Introduction
Ischemia-reperfusion injury (IRI) can induce acute
hepatic injury, which is associated with a high level of mortality
(1,2). The liver is subjected to IRI in
different medical conditions, including hepatic resection and liver
transplantation (3). Notably, IRI
is not only a pathophysiological process involving liver injury; it
also affects multiple organs (4).
However, the exact intrinsic mechanism of hepatic injury caused by
IRI is still unknown.
Recent advances have demonstrated that complement
activation is responsible for the pathogenesis of IRI-induced acute
hepatic injury (5). The membrane
attack complex C5b-9 is formed during complement pathway activation
in IRI-induced acute hepatic injury, leading to lysis and
destruction of target cells. Additionally, anaphylatoxins C3a and
C5a are released and interact with their receptors, causing
activation of the inflammatory and apoptotic pathways (6,7).
Inhibition of abnormal complement activation potentially alleviates
IRI-induced acute hepatic injury (8).
Cobra venom factor (CVF) is a stable anticomplement
protein from cobra venom. It is a structural and functional analog
of the human complement component C3b (the active fragment of C3).
Similar to C3b, CVF binds to factor B, which is then cleaved by
factor D to form the bimolecular complex CVF/Bb; and CVF/Bb is a
C3/C5 convertase that cleaves both complement components C3 and C5
(9–11). Therefore, CVF/Bb can continuously
activate C3 and C5 to deplete complement components and inhibit its
activation, achieving the desired anticomplement effect.
Several studies have demonstrated that CVF
pretreatment can alleviate acute lung injury, myocardial injury and
cerebral injury induced by IRI, and prolong the survival time
following organ transplantation (12–15).
The aim of the current study was to investigate the effect and
mechanism of CVF pretreatment on IRI-induced acute hepatic injury
in rats. The results indicated that CVF pretreatment significantly
attenuated hepatic injury through depletion of completion
activation and subsequent inhibition of inflammatory mediator
release and hepatic cell apoptosis. However, further studies are
required to determine whether this therapy may be a potential agent
for the treatment of IRI injuries in clinical settings.
Materials and methods
Establishment of the rat model of
IRI-induced hepatic injury
All of the experiments were performed as approved by
the Laboratory Animal Care Committee of Tianjin First Center
Hospital (approval no. 2015013Z). One hundred and seventy six-week
old male and female Sprague Dawley rats (200–250 g; 1:1 ratio) were
obtained from the Experimental Animal Center, Academy of Military
Medical Sciences (Beijing, China; Animal qualified batch number:
SCXK-2007-004). These rats were housed in the rooms of the
Laboratory Animal Care of Tianjin First Center Hospital with
specified pathogen-free conditions. The room was maintained at room
temperature under relative humidity 40–60%. Food and water were
provided ad libitum.
To simulate a clinical traumatic hemorrhagic shock
situation, the rat model of IRI-induced acute hepatic injury was
successfully established, as described previously (16). Briefly, rats were anesthetized by
intraperitoneal injection of chloral hydrate (200 mg/kg), as
described previously (17,18). An open mid-diaphyseal transverse
fracture was created in the left femur to induce trauma, after
which a mean arterial blood pressure of 30±5 mmHg was maintained
for 90 min via hemorrhage at 2.5 ml/100 g. Subsequently, the rats
were resuscitated for a period of 20 min with Ringer's lactate
solution at a constant rate to manage the shock. The volume of
Ringer's solution was twice that of the blood loss. Finally, the
catheters were removed, and the vessels were ligated.
Treatment design
The experimental rats were randomly and equally
divided into five groups: i) The control group underwent the same
anesthetic process without surgical procedures; and the following
four groups all have acute hepatic injury was induced by bone
fracture to simulate trauma, followed by hemorrhage, and then were
resuscitated of reperfusion as described above: ii) the IRI group;
iii) the CVF1 group received a high-dose (100 µg/kg) CVF (cat. no.
C8406, Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) pretreatment
at 24 h before the surgery + IRI; iv) the CVF2 group received
low-dose (50 µg/kg) CVF pretreatment at 24 h before the surgery +
IRI; and v) the CVF3 group received low-dose (50 µg/kg) CVF at day
2 after IRI. The CVF frozen powder was dissolved in normal
saline.
Survival time analyses
A total 170 rats were used for survival time
assessment and the intrinsic mechanism determination. Firstly, the
effect of CVF administration on survival time was assessed in the
five groups described above (n=10 per group), and survival was
defined as the time between the first treatment and the time of rat
death or 48 h follow-up for censored rats. Subsequently, 120 rats
were used for intrinsic mechanism determined among the control,
IRI, and CVF2 + IRI groups. In each group, 10 rats were sacrificed
at 1, 3, 6, and 24 h after IRI, and the hepatic tissues and at
least 4 ml blood samples were collected and analyzed.
Histological analyses
For hematoxylin-eosin (H&E) staining, the
hepatic tissues were fixed with at least 15 ml 10% neutral
formaldehyde solution for ≥24 h. Subsequently, the samples were
dehydrated, embedded in paraffin, sectioned into 4-µm-thick slices,
and mounted onto slides. Following heating at 60°C for 2 h, slicing
and dewaxing at room temperature for 10 min, H&E staining was
conducted at room temperature for 10 min, and the slides were
observed under an Olympus BX40F light microscope (Olympus
Corporation, Tokyo, Japan).
For the immunohistochemical assay, the 4-µm-thick
hepatic tissue sections were deparaffinized, rehydrated, and
treated with 3% H2O2 at 20°C for 10 min.
Antigen retrieval was performed by boiling the sections in
Tris/EDTA Antigen Retrieval Solution (pH 8.0) for 2 min.
Nonspecific antibody binding was blocked using 3% bovine serum
albumin at 20°C for 30 min. Subsequently, the tissue sections were
incubated with anti-C5b-9 antibody (cat. no. ab55811; 1:500),
anti-Bcl-2 associated X, apoptosis regulator (Bax) antibody (cat.
no. ab32503; 1:500), anti-Bcl-2 apoptosis regulator (Bcl-2)
antibody (cat. no. ab59348; 1:500; all Abcam, Cambridge, UK), or
control IgG overnight at 4°C. The bound antibodies were detected
with horseradish peroxidase (HRP)-conjugated secondary antibodies
(cat. no. ab205718; 1:100; Abcam) at room temperature for 1 h, and
the products were visualized using a 3, 30′-diaminobenzidine
tetrahydrochloride (DAB) staining kit (Tiangen Biotech Co., Ltd.,
Beijing, China) at room temperature for 2 min and counterstained
with hematoxylin at room temperature for 5 min. The specimens were
examined with an inverted fluorescence microscope (IX50; Olympus
Corporation, Tokyo, Japan).
In addition, the expression levels of C5b-9
deposition in specimens were assessed by two independent
researchers who were double-blinded manner. Their conclusions were
in complete agreement for 85% of specimens indicating this scoring
method was highly reproducible. If two agreed with the scoring
results, the value was selected. If there was a disagreement, then
the third researcher worked collaboratively to confirm the score.
For evaluation of C5b-9 deposition, a semi-quantitative scoring
criterion was used, in which both staining intensity and positive
areas were recorded from 0 to 100.
ELISA
For the ELISA, the serum samples were centrifuged at
3,000 r/min (12,000 × g) for 5 min at 4°C and were stored at −20°C
until use. Alanine transaminase (ALT; cat. no. EIA-325), aspartate
transaminase (AST; cat. no. EIA-326), total hemolytic complement
(CH50; cat. no. EIA-121), C5b-9 (cat. no. EIA-765), tumor necrosis
factor (TNF)-α (cat. no. SRTA00), interleukin (IL)-1β (cat. no.
DY501) and C5a (cat. no. DY2150) were measured using the monoclonal
anti-rat capture antibody (ZSGB-BIO, Inc., Beijing, China and
R&D Systems, Inc., Minneapolis, MN, USA), according to the
manufacturer's instructions. The colorimetric reaction was measured
using a Benchmark microplate reader (Benchmark Electronics, Inc.,
Scottsdale, AZ, USA).
Semi-quantitative reverse
transcriptase-polymerase chain reaction (RT-PCR)
Guided by the manufacturer's instructions, total RNA
was extracted using the TRIzol reagent (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Following measurement of RNA
concentration, reverse transcription was performed using a
SuperScript RT kit (Takara Biotechnology Co., Ltd., Dalian, China)
under the following conditions: 37°C for 15 min, 85°C for 5 sec,
and then maintained at 4°C. RT-PCR was performed in a 25 µl
reaction volume: 1 µl diluted cDNA, 1 µl sense primer (10 pmol/µl),
1 µl antisense primer (10 pmol/µl), MgCl2 0.3 µl, 12.5
µl Premix EX Taq II (2X), and 9.2 µl RNase-free d H2O.
Following mixing, the cDNAs were synthesized under the following
conditions: 94°C for 2 min, 94°C for 30 sec, 60°C for 1 min at 40
cycles, 72°C for 30 sec, 72°C for 10 min, and 10°C for 10 min by
PCR. The primers for gene amplification were synthesized by
Shenggong Biological Engineering Technology and Services (Shanghai,
China): Bax sense, 5′-ACAGATCATGAAGACAGGGG-3′ and antisense,
5′-CAAAGTAGAAGAGGGCAACC-3; Bcl-2 sense, 5′-TTCTCCTTCCAGCCTGAGAGC-3′
and antisense, 5′-ATGACCCCACCGAACTCAAAG-3′; GAPDH sense,
5′-GGTGAAGGTCGGTGTGAACG-3′ and antisense,
5′-CTCGCTCCTGGAAGATGGTG-3′. GAPDH was used as an internal control.
The fidelity of all RT-PCR assays was assessed by 1.5% agarose gel
electrophoresis containing 20 g/l TAE buffer, and the relative gene
expression levels were calculated relative to GAPDH.
Protein extraction and western
blotting
Western blot analysis was executed to measure the
protein levels of C5a, caspase-3 and cleaved caspase-3. Briefly,
the hepatic tissues were crushed, and the total protein was
extracted from the cell lysate using T-PER™ Tissue Protein
Extraction Reagent (cat. no. 78510; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), which was prepared with an inhibitor cocktail
against proteases and phosphatases. Then the protein concentration
in samples was measured using the Pierce™ BCA Protein Assay kit
(cat. no. 23227; Thermo Fisher Scientific, Inc.). An equivalent
amounts of protein (80 µg) were separated SDS-PAGE on 15% gels and
transferred to a polyvinylidene difluoride membrane at 4°C.
Following this, the membranes were blocked for 2 h at room
temperature using Tris-buffered saline containing 0.1% Tween-20 and
5% skimmed milk at room temperature. The membrane was then
incubated at 4°C overnight with anti-C5a antibody (cat. no.
ab194637; Abcam; 1:500), caspase-3 antibody (cat. no. ab13847;
1:100), cleaved caspase-3 antibody (cat. no. ab2302; 1:500), or
anti-GAPDH antibody (cat. no. ab49822; 1:500; all Abcam).
Subsequently, the membranes were incubated with the goat anti-mouse
(cat. no. ab6789; 1:5,000) and anti-rabbit secondary antibodies
(cat. no. ab191866; 1:500; both Abcam) for 2 h at room temperature.
Antibody binding was detected using an electrochemiluminescence
detection kit to produce a chemiluminescence signal, which was
captured on X-ray film. Relative protein expression levels were
calculated relative to GAPDH.
Terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate nick end labeling
(TUNEL) assay
A TUNEL assay kit was used to detect hepatic cell
apoptosis as recommended by the manufacturer (Roche Diagnostics,
Basel, Switzerland). The number of apoptotic liver cells was
counted in five random fields at magnification, ×400 and five
consecutive sections per liver tissue.
Statistical analysis
Quantitative data are expressed as the mean ±
standard deviation. Two groups of comparison were analyzed using
the unpaired Student's t-test, and multigroup comparisons were
performed by one-way analysis of variance) test with post hoc
contrasts by the Student-Newman-Keuls method. Survival time curves
were estimated using Kaplan-Meier analysis and compared using the
stratified log-rank test. Data were analyzed using SPSS 22.0 for
Windows (IBM Corp., Armonk, NY, USA). P<0.05 (two-tailed) was
considered to indicated a statistically significant difference.
Results
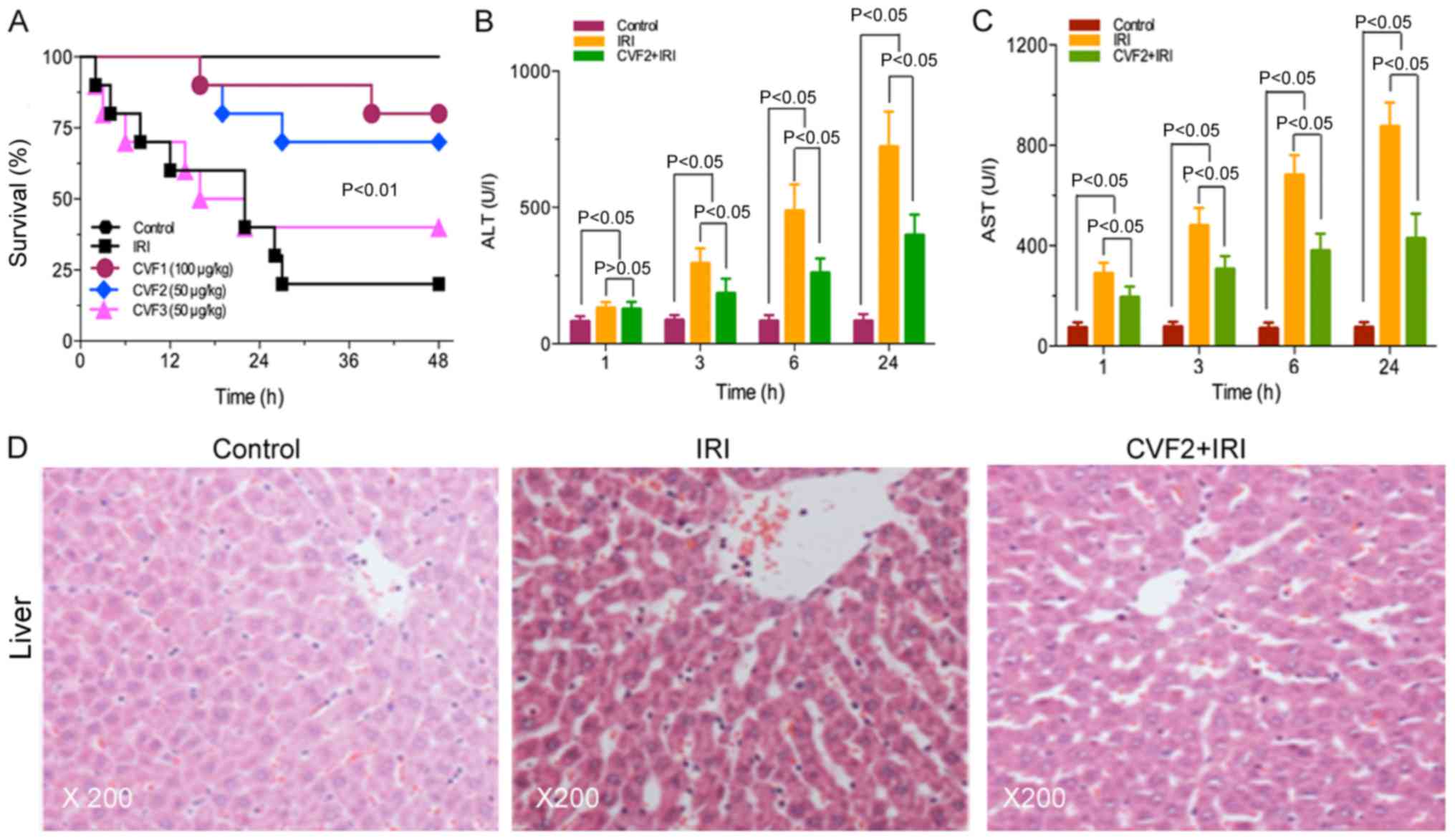
CVF pretreatment improves the
prognosis of rats with IRI
No rats in the control group died. The survival
times were significantly prolonged in the high-dose and low-dose
CVF groups (CVF1 and CVF2, respectively), compared with the IRI
group (all P<0.01). However, no difference was observed between
the CVF1 and CVF2 groups following IRI (P>0.05). Additionally,
when CVF was administered at day 2 (CVF3 group) after IRI, the
survival time was not improved compared with the IRI group
(P>0.05) (Fig. 1A).
CVF pretreatment attenuates the
hepatic injury of rats with IRI
The low-dose CVF pretreatment (CVF2) was used for
subsequent experiments because the survival times were not
significantly difference between the high-dose and low-dose CVF
groups following IRI (P>0.05). In the CVF2 + IRI group, the
levels of ALT and AST were significantly attenuated, compared with
the IRI group at 3, 6, and 24 h (all P<0.05); however, the
levels of ALT and AST in the CVF2 + IRI group remained
significantly increased compared with the control group (all
P<0.05) (Fig. 1B and C).
Furthermore, the H&E staining results of the
hepatic tissues of the control, IRI, and CVF2 + IRI groups (24 h)
are presented in Fig. 1D. No
hepatic cell injury was detected in the control group. However, the
IRI group exhibited albuminoid and vacuolar degeneration,
eosinophilic variants, spotty liver necrosis, inflammatory cell
infiltration and Kupffer cell proliferation; while only focal
eosinophilic variants of hepatic cells were observed in the CVF2 +
IRI group.
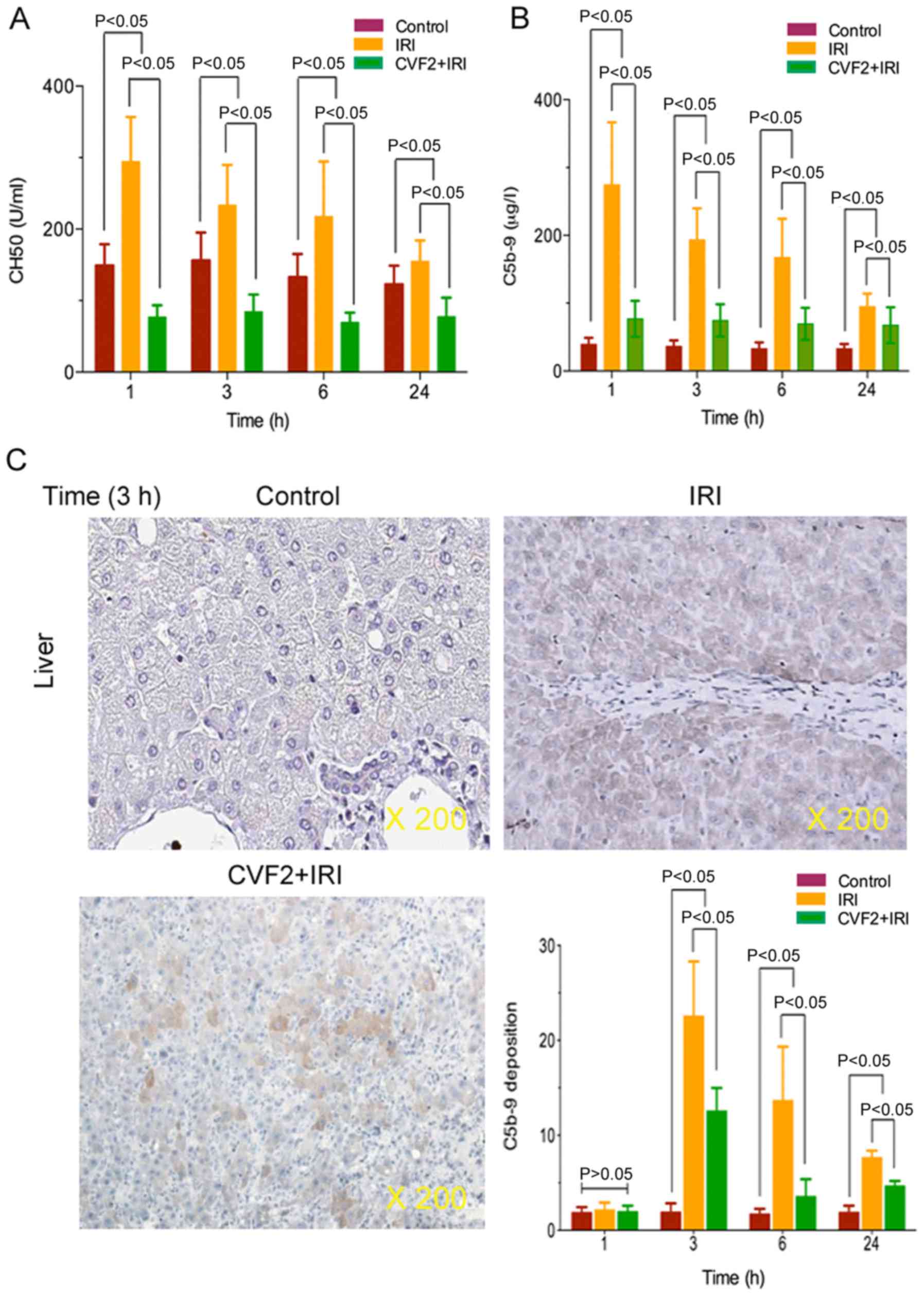
CVF pretreatment results in complement
and membrane attack complex depletion
To verify that CVF pretreatment induced complement
and membrane attack complex depletion, plasma complement CH50 and
C5b-9 activities were determined in the control, IRI and CVF2 + IRI
groups. The ELISA results demonstrated that IRI significantly
increased complement CH50 and C5b-9 expression, and the expression
levels reached a peak at 1 h after IRI initiation, compared with
the control group (all P<0.05) (Fig. 2A and B). When IRI rats received
CVF2 pretreatment, the CH50 and C5b-9 activities were significantly
decreased compared with those of the rats in the IRI group within
24 h (all P<0.05) (Fig. 2A and
B); however, there was no significant difference in CH50 and
C5b-9 levels between the IRI group and the CVF2 + IRI group at
>24 h.
Additionally, immunohistochemistry assay
demonstrated that the level of C5b-9 deposition was significantly
reduced in the CVF2 + IRI group at 3, 6, and 24 h, compared with
the IRI group. However, the level of C5b-9 deposition was decreased
in the CVF2 + IRI group compared with IRI rats; however, the C5b-9
deposition remained significantly higher than the control group
(all P<0.05) (Fig. 2C).
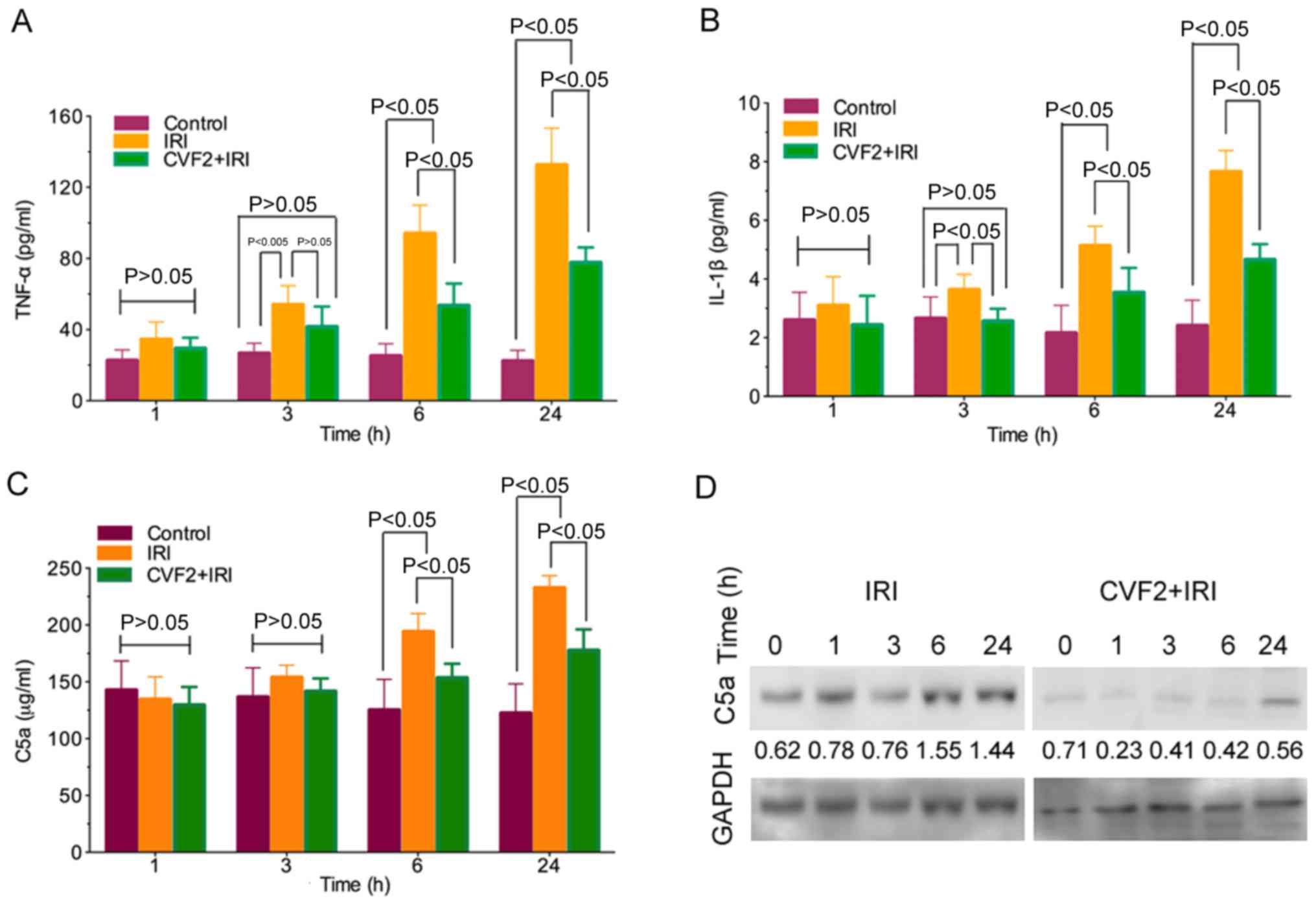
CVF pretreatment reduces the release
of inflammatory mediators
The ELISA results demonstrated that TNF-α and IL-1β
release was significantly increased in the IRI group compared with
the control group (all P<0.05). Following CVF2 pretreatment, the
levels of inflammatory mediator release were significantly reduced
at 3, 6, and 24 h (all P<0.05) (Fig. 3A and B).
Activation of the complement system generates
anaphylatoxin C5a. In the IRI group, C5a expression was
significantly increased compared with the control group at 6 and 24
h (all P<0.05). By contrast, pretreatment with CVF2 attenuated
the elevation of C5a expression in the IRI group at 6 and 24 h
(P<0.05) (Fig. 3C). Consistent
with the ELISA outcomes, the C5a protein level in the liver tissues
within 24 h were also significantly reduced by CVF2 pretreatment,
compared with the IRI group, according to western blot analysis
(Fig. 3D).
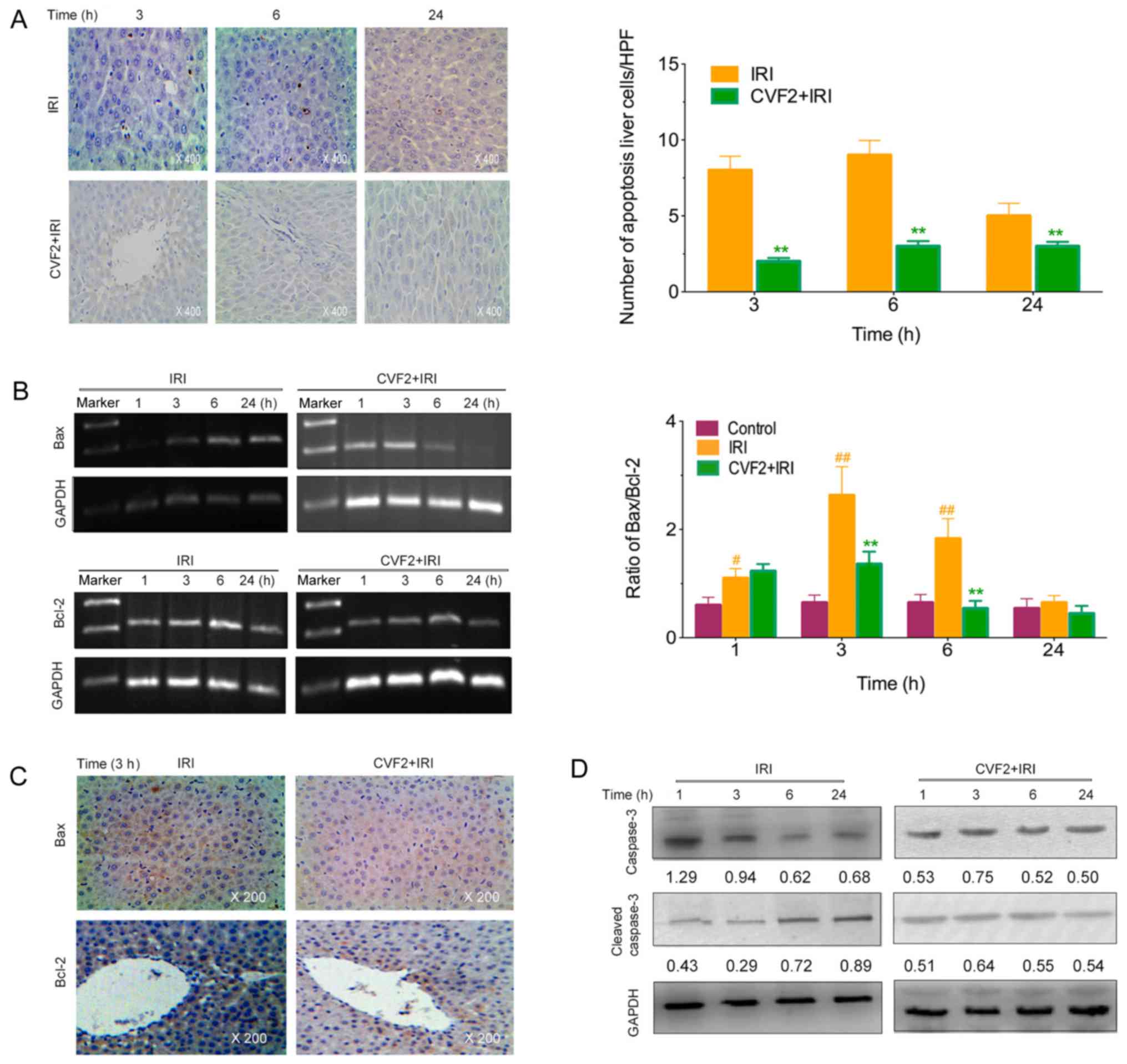
CVF pretreatment alleviates the
apoptosis of liver cells
The levels of apoptotic hepatic cells were observed
using a TUNEL assay. No apoptotic cells were detected in the
control group. However, the IRI rats exhibited a marked increase in
apoptotic liver cells; whereas CVF2 pretreatment significantly
reduced the hepatic cell apoptosis at 3, 6 and 24 h (all P<0.05)
(Fig. 4A).
Compared with the IRI group, the Bax expression and
Bax/Bcl-2 ratio were significantly lower in the CVF2 group within
24 h (all P<0.05; Fig. 4B and
C). Consistent with the reduced Bax expression and Bax/Bcl-2
ratio, pretreatment with CVF2 markedly attenuated the elevation of
cleaved caspase-3 expression within 24 h (Fig. 4D).
Discussion
IRI can induce acute hepatic injury, which is
associated with high mortality rates. The liver is subjected to IRI
in several medical conditions, including hepatic resection and
liver transplantation. Increasing evidence has demonstrated that
complement activation is required for IRI-induced hepatic damage
(5–8), and CVF can deplete the complements of
the component system (9–11). The current study investigated the
effect and intrinsic mechanism of CVF pretreatment on acute hepatic
injury induced by IRI in rats, and the results indicated that CVF
pretreatment ameliorates IRI-induced acute hepatic injury. However,
further studies are required to determine whether this therapy
could be a potential agent for the treatment of IRI injuries in
clinical settings.
In the present study, the survival time of rats was
significantly prolonged in the high-dose and low-dose CVF
pretreatment groups (CVF1 and CVF2 groups, respectively), compared
with the IRI group. CVF administration at day 2 (CVF3 group) after
IRI did not improve the survival time, compared with the IRI group.
These results suggested the involvement of complement activation at
the early stage, not the late stage, following IRI. In addition,
either high-dose or low-dose CVF pretreatment improved the survival
time of rats with IRI.
In the current study, when IRI was initiated, the
liver function significantly deteriorated compared to the control
group; while CVF pretreatment significantly improved the liver
function. However, the exact intrinsic mechanism of CVF
pretreatment in attenuating hepatic injury following IRI is remains
unknown. In the present study, the CH50 and C5b-9 levels were
significantly increased, and their expression levels reached a peak
at 1 h after IRI initiation. These results were in agreement with
previous studies (14), suggesting
that CVF pretreatment results in complement and membrane attack
complex depletion, and that complement activation is involved in
the pathogenesis of hepatic injury following IRI.
Notably, when the IRI rats received CVF
pretreatment, the CH50 and C5b-9 activities were significantly
decreased within 24 h, but not at >24 h, compared with the rats
in the IRI group. These results suggested that systemic complement
activity was gradually restored. The return of complement activity
may have been due to the development of antibodies against CVF or
due to complement regeneration in the liver. Therefore, the window
of time available for therapeutic intervention to block
complement-mediated events in the post-IRI period may be
important.
The mechanism by which complement activity
contributes to hepatic injury remains unresolved. Figueroa et
al (13) has demonstrated that
CVF administration may allow accommodation to take place by
impairing the ability of anaphylatoxin C5a to form. Given that the
release of inflammatory mediators such as TNF-α and IL-1β was
significantly decreased in the IRI rats receiving CVF pretreatment,
inflammatory mediator release is potentially actively involved in
complement activation (12). In
addition, considering that the number of apoptotic of liver cells
was significantly increased in the rats with IRI, it is likely that
apoptosis may participate in complement activation by mediating
caspase activity, as reported in Gorsuch et al study
(5).
In conclusion, CVF pretreatment ameliorated
IRI-induced acute hepatic injury in rats. The results of this study
were consistent with those of previous research (12–15)
may result in novel therapies based on the strategy of complement
inhibition to ameliorate multiple organ injury resulting from IRI.
Several clinically applicable complement inhibitors are in
development or in preclinical trials for various disorders
(19). Continued research may
yield data justifying clinical trials of one or more complement
inhibitors administered during or shortly after events
characterized by IRI.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the
Foundation of Tianjin Heath and Family Planning Commission (grant
nos. 14KG101 and 2014KR07), and the National Clinical Key Specially
Project Foundation of the Ministry of Health in China (grant no.
2011-873) to Professor Yong-Qiang Wang.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
BW carried out conception and design, performed
experimental study, collection and assembly of data, data analysis
and interpretation, manuscript writing. HX, JL and HMG performed
the experimental study, collection and assembly of data, data
analysis and interpretation. YHX, ZL and HJL performed collection
and assembly of data, data analysis and interpretation. YQW and SHC
carried out conception and design, collection and assembly of data,
data analysis and interpretation, manuscript writing. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were performed according
to the Animal Care Committee guidelines, and the experimental
protocol was approved by the Ethics Committee of Tianjin First
Center Hospital (approval no. 2015013Z).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cannistrà M, Ruggiero M, Zullo A, Gallelli
G, Serafini S, Maria M, Naso A, Grande R, Serra R and Nardo B:
Hepatic ischemia reperfusion injury: A systematic review of
literature and the role of current drugs and biomarkers. Int J
Surg. 33 Suppl 1:S57–S70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peralta C, Jiménez-Castro MB and
Gracia-Sancho J: Hepatic ischemia and reperfusion injury: Effects
on the liver sinusoidal milieu. J Hepatol. 59:1094–1106. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abe Y, Hines IN, Zibari G, Pavlick K, Gray
L, Kitagawa Y and Grisham MB: Mouse model of liver ischemia and
reperfusion injury: Method for studying reactive oxygen and
nitrogen metabolites in vivo. Free Radic Biol Med. 46:1–7. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pantazi E, Bejaoui M, Folch-Puy E, Adam R
and Roselló-Catafau J: Advances in treatment strategies for
ischemia reperfusion injury. Expert Opin Pharmacother. 17:169–179.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gorsuch WB, Chrysanthou E, Schwaeble WJ
and Stahl GL: The complement system in ischemia-reperfusion
injuries. Immunobiology. 217:1026–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bajic G, Degn SE, Thiel S and Andersen GR:
Complement activation, regulation, and molecular basis for
complement-related diseases. EMBO J. 34:2735–2757. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Melis JP, Strumane K, Ruuls SR, Beurskens
FJ, Schuurman J and Parren PW: Complement in therapy and disease:
Regulating the complement system with antibody-based therapeutics.
Mol Immunol. 67:117–130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krishnan V, Ponnuraj K, Xu Y, Macon K,
Volanakis JE and Narayana SV: The crystal structure of cobra venom
factor, a cofactor for C3- and C5-convertase CVFBb. Structure.
17:611–619. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vogel CW and Fritzinger DC: Cobra venom
factor: Structure, function, and humanization for therapeutic
complement depletion. Toxicon. 56:1198–1222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vogel CW, Finnegan PW and Fritzinger DC:
Humanized cobra venom factor: Structure, activity, and therapeutic
efficacy in preclinical disease models. Mol Immunol. 61:191–203.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mao YF, Yu QH, Zheng XF, Liu K, Liang WQ,
Wang YW, Deng XW and Jiang L: Pre-treatment with cobra venom factor
alleviates acute lung injury induced by intestinal
ischemia-reperfusion in rats. Eur Rev Med Pharmacol Sci.
17:2207–2217. 2013.PubMed/NCBI
|
|
12
|
Gorsuch WB, Guikema BJ, Fritzinger DC,
Vogel CW and Stahl GL: Humanized cobra venom factor decreases
myocardial ischemia-reperfusion injury. Mol Immunol. 47:506–510.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Figueroa E, Gordon LE, Feldhoff PW and
Lassiter HA: The administration of cobra venom factor reduces
post-ischemic cerebra injury in adult and neonatal rats. Neurosci
Lett. 380:48–53. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vogel CW, Fritzinger DC, Gorsuch WB and
Stahl GL: Complement depletion with humanized cobra venom factor:
Efficacy in preclinical models of vascular disease. Thromb Haemost.
113:548–552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun QY, Chen G, Guo H, Chen S, Wang WY and
Xiong YL: Prolonged cardiac xenograft survival in guinea pig-to-rat
model by a highly active cobra venom factor. Toxicon. 42:257–262.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui YL, Qiu LH, Zhou SY, Li LF, Qian ZZ,
Liu XM, Zhang HL, Ren XB and Wang YQ: Necroptosis as a potential
therapeutic target in multiple organ dysfunction syndrome.
Oncotarget. 8:56980–56990. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silverman J and Muir WW III: A review of
laboratory animal anesthesia with chloral hydrate and chloralose.
Lab Anim Sci. 43:210–216. 1993.PubMed/NCBI
|
|
18
|
Zimpfer M, Silt SP and Vatner SF: Effect
of anesthesia on the canine carotid chemoreceptor reflex. Circ Res.
48:400–406. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bahde R and Spiegel HU: Hepatic
ischemia-reperfusion injury from bench to beside. Br J Surg.
97:1461–1475. 2010. View
Article : Google Scholar : PubMed/NCBI
|