGCTCATGTCCTCATCC |
| Mouse-TNF-α | Forward
ACGGCATGGATCTCAAAGAC |
|
| Reverse
GTGGGTGAGGAGCACGTAGT |
| Mouse-IL-6 | Forward
CCAGTTGCCTTCTTGGGACT |
|
| Reverse
GGTCTGTTGGGAGTGGTATCC |
| Mouse-β-actin | Forward
GTGCTATGTTGCTCTAGACTTCG |
|
| Reverse
ATGCCACAGGATTCCATACC |
| Rat-IL-1β | Forward
AAAAATGCCTCGTGCTGTCT |
|
| Reverse
TCGTTGCTTGTCTCTCCTTG |
| Rat-IL-6 | Forward
AGTTGCCTTCTTGGGACTGA |
|
| Reverse
ACTGGTCTGTTGTGGGTGGT |
| Rat-β-actin | Forward
TGTCACCAACTGGGACGATA |
|
| Reverse
GGGGTGTTGAAGGTCTCAAA |
ELISA assay for cytokines
IL-1β, tumor necrosis factor (TNF)-α and IL-6
expression levels in the BAL fluid or the supernatant of treated
cells were measured by ELISA assays (IL-1β: Mouse: MLB00C, rat:
RLB00; TNF-α: Mouse: MTA00B; IL-6: Mouse: M6000B, rat: R6000B;
R&D Systems Inc, Minneapolis, MN, USA) according to the
manufacturer's protocols.
Immunofluorescence staining
RAW264.7 (1×104 cells/ml) were stimulated
with 1 µg/ml LPS for 4, 8, 12 and 24 h. Following treatment, the
cells were incubated with fixing buffer and were blocked with
blocking buffer (Shanghai Beyotime Institute of Biotechnology) for
30 min at room temperature. Subsequently, the cells were incubated
with primary antibodies against rabbit anti-NLRP3 (1:1,000;
NBP2-12446; Novus Biologicals, LLC) and mouse anti-caspase-1
(1:1,000; sc-514; Santa Cruz Biotechnology, Inc.) for 2 h at 4°C.
The cells were incubated with secondary antibodies Cy3-conjugated
anti-rabbit IgG (1:1,000; A0516; Shanghai Beyotime Institute of
Biotechnology) and FITC-conjugated anti-mouse IgG (1:1,000; A0568;
Shanghai Beyotime Institute of Biotechnology) for 1 h at room
temperature following incubation with the primary antibody and
washing. DAPI was used for staining nuclei for 10 min at room
temperature. Finally, the stained cells were visualized under a
fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometry analysis
Pyroptotic cells were characterized by greater
expression of cleaved caspase-1 and positive propidium iodide (PI)
staining (26) and early apoptotic
cells were Annexin V+PI- (27).
Detection of apoptotic and pyroptotic cells by flow cytometry was
performed with Annexin V: FITC Apoptosis Detection kit I (556547;
BD Pharmingen; BD Biosciences, San Jose, CA, USA) and FLICA
660-YVAD-FMK far-red caspase-1 reagent (9122; ImmunoChemistry
Technologies, LLC, Bloomington, MN, USA) according to the
manufacturer's protocols in vitro (BD Pharmingen; BD
Biosciences). Briefly, the cells (1×105 cells/ml) were
resuspended in 100 µl 1X binding buffer and incubated with 5 µl
FITC-Annexin V (FL1 channel) and 5 µl PI in the dark for 20 min at
room temperature. The stained cells were washed with PBS twice and
fixed in 4% paraformaldehyde. To analyze pyroptotic cells, a 150 µl
single cell suspension was incubated with 5 µl FITC-conjugated
Annexin V, 5 µl PI (FL2 channel) and 5 µl FLICA 660-YVAD-FMK
far-red caspase-1 reagent (FL4 channel) in the dark at room
temperature for 20 min, then washed with PBS and fixed with 4%
paraformaldehyde for 10 min at room temperature. Analysis was
performed on a FACScan cytometer (Becton Dickinson; BD
Biosciences), and data were analyzed with FlowJo software version
7.6 (FlowJo LLC, Ashland, OR, USA).
Statistical analysis
All experimental values were presented as the mean ±
standard error of the mean. Statistical comparison between two
groups was performed by Student's t-test, and one-way analysis of
variance followed by Bonferroni post hoc analyses was performed
among multiple groups for parametric data. P<0.05 was considered
to indicate a statistically significant difference.
Results
Intraperitoneal injection of p38 MAPK
inhibitor SB203580 suppresses murine acute lung injury and
inflammation
To establish an acute lung injury mouse model, 8- to
10-week-old male mice were injected with 5 mg/kg LPS
intratracheally (i.t.). The mice injected with PBS were used as
controls. Massive inflammatory infiltrates, perivascular edema and
severe alveolar space destruction were observed compared to the
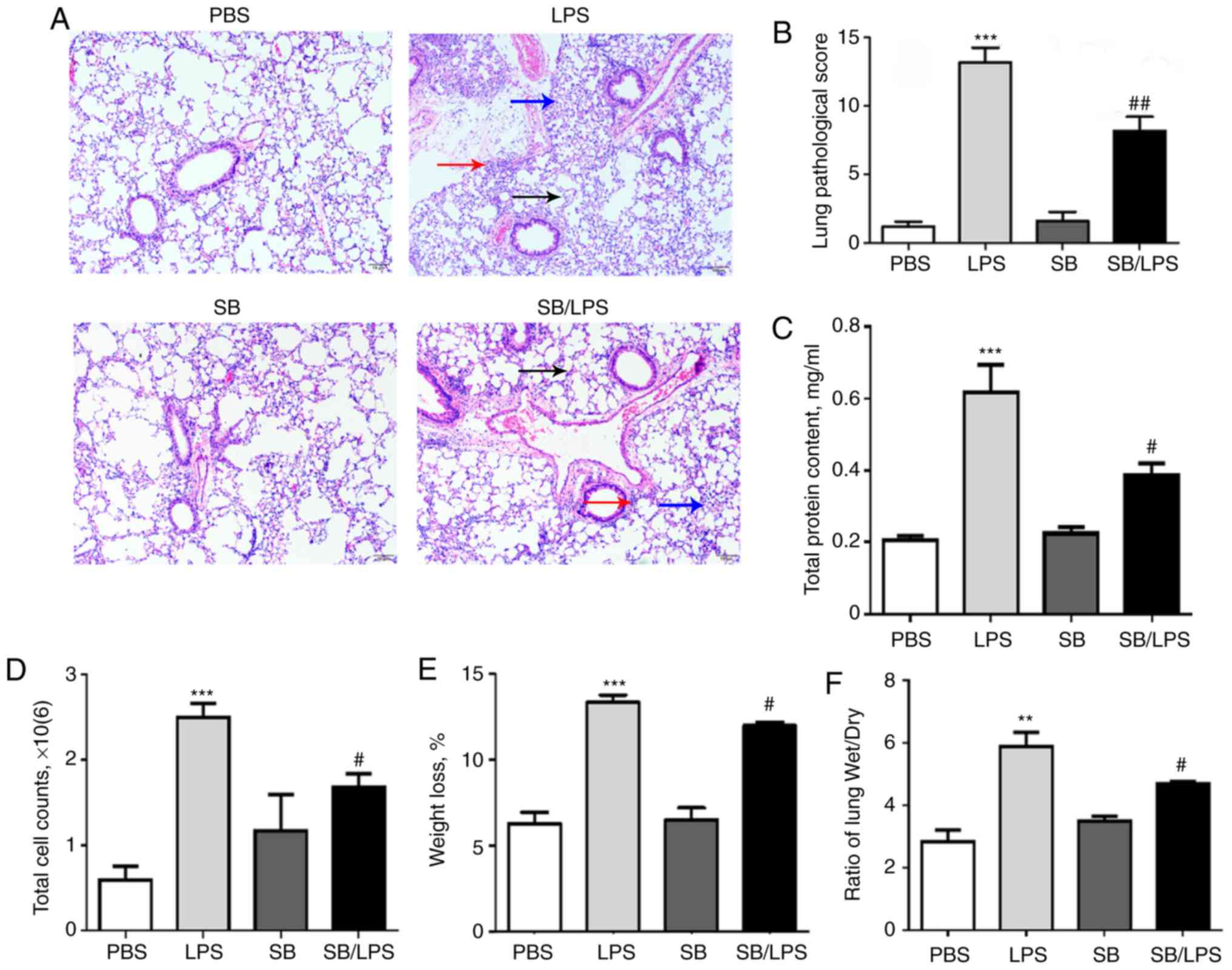
PBS-treated group 24 h following LPS injection (Fig. 1A). However, pretreatment with the
p38 MAPK inhibitor SB203580 significantly reversed the severity of
acute lung injury compared with the mice that received LPS
treatment alone (Fig. 1A and B).
Additionally, LPS treatment significantly induced more total BAL
protein content (Fig. 1C) and cell
counts (Fig. 1D). The weight loss
(Fig. 1E) and lung wet/dry ratio
(Fig. 1F) were also significantly
elevated after i.t. LPS injection. The beneficial effects were
consistent with the reduced total BAL protein content (Fig. 1C), cell counts (Fig. 1D), weight loss (Fig. 1E) and lung wet/dry ratio (Fig. 1F) in the group of mice that
received both SB203580 and LPS.
Blockage of the p38 MAPK signaling
pathway attenuates the expression of proinflammatory cytokines and
the NLRP3 inflammasome in the inflamed murine lung tissues
To determine whether the reduced acute lung injury
and inflammation in the SB203580-treated mice was supported by the
decreased expression of proinflammatory cytokines, IL-1β, tumor
necrosis factor (TNF)-α and IL-6 expression levels in BAL and lung
tissues were measured by ELISA and RT-qPCR analysis. Supporting the
observation above, it was demonstrated that the expression levels
of IL-1β (Fig. 2A and B), TNF-α
(Fig. 2C and D) and IL-6 (Fig. 2E and F) were greatly elevated at
the mRNA and protein levels in the mice treated with LPS. However,
blockage of p38 MAPK signaling pathway by SB203580 pretreatment
significantly attenuated their expression (Fig. 2).
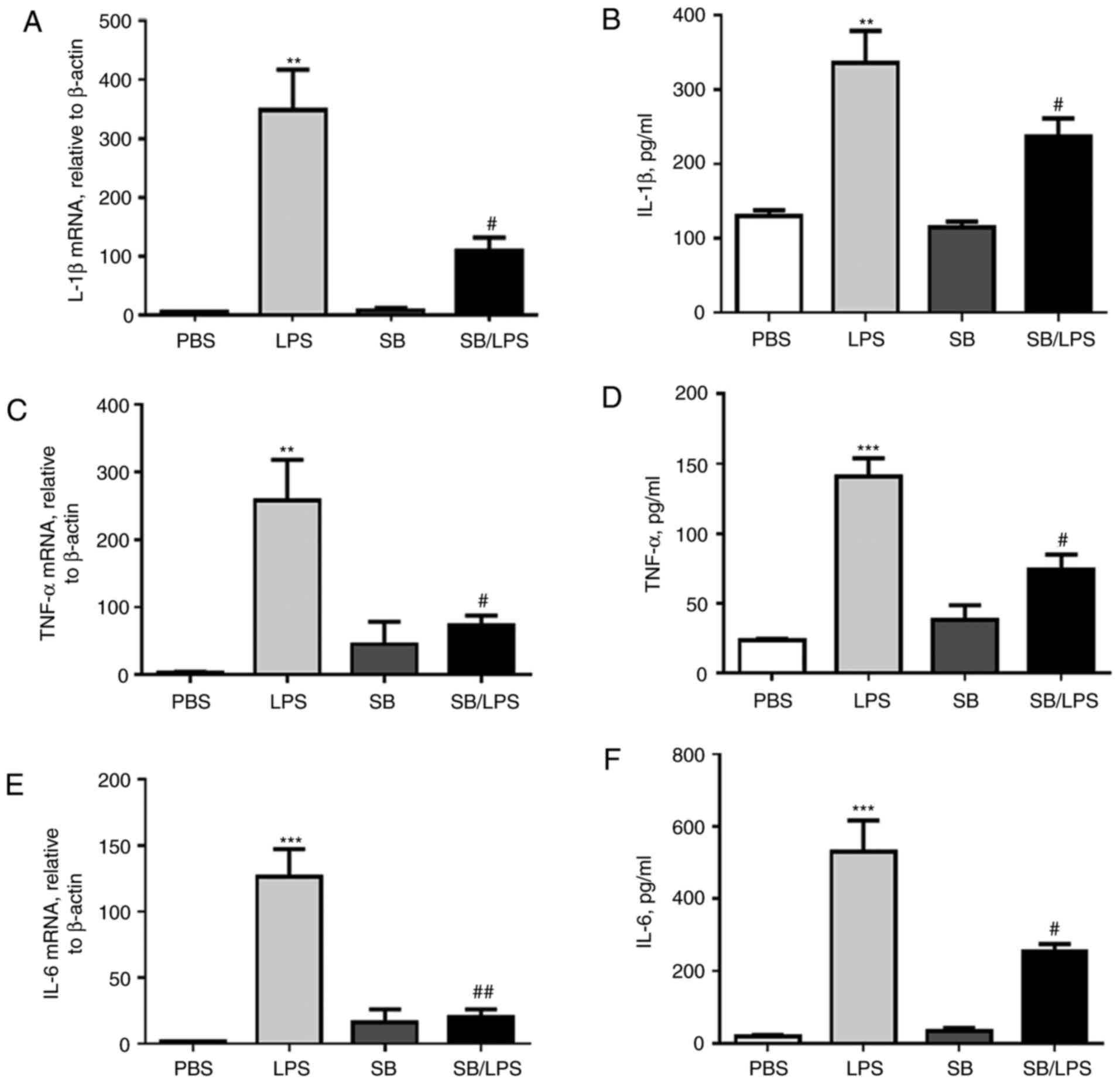 | Figure 2.p38 MAPK inhibitor SB203580
suppresses IL-1β, TNF-α and IL-6 expression in mice following the
i.t. injection of LPS. Twenty-four hours following SB203580 i.p.
and LPS i.t. treatment, the lung and BAL fluids were collected.
IL-1β (A) mRNA and (B) protein expression levels; TNF-α (C) mRNA
and (D) protein expression levels and IL-6 (E) mRNA and (F) protein
expression levels were analyzed by RT-qPCR analysis and by ELISA
analysis, respectively. Data are presented as the mean ± standard
error of the mean. **P<0.01, ***P<0.001 vs. PBS group;
#P<0.05, ##P<0.01 vs. LPS group, n=5
mice per group. BAL, bronchoalveolar lavage fluid; i.p.,
intraperitoneal; i.t., intratracheal; IL, interleukin; LPS,
lipopolysaccharide; TNF, tumor necrosis factor. |
IL-1β is a downstream proinflammatory cytokine of
the NLRP3 inflammasome and caspase-1 activation (28). To further investigate the effects
of p38 MAPK signaling pathway blockage on the NLRP3 inflammasome,
the relative protein expression levels of NLRP3, pro-caspase-1,
cleaved-caspase-1 (Fig. 3A) and
TLR2 (Fig. 3B) was analyzed by
western blot analysis and the mRNA of NLRP3 (Fig. 3C) and TLR2 (Fig. 3D) by RT-qPCR. SB203580 pretreatment
significantly reversed the LPS-induced upregulation of NLRP3,
cleaved caspase-1 and TLR2, indicating a beneficial role of
blockage of the p38 MAPK signaling pathway in reducing the
formation of the NLRP3 inflammasome and pyroptosis.
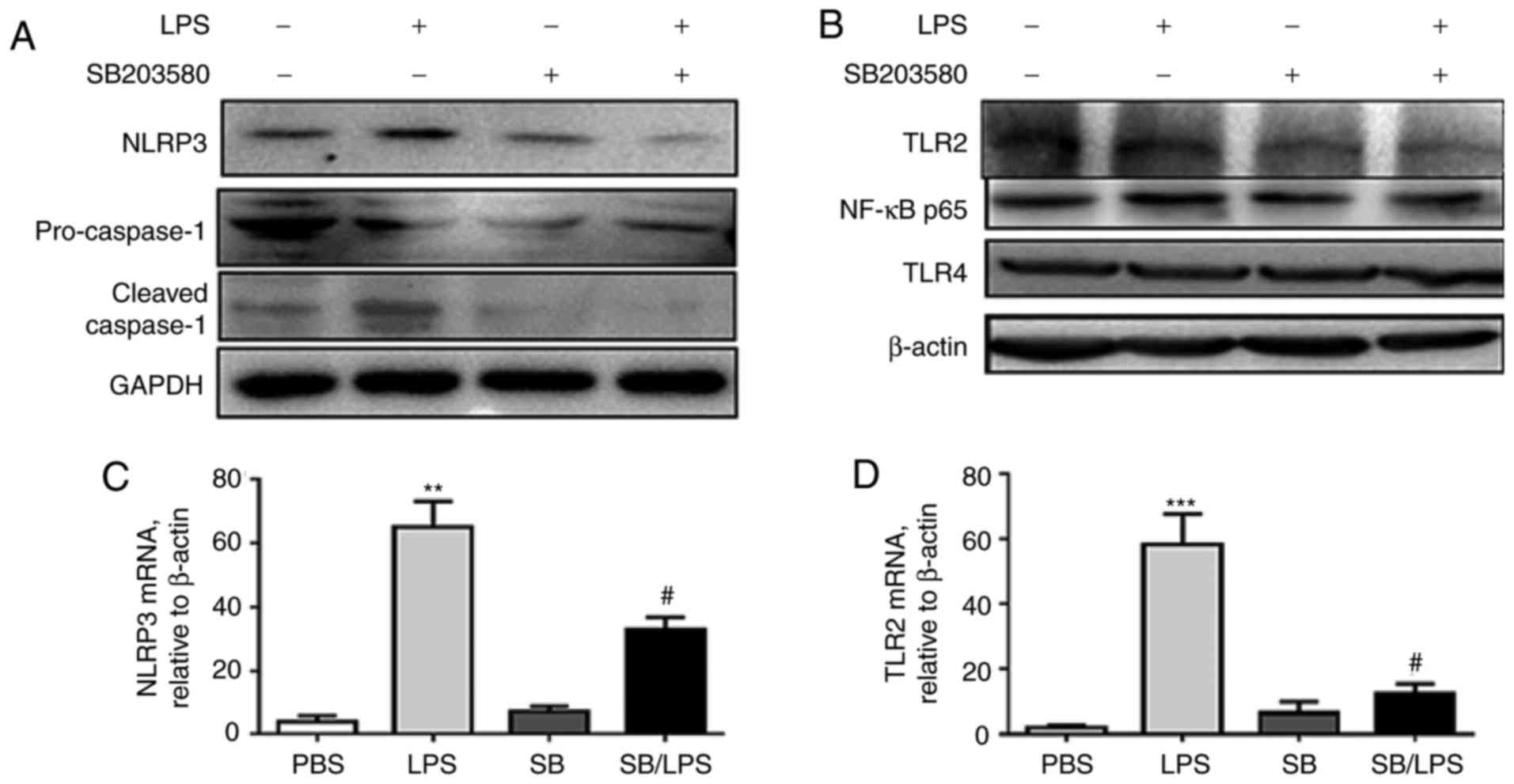 | Figure 3.p38 MAPK inhibitor SB203580
suppresses caspase-1, NLRP3 and TLR2 expression in mice following
i.t. injection of LPS. The pro-caspase-1, cleaved caspase-1, NLRP3
(A), NF-κB and TLR2 and TLR4 (B) were analyzed by western blot
analysis following LPS treatment. NLRP3 (C) and TLR2 (D) mRNA
expression levels were analyzed by RT-qPCR analysis. **P<0.01,
***P<0.001 vs. PBS group; #P<0.05 vs. LPS group,
n=5 per group. IL, interleukin; i.t., intratracheal; LPS,
lipopolysaccharide; MAPK, mitogen-activated protein kinase; NF-κB,
nuclear factor-κ-light-chain-enhancer of activated B cells; NLRP,
nucleotide-binding domain, leucine-rich-containing family, pyrin
domain-containing; TLR, Toll-like receptor. |
LPS treatment increases the expression
of NLRP3 and caspase-1 in macrophages
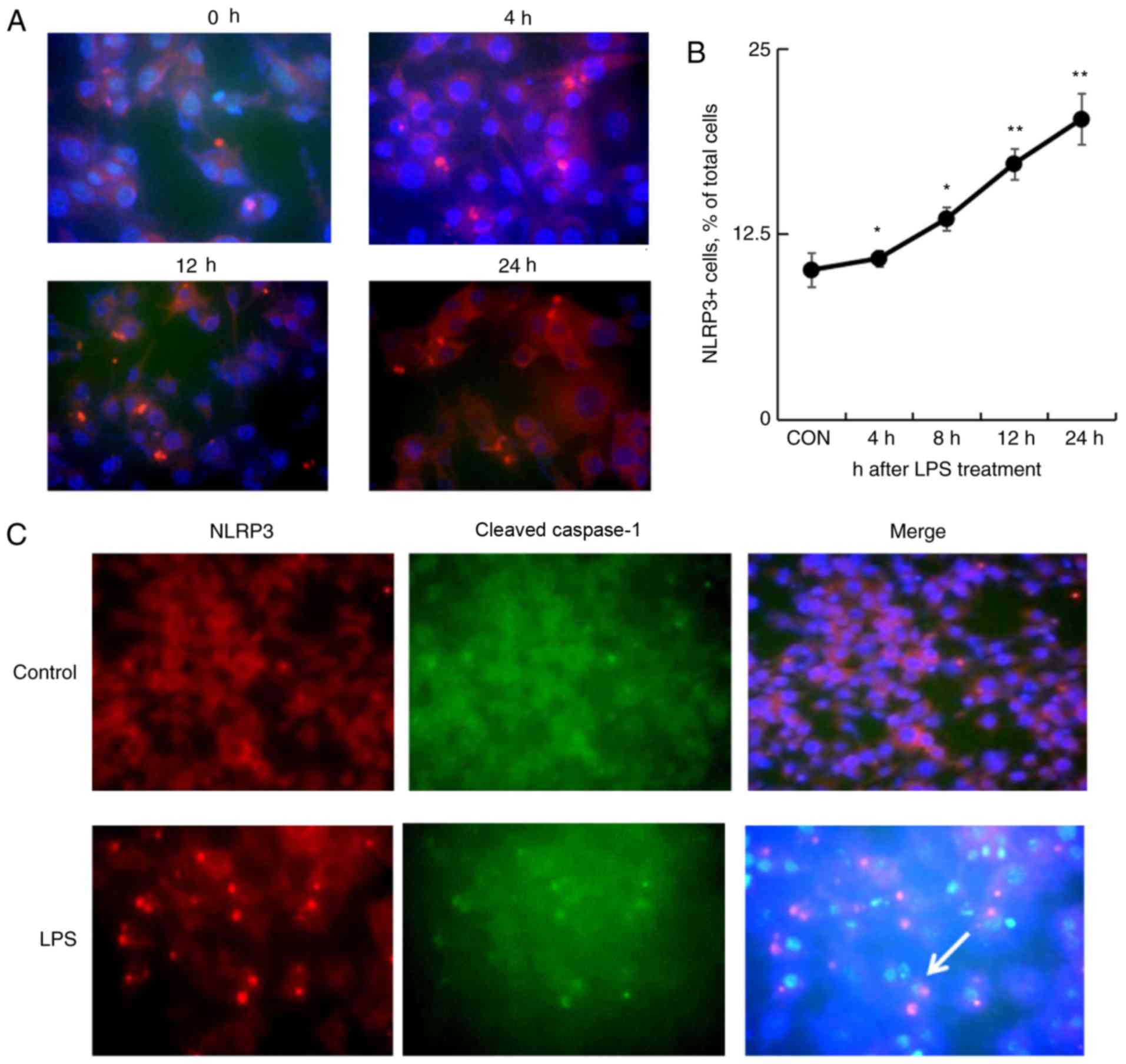
To further confirm the effects of LPS in inducing
macrophage pyroptosis, the mouse macrophage cell line, RAW264.7
cells were treated, with 1 µg/ml LPS for 4, 8, 12, and 24 h. The
NLRP3 expression was analyzed by immunofluorescence staining. As a
result, the NLRP3-expressing cells were increased 4 h following LPS
treatment that was gradually increased from 10.1±1.1% untreated
cells to a maximal 20.2±1.7% cells treated for 24 h (P<0.01;
Fig. 4A and B). In addition, the
increasing number of NLRP3+ and cleaved caspase-1+ cells was
observed by immunofluorescence staining following LPS treatment,
indicating a role of LPS in NLRP3 inflammasome formation and
activation (Fig. 4C).
Blockage of p38 MAPK signaling pathway
reverses the LPS-induced expression of NLRP3 and caspase-1 in
macrophages
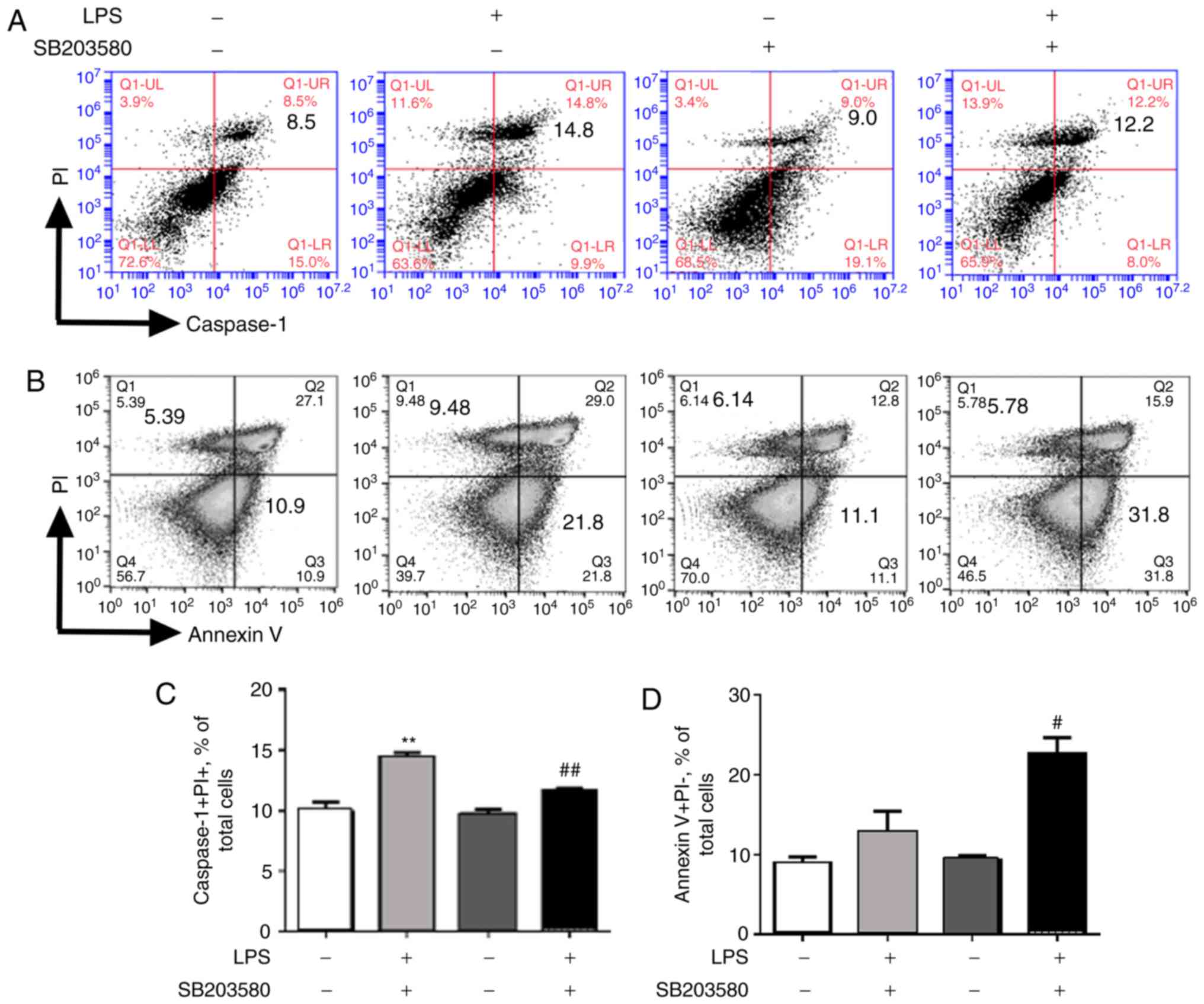
Pyroptosis and early apoptosis of NR8383 after
treatment were determined by flow cytometry. Further analysis
indicated that LPS treatment resulted in 1.7-fold increases in
caspase-1+PI+ pyroptotic cells as well as Annexin V+PI- early
apoptotic cells (Fig. 5A and B).
The present analysis further indicated that pretreatment with
SB203580 significantly reversed LPS-induced caspase-1+PI+
pyroptotic cells (Fig. 5A and C)
but enhanced LPS-induced Annexin V+PI- apoptotic cells (Fig. 5B and D). Thus, SB203580 treatment
reduced cell pyroptosis but increased cell apoptosis by blockage of
the p38 MAPK signaling pathway. LPS induced uncontrolled lung
inflammation, possibly by inducing more cell pyroptosis and less
cell apoptosis through p38 MAPK signaling, but this needs to be
investigated further.
Additional analysis by western blot revealed that
the blockage of p38 MAPK signaling pathway suppressed LPS-induced
p38 MAPK activation (Fig. 6A) and
inhibited the LPS-induced upregulation of NLRP3 and TLR2 proteins
(Fig. 6B), consistent with the
results of pyroptosis analyzed by flow cytometry (Fig. 5). However, higher expression levels
of cleaved caspase-3 were detected in the cells treated with both
SB203580 and LPS (Fig. 6C), which
was in line with the results analyzed by flow cytometry,
demonstrating a larger Annexin V+PI- apoptotic cell population
following SB203580 pretreatment (Fig.
5B and D).
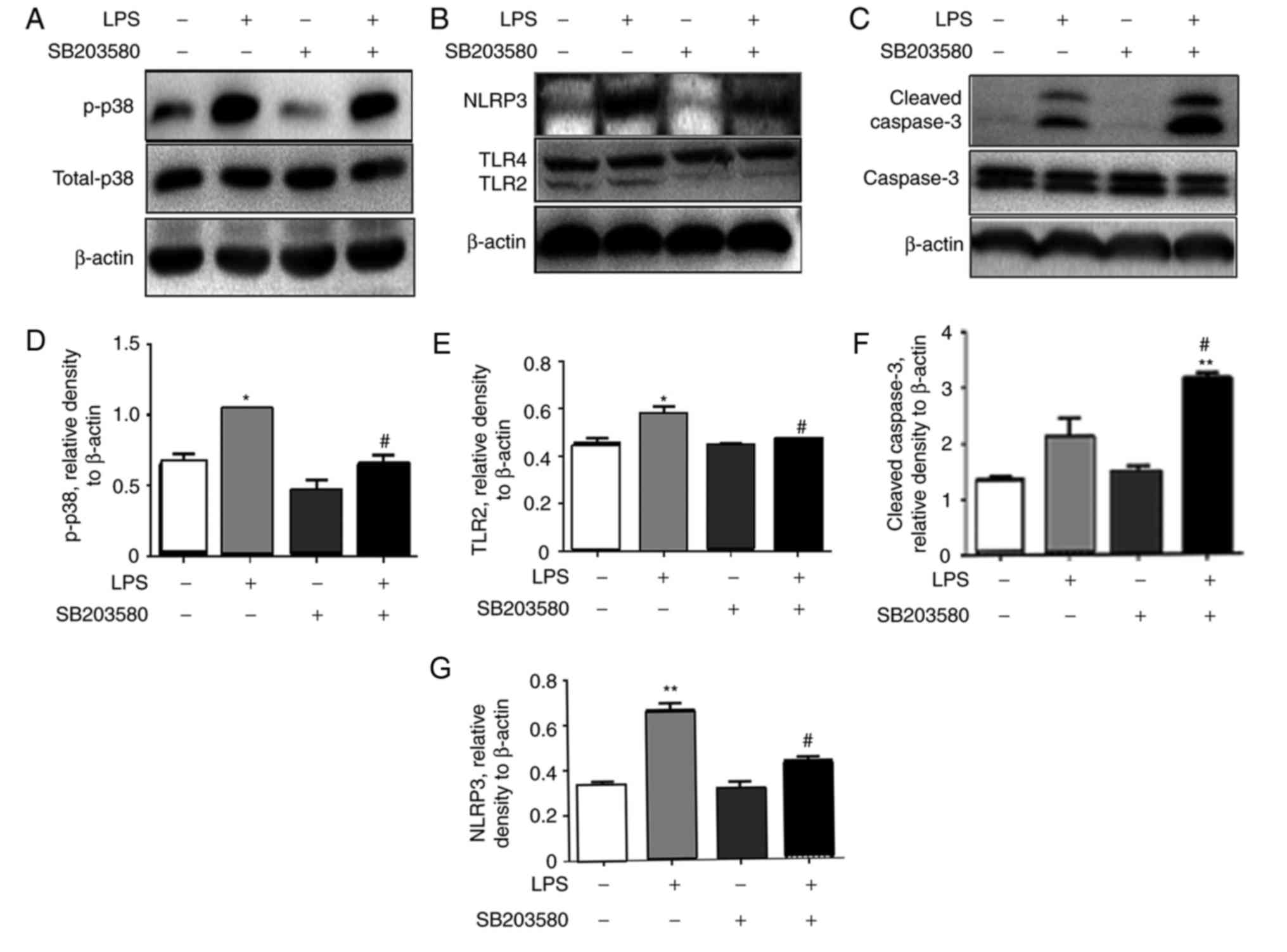 | Figure 6.p38 MAPK inhibitor SB203580
suppresses the expression of NLRP3 and cleavage of caspase-1 in
vitro. The rat macrophage cell line NR8383 was treated with 1
µg/ml LPS for 4 h with/without pretreatment with 10 µM SB203580.
Protein expression levels of (A) p-p38, (B) NLRP3 and TLR2/TLR4 and
(C) caspase-3 and cleaved-caspase-3, were analyzed by western blot
analysis. Quantitative analysis for (D) p-p38, (E) TLR2, (F)
cleaved-caspase-3 and (G) NLRP3 was performed, and data are
presented as the mean ± standard error of the mean. *P<0.05,
**P<0.01 vs. the untreated control. #P<0.05 vs.
the LPS treated group. n=3 per sample. LPS, lipopolysaccharide;
MAPK, mitogen-activated protein kinase; NLRP, nucleotide-binding
domain, leucine-rich-containing family, pyrin domain-containing; p,
phosphorylated; TLR, Toll-like receptor. |
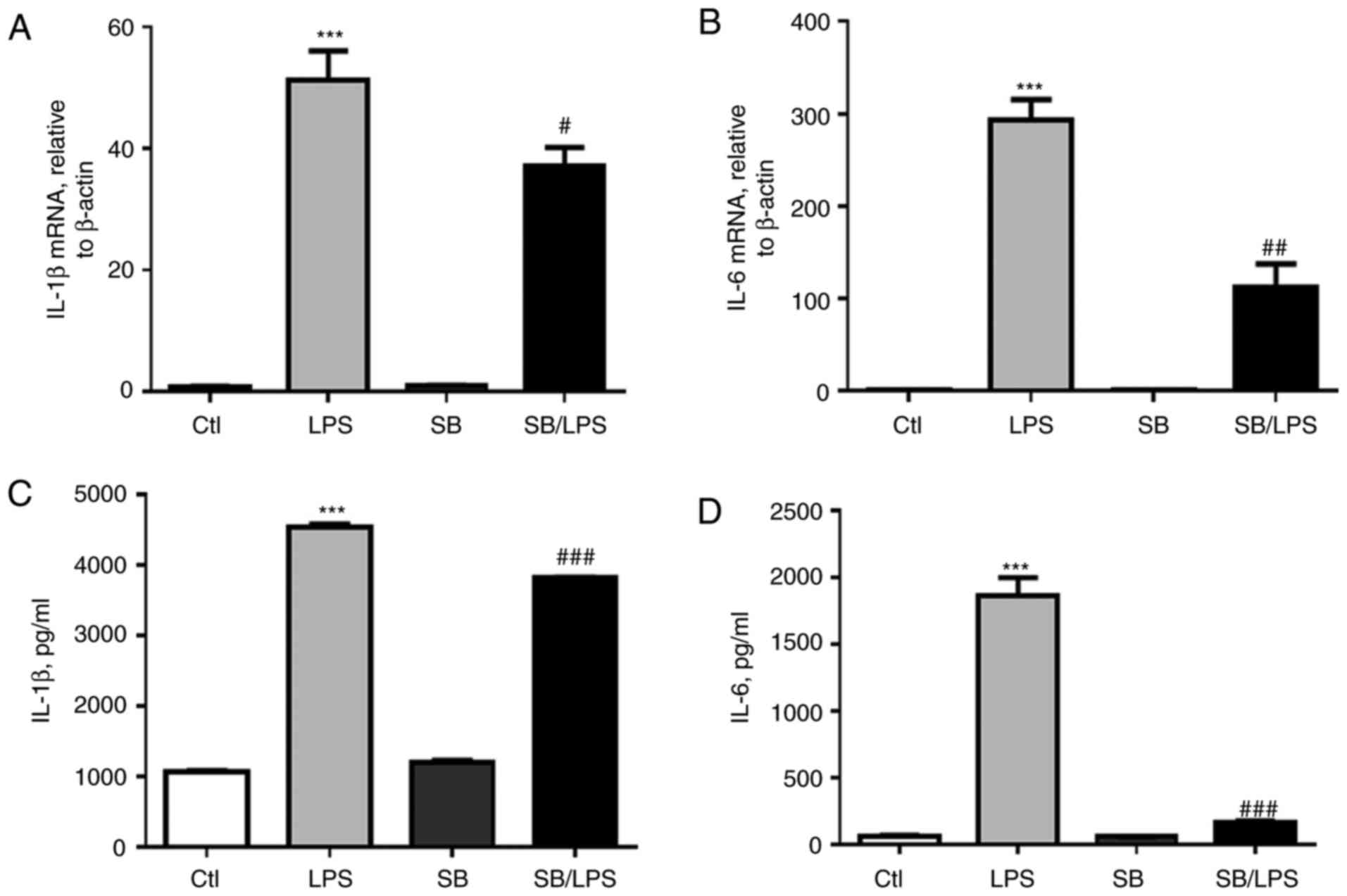
Further analysis of cytokines in the supernatant and
the cell lysate of the treated cells indicated that LPS treatment
induced higher mRNA expression levels of IL-1β and IL-6 (Fig. 7A and B, respectively) and higher
protein expression levels (Fig. 7C and
D). However, the elevated expression of IL-1β and IL-6 were
significantly suppressed by SB203580 pretreatment (Fig. 7; P<0.05), further supporting the
effects of p38 MAPK blockage in reducing macrophage pyroptosis
in vitro, but this needs to be validated further.
Discussion
Our previous study revealed that LPS can upregulate
the expression of Toll-like receptor 4 (TLR4), myeloid
differentiation protein 2 (MD-2), TNF-α, IL-6 and IL-1β in the rat
alveolar macrophage cell line NR8383 (29,30)
because IL-1β is mainly released from activated macrophages
(31) and is a downstream cytokine
of activation of inflammasome and pyroptosis (28). Therefore, it was hypothesized that
LPS may induce release of IL-1β through macrophage pyroptosis,
which is partially responsible for the uncontrolled lung
inflammation in ALI/ARDS. To address this issue, in the present
study, the effects of LPS on macrophage pyroptosis and lung
inflammation was investigated in vitro and in vivo.
The in vivo results indicated that LPS i.t. treatment
significantly increased excessive lung inflammation and neutrophil
infiltration, in association with increased caspase-1 cleavage and
active IL-1β release. A similar result was also observed in
RAW264.7 and NR8383 macrophage cell lines, in which NLRP3 and
cleaved caspase-1 were upregulated and colocalized within the
intracellular NLRP3 inflammasome following LPS treatment. Thus, LPS
not only induced excessive lung inflammation through increasing
macrophage and neutrophil recruitment but also caused a
pyroptosis-biased macrophage death and subsequent IL-1β
release.
The p38 MAPK signaling pathway has been suggested to
be involved in the inflammatory response of ALI/ARDS (20,21).
The production of many cytokines, including IL-1β, TNF-α and IL-6,
which serve a key role in ALI/ARDS is mainly via the p38 MPAK
signaling pathway (32). A number
of anti-inflammatory medications work by targeting p38 MAPK
(33,34). Inhibition of p38 MAPK downregulated
the IL-1β and reduced acute injury in intestinal ischemia
reperfusion rats (35). In
addition, blocking p38 downregulated the apoptosis of endothelia or
epithelial cells producing protective effects on pulmonary
alveolar-capillary barrier permeability (36–38).
However, the exact mechanism of the p38 MAPK signaling pathway in
inflammatory response and regulation remains to be elucidated.
There are relatively few studies on the regulation of p38 MAPK for
NLRP3 inflammasome and alveolar macrophage pyroptosis in the
ALI/ARDS. In a recent study, NLRP3 gene knockout blocked NF-κB and
the MAPK signaling pathway in a chronic unpredictable mild
stress-induced depression mouse model (39). The results in the present study
indicated that LPS treatment activated NLRP3 and IL-1β expression
and activation through the p38 MAPK signaling pathway because
blocking the p38 MAPK signaling pathway through SB203580
pretreatment reversed the LPS-induced upregulation of TLR2, NLRP3,
caspase-1, IL-1β and IL-6 in NR8383 cells. The suppressed p38 MAPK
signaling pathway was associated with less caspase-1+PI+ pyroptotic
cell population as analyzed by flow cytometry.
Thus, the blockage of p38 MAPK signaling pathway has
the potential to suppress excessive lung inflammation by inhibiting
inflammatory macrophage pyroptosis-biased cell death. This finding
may encourage further studies of the beneficial role of p38 MAPK
signal blockade in inhibiting uncontrolled lung inflammation in the
ALI/ARDS animal model. I.p. pretreatment with SB203580
significantly suppressed acute lung injury, which was characterized
by reduced alveolar edema, hemorrhage, alveolar septal thickening
and infiltration of polymorphonuclear leukocytes compared with the
mice that received LPS treatment alone. Consistent with the
pathological observation, NLRP3 and TLR2 expression and caspase-1
cleavage in lung tissue were significantly reduced following
SB203580 pretreatment. In addition, IL-1β, TNF-α and IL-6
expression levels were attenuated, supporting the role of p38 MAPK
signaling in LPS-induced acute lung injury and excessive lung
inflammation. Previous studies indicated that TLR2/TLR3/TLR4/NF-κB
signaling pathway was involved in NLRP3 inflammasome activation in
LPS-induced macrophages (40,41).
However, notable decreases in TLR4 expression levels was not
evident, suggesting the possibility of reduced involvement of the
TLR4/NF-κB signaling pathway in p38 MAPK blockade.
It should be noted that greater cell apoptosis was
observed following treatment with both LPS and p38 MAPK inhibitor
compared with LPS treatment alone. The results were demonstrated by
a larger Annexin V+PI- cell population and greater cleavage of
pro-caspase-3 in the apoptotic cells treated with both p38 MAPK
inhibitor and LPS. Apoptosis can prevent the leakage of
pro-inflammatory mediator, proteases, reactive oxygen species
production and lysozymes which contribute to promote and maintain
self-tolerance and the resolution of lung inflammation (42,43).
Alveolar macrophages release the cytokines IL-8, TNF-α and IL-6
which not only directly induce lung injury but also recruit the
neutrophils into the lung tissue as chemokines to aggravate the
damage (44,45). In contrast to the results in
endothelial or epithelial cells (36–38),
in macrophages it has been demonstrated that blocking the p38 MAPK
signaling pathway enhances macrophage apoptosis and cleavage of
caspase-3. The results suggested that inhibition of the p38 MAPK
signaling pathway may promote a shift in macrophage cell death from
proinflammatory pyroptosis towards a noninflammatory apoptosis
process which may contribute to the attenuated acute lung injury
and excessive inflammation in ALI/ARDS. However, it is important to
maintain an optimal balance between pyroptosis and apoptosis during
disease progression and this needs further research.
In conclusion, blocking the p38 MAPK signaling
pathway may ameliorate acute lung injury and lung inflammation,
partially through suppressing macrophage pyroptosis (Fig. 8). The results of the present study
may provide a rationale for ALI/ARDS immunotherapy through
modulation of p38 MAPK signaling pathway and macrophage
pyroptosis.
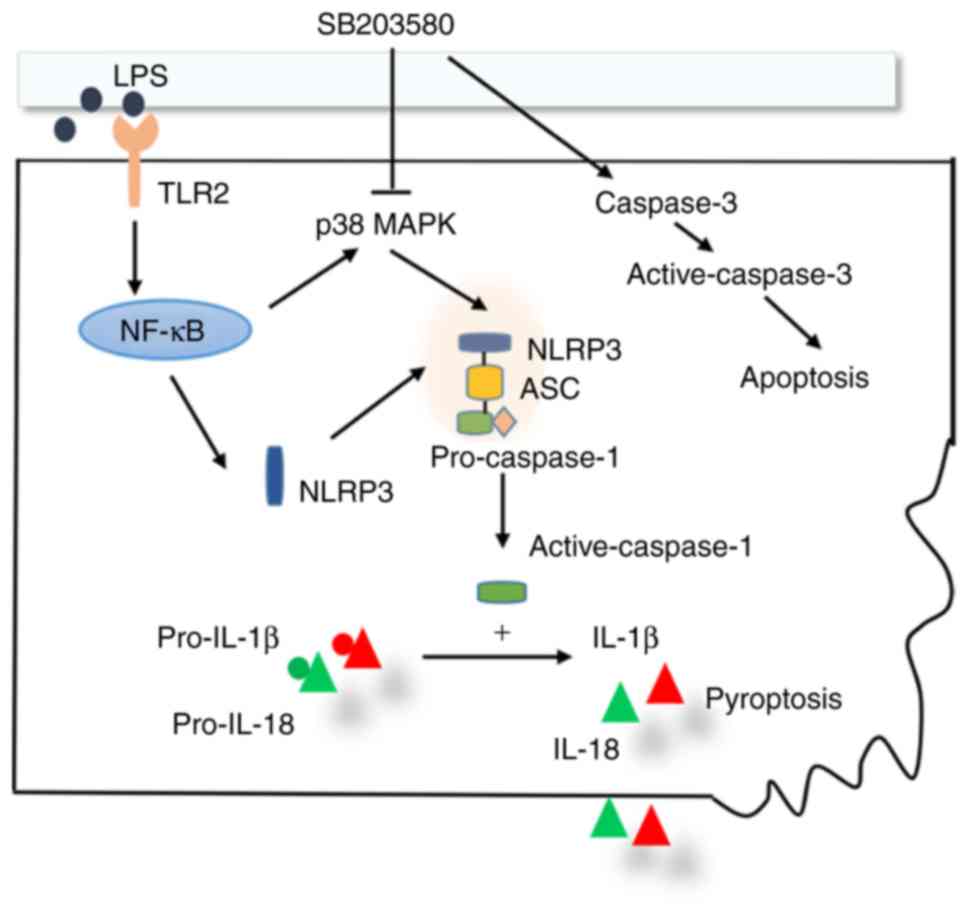 | Figure 8.Schematic diagram of the p38 MAPK
signaling pathway in the regulation of the NLRP3 inflammasome and
macrophage pyroptosis. LPS activates p38 MAPK signaling pathway in
macrophages through the TLR2/NF-κB signaling pathway, subsequently
upregulating protein expression and promoting the formation of the
NLRP3/caspase-1/ASC protein complex (NLRP3 inflammasome). The
activated NLRP3 inflammasome cleaves pro-IL-1β and pro-IL-18 to
become active IL-1β and IL-18, which ultimately induces macrophage
pyroptosis. The signaling pathway is suppressed by p38 MAPK
inhibitor SB203580. In addition, SB203580 upregulates caspase-3 and
enhances macrophage apoptosis. ASC, apoptosis-associated speck-like
protein; IL, interleukin; LPS, lipopolysaccharide; MAPK,
Mitogen-Activated Protein Kinase; NF-κB, nuclear
factor-κ-light-chain-enhancer of activated B cells; NLRP
leucine-rich-containing family, pyrin domain-containing; TLR,
Toll-like receptor. |
Acknowledgements
The authors would like to thank the Experimental
Center of Fudan University, Zhongshan Hospital (Shanghai, China)
for their assistance and cooperation during the present study.
Funding
The present study was supported by a research grant
from the National Natural Science Foundation of China and the
Shanghai Three-Year Plan of the Key Subjects Construction in Public
Health-Infectious Diseases and Pathogenic Microorganism to LZ
(grant nos. 81270137 and 81300054) and support from Zhongshan
Hospital, Fudan University in China to ZJ (grant no.
A654116001).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
DL performed the experiments, experimental design,
data assembly, analysis and manuscript writing. ZJ participated in
experimental design, data interpretation and manuscript writing. WR
participated in the generation of ideas and data interpretation. LZ
participated in the generation of the hypothesis and data
interpretation and was responsible for the overall direction of the
work. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocols were approved by the
Laboratory Animal Care and Use Committee at the Medical College of
Fudan University, Zhongshan Hospital (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
ARDS Definition Task Force, ; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin Definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
2
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Z, Chen L and Ni H: The
effectiveness of corticosteroids on mortality in patients with
acute respiratory distress syndrome or acute lung injury: A
secondary analysis. Sci Rep. 5:176542015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Frank JA, Wray CM, McAuley DF, Schwendener
R and Matthay MA: Alveolar macrophages contribute to alveolar
barrier dysfunction in ventilator-induced lung injury. Am J Physiol
Lung Cell Mol Physiol. 291:L1191–L1198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Machado-Aranda D, V Suresh M, Yu B,
Dolgachev V, Hemmila MR and Raghavendran K: Alveolar macrophage
depletion increases the severity of acute inflammation following
nonlethal unilateral lung contusion in mice. J Trauma Acute Care
Surg. 76:982–990. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Niesler U, Palmer A, Fröba JS, Braumüller
ST, Zhou S, Gebhard F, Knöferl MW and Seitz DH: Role of alveolar
macrophages in the regulation of local and systemic inflammation
after lung contusion. J Trauma Acute Care Surg. 76:386–393. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
D'Alessio FR, Craig JM, Singer BD, Files
DC, Mock JR, Garibaldi BT, Fallica J, Tripathi A, Mandke P, Gans
JH, et al: Enhanced resolution of experimental ARDS through
IL-4-mediated lung macrophage reprogramming. Am J Physiol Lung Cell
Mol Physiol. 310:L733–L746. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang Z, Zhou Q, Gu C, Li D and Zhu L:
Depletion of circulating monocytes suppresses IL-17 and HMGB1
expression in mice with LPS-induced acute lung injury. Am J Physiol
Lung Cell Mol Physiol. 312:L231–L242. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu Z, Hu J, Van den Steen PE and
Opdenakker G: Targeting matrix metalloproteinases in acute
inflammatory shock syndromes. Comb Chem High Throughput Screen.
15:555–570. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gustot T: Multiple organ failure in
sepsis: Prognosis and role of systemic inflammatory response. Curr
Opin Crit Care. 17:153–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovarova M, Hesker PR, Jania L, Nguyen M,
Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP and Koller
BH: NLRP1-dependent pyroptosis leads to acute lung injury and
morbidity in mice. J Immunol. 189:2006–2016. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang J, Jung Y, Tighe RM, Xie T, Liu N,
Leonard M, Gunn MD, Jiang D and Noble PW: A macrophage
subpopulation recruited by CC chemokine ligand-2 clears apoptotic
cells in noninfectious lung injury. Am J Physiol Lung Cell Mol
Physiol. 302:L933–L940. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Q, Imamura R, Motani K, Kushiyama H,
Nagata S and Suda T: Pyroptotic cells externalize eat-me and
release find-me signals and are efficiently engulfed by
macrophages. Int Immunol. 25:363–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Compan V, Martín-Sánchez F, Baroja-Mazo A,
López-Castejón G, Gomez AI, Verkhratsky A, Brough D and Pelegrín P:
Apoptosis-associated speck-like protein containing a CARD forms
specks but does not activate caspase-1 in the absence of NLRP3
during macrophage swelling. J Immunol. 194:1261–1273. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lamkanfi M, Sarkar A, Vande Walle L,
Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD and Dixit
VM: Inflammasome-dependent release of the alarmin HMGB1 in
endotoxemia. J Immunol. 185:4385–4392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kamo N, Ke B, Ghaffari AA, Shen XD,
Busuttil RW, Cheng G and Kupiec-Weglinski JW: ASC/caspase-1/IL-1β
signaling triggers inflammatory responses by promoting HMGB1
induction in liver ischemia/reperfusion injury. Hepatology.
58:351–362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Allam R, Kumar SV, Darisipudi MN and
Anders HJ: Extracellular histones in tissue injury and
inflammation. J Mol Med (Berl). 92:465–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haggadone MD, Grailer JJ, Fattahi F,
Zetoune FS and Ward PA: Bidirectional crosstalk between C5a
receptors and the NLRP3 inflammasome in macrophages and monocytes.
Mediators Inflamm. 2016:13401562016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiong LL, Tan Y, Ma HY, Dai P, Qin YX,
Yang RA, Xu YY, Deng Z, Zhao W, Xia QJ, et al: Administration of
SB239063, a potent p38 MAPK inhibitor, alleviates acute lung injury
induced by intestinal ischemia reperfusion in rats associated with
AQP4 downregulation. Int Immunopharmacol. 38:54–60. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma L, Zhao Y, Wang R, Chen T, Li W, Nan Y,
Liu X and Jin F: 3,5,4′-Tri-O-acetylresveratrol attenuates
lipopolysaccharide-induced acute respiratory distress syndrome via
MAPK/SIRT1 pathway. Mediators Inflamm. 2015:1430742015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McGuigan RM, Mullenix P, Norlund LL, Ward
D, Walts M and Azarow K: Acute lung injury using oleic acid in the
laboratory rat: Establishment of a working model and evidence
against free radicals in the acute phase. Curr Surg. 60:412–417.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Geng Y, Ma Q, Liu YN, Peng N, Yuan FF, Li
XG, Li M, Wu YS, Li BL, Song WB, et al: Heatstroke induces liver
injury via IL-1β and HMGB1-induced pyroptosis. J Hepatol.
63:622–633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the nomenclature committee on cell
death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lamkanfi M and Dixit VM: Mechanisms and
functions of inflammasomes. Cell. 157:1013–1022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ren WY, Zhu L, Hua F, Jin JJ and Cai YY:
The effect of lipopolysaccharide on gene expression of TLR4 and
MD-2 in rat alveolar macrophage and its secretion of inflammation
cytokines. Zhonghua Jie He He Hu Xi Za Zhi. 33:367–371. 2010.(In
Chinese). PubMed/NCBI
|
|
30
|
Ren W, Hu L, Hua F, Jin J, Wang Y and Zhu
L: Myeloid differentiation protein 2 silencing decreases
LPS-induced cytokine production and TLR4/MyD88 pathway activity in
alveolar macrophages. Immunol Lett. 141:94–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Netea MG, Simon A, van de Veerdonk F,
Kullberg BJ, Van der Meer JW and Joosten LA: IL-1beta processing in
host defense: Beyond the inflammasomes. PLoS Pathog.
6:e10006612010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bode JG, Ehlting C and Häussinger D: The
macrophage response towards LPS and its control through the
p38(MAPK)-STAT3 axis. Cell Signal. 24:1185–1194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu W, Jiang HL, Cai LL, Yan M, Dong SJ
and Mao B: Tanreqing injection attenuates
lipopolysaccharide-induced airway inflammation through MAPK/NF-κB
signaling pathways in rats model. Evid Based Complement Alternat
Med. 2016:52923462016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen CC, Lin MW, Liang CJ and Wang SH: The
anti-inflammatory effects and mechanisms of eupafolin in
lipopolysaccharide-induced inflammatory responses in RAW264.7
macrophages. PLoS One. 11:e01586622016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng DY, Zhou M, Jin J, He M, Wang Y, Du
J, Xiao XY, Li PY, Ye AZ, Liu J and Wang TH: Inhibition of P38 MAPK
downregulates the expression of IL-1β to protect lung from acute
injury in intestinal ischemia reperfusion rats. Mediators Inflamm.
2016:93480372016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bai X, Fan L, He T, Jia W, Yang L, Zhang
J, Liu Y, Shi J, Su L and Hu D: SIRT1 protects rat lung tissue
against severe burn-induced remote ALI by attenuating the apoptosis
of PMVECs via p38 MAPK signaling. Sci Rep. 5:102772015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Q, Wang J, Hu M, Yang Y, Guo L and Xu
J, Lei C, Jiao Y and Xu J: Uncoupling protein 2 increases
susceptibility to lipopolysaccharide-induced acute lung injury in
mice. Mediators Inflamm. 2016:91542302016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu X, Zhu Q, Niu F, Zhang R, Wang Y, Wang
W, Sun D, Wang X and Wang A: A2BAR activation attenuates acute lung
injury by inhibiting alveolar epithelial cell apoptosis both in
vivo and in vitro. Am J Physiol Cell Physiol. Jun 13–2018.(Epub
ahead of print). View Article : Google Scholar
|
|
39
|
Su WJ, Zhang Y, Chen Y, Gong H, Lian YJ,
Peng W, Liu YZ, Wang YX, You ZL, Feng SJ, et al: NLRP3 gene
knockout blocks NF-κB and MAPK signaling pathway in CUMS-induced
depression mouse model. Behav Brain Res. 322:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiang P, Chen T, Mou Y, Wu H, Xie P, Lu G,
Gong X, Hu Q, Zhang Y and Ji H: NZ suppresses TLR4/NF-κB signalings
and NLRP3 inflammasome activation in LPS-induced RAW264.7
macrophages. Inflamm Res. 64:799–808. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Borges PV, Moret KH, Raghavendra NM,
Maramaldo Costa TE, Monteiro AP, Carneiro AB, Pacheco P, Temerozo
JR, Bou-Habib DC, das Graças Henriques M and Penido C: Protective
effect of gedunin on TLR-mediated inflammation by modulation of
inflammasome activation and cytokine production: Evidence of a
multitarget compound. Pharmacol Res. 115:65–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Robb CT, Regan KH, Dorward DA and Rossi
AG: Key mechanisms governing resolution of lung inflammation. Semin
Immunopathol. 38:425–428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Linkermann A, Stockwell BR, Krautwald S
and Anders HJ: Regulated cell death and inflammation: An
auto-amplification loop causes organ failure. Nat Rev Immunol.
14:759–767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chollet-Martin S, Jourdain B, Gibert C,
Elbim C, Chastre J and Gougerot-Pocidalo MA: Interactions between
neutrophils and cytokines in blood and alveolar spaces during ARDS.
Am J Respir Crit Care Med. 154:594–601. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams AE and Chambers RC: The mercurial
nature of neutrophils: Still an enigma in ARDS? Am J Physiol Lung
Cell Mol Physiol. 306:L217–L230. 2014. View Article : Google Scholar : PubMed/NCBI
|