Introduction
Nephrotic syndrome (NS) is a renal disorder
characterized by a tetrad of clinical conditions, including
proteinuria, hypoalbuminemia, hyperlipidemia and edema (1). It is one of the most prevalent
defective states of the glomerular filtration barrier, whereby
macromolecules, including albumin, are excreted in the urine.
Worldwide prevalence is reported to be ~1–3 out of 100,000 children
under the age of 16 (2,3). Congenital nephrotic syndrome (CNS)
represents a dysfunctional glomerular state that is presented in
utero or during the first three months of life; contrary to
infantile nephrotic syndrome, in which disease symptoms appear at
4–12 months of age (4). Cases in
which the symptoms of NS manifest in children aged between 2–5
years old are classified as childhood NS.
As patients with NS typically respond well to
steroid therapy, NS is characterized as either steroid resistant
nephrotic syndrome (SRNS), which occurs in ~20% of children with
NS, or as steroid susceptible nephrotic syndrome, occurring in ~80%
of patients (5,6). SRNS manifests as a glomerular
disorder that may lead to end-stage renal disease (7–9). A
great deal of pathogenic genetic defects for CNS have been reported
since 1998 (10). Over the last 15
years, mutations in >39 dominant or recessive genes have been
revealed to cause NS or SRNS, including KN motif and ankyrin repeat
domains 1, 2 and 4; nephrin, podocin, nucleoporin 93, 107 and 205;
and Wilms- tumor 1 (11–14).
Next generation sequencing (NGS) is a technique that
can effectively identify genetic defects underlying inherited
disorders with high precision and reliability, which consequently
increases the amount of known genetic mutations. Whole exome
sequencing (WES) in NGS is a powerful tool used to identify
underlying genetic elements (15,16)
without any prior knowledge of the gene or genes involved in the
pathogenesis (17). The
pathological genetic mutations in a variety of human genetic
disorders have been identified by WES (18,19).
However, there are a limited number of reports that have identified
genetic mutations that result in NS in the Saudi population, using
microarray and direct sequencing techniques (20,21).
The aim of the present study was to utilize WES in a Saudi
consanguineous family suffering from SRNS to identify the
underlying genetic defects, in order to broaden the disease
understanding and improve disease management. To the best of our
knowledge, this is the first report from Saudi Arabia in which WES
was performed to identify novel mutations in the phospholipase C
ε-1 (PLCE1) gene in a family with segregating SRNS.
Patients and methods
Ethical approval and patient
recruitment
The Ethical Review Committee of Taibah University
(Medina, Saudi Arabia) approved the protocol of the present study
(approval no. TU-REC-2016018). Informed written consent was
obtained from all participants prior to study commencement.
Experiments were performed in the Centre for Genetics and Inherited
Diseases, Taibah University (Medina, Saudi Arabia).
A Saudi family was recruited in September 2015. In
total, the family included five members (IV:1, IV:2, IV:3, IV:5 and
IV:7) with SRNS. All individuals with SRNS as well as seven healthy
individuals (III:1, III:2, III:3, III:4, IV:4, IV:6, IV:8)
belonging to the family were clinically examined by a nephrologist
in the Department of Nephrology, Madinah Maternity and Children
Hospital (Medina, Kingdom of Saudi Arabia). Clinical reports of
three affected individuals (IV:1, IV:3, IV:7) were obtained;
however, DNA samples were not available, as these individuals
succumbed to mortality prior to the initiation of the genetic
studies. The family pedigree was drawn upon querying the elders of
the family. The following parameters were used to diagnose
patients: Proteinuria or spot urine protein:creatinine ratio, serum
albumin, peripheral edema on clinical examination and
hyperlipidemia (total blood cholesterol).
Genomic DNA extraction
Blood samples were obtained for genetic analysis
from the affected individuals (IV:2, IV:5), asymptomatic siblings
(IV:4, IV:6) and the parents (III:1, III:2). Furthermore, blood
samples (3 ml) were collected from 30 healthy individuals (aged
5–27 years old; 15 males and 15 females) in the Centre for Genetics
and Inherited Diseases, Taibah University (Medina, Saudi Arabia).
These individuals were not related to the family; however, they
were from the same population. Blood vacutainers containing EDTA
were used to collect the blood specimens. QIAquick DNA extraction
kits (Qiagen, Inc., Valencia, CA, USA) were used to extract genomic
DNA from the whole blood samples. DNA quantification was performed
with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and Qubit fluorometer (Thermo Fisher Scientific,
Inc.). DNA integrity was resolved through 1% agarose gel
electrophoresis.
Whole exome sequencing (WES)
WES was performed using DNA from the two affected
individuals (IV:2 and IV:5) of the family. Library preparation and
exome enrichment was performed using the Nextra Rapid Capture Exome
kit (Illumina, Inc., San Diego, CA, USA), which captured 214,405
exons and splice sites with 98.3% RefSeq coverage. Subsequently,
the Illumina NextSeq500 instrument (Illumina, Inc.) was used to
produce clusters and DNA sequence reads, as described previously
(22).
The Illumina NextSeq500 instrument generated bcl
files that were converted to fastq files using the BCL2FASTQ tool
(Illumina, Inc.). BaseSpace (Illumina, Inc.; basespace.illumina.com/home/index) was used to align
fastq files to the reference genome using the Burrows-Wheeler
Aligner-MEM algorithm (23).
Variant calling was performed with the genome analysis toolkit
(24). Finally, VariantStudio
software (version 3.0; Illumina, Inc.) was used for the annotation
of the variants obtained. Variant filtration and prioritization was
performed as described previously (18).
Sanger sequencing
Genetic variants that were exposed by WES were
further confirmed by Sanger sequencing as described by Basit et
al (25). The genomic sequence
of human PLCE1 gene (12,024 bp; ENST00000371380.7) was downloaded
from the Ensembl genome browser (asia.ensembl.org/index.html). In order to amplify and
validate the targeted sequence variant with their flanking sites,
primers (forward, 5-GAATAAGTTGTGCCGTTGCC-3- and reverse,
5-TTCAGGGAGGTTGTGAGTGG-3′) were designed using Primer3 software
(version 0.4.0; frodo.wi.mit.edu/primer3/). PCR master mix (Promega
Corporation, Madison, WI, USA) was used to amplify the target
region. Following this, amplified regions were resolved on a 2%
agarose gel and visualized using a Bio-Rad gel documentation system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The amplified
sequences were analyzed using BIOEDIT sequence alignment editor
(version 6.0.7; Ibis Biosciences, Inc., Carlsbad, CA, USA).
Thermocycling conditions used were as follows: 95°C for 1 min;
followed by 30 cycles of 95°C for 35 sec, 60°C for 35 sec and 70°C
for 3.5 min; followed by a single incubation at 70°C for 10
min.
Mutation prediction analysis
The Simple Modular Architecture Research Tool
(SMART) (26) was used to identify
the predicted effect of the identified single base pair insertion
(c.6272_6273insT) on the product of PLCE1 enzyme.
Results
Clinical evaluation of patients
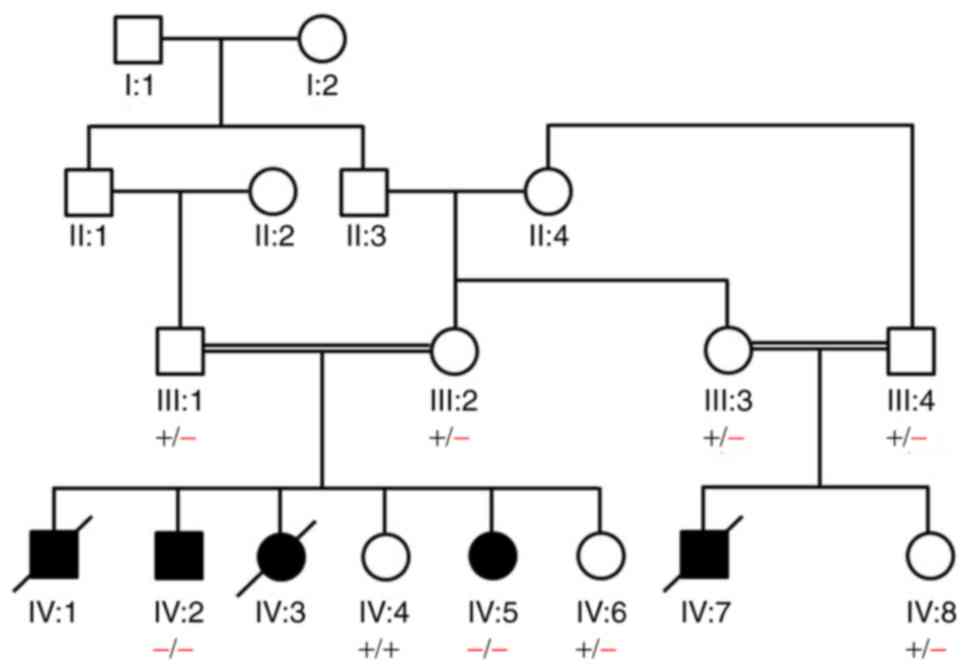
The family pedigree is presented in Fig. 1. The affected individuals were
clinically examined by a pediatric nephrologist in Madinah
Maternity and Children Hospital (Medina, Saudi Arabia). Blood and
urine investigations were used to diagnose patients: Proteinuria
(>3-3.5 g/24 h) or spot urine protein:creatinine ratio
(>300–350 mg/mmol); serum albumin (<25 g/l); and peripheral
edema on clinical examination and hyperlipidemia (total blood
cholesterol >10 mmol/l).
The affected son (IV:2) was evaluated at the age of
11 months and was determined to have severe pulmonary edema,
sarcoidosis, oliguria and severe hypertension. A kidney biopsy
revealed vasculitis. The overall presentation of this case was
diffuse mesangial sclerosis (DMS). Renal transplantation was
eventually performed for this patient, who is 21 years old at
present. The subject is living a normal life without disease
recurrence.
The affected daughter (IV:5) was admitted to the
hospital at the age of 2 years with a history of persistent
vomiting for two days and a low-grade fever without convulsion.
Furthermore, the patient had been unwell for the last two years
with a history of abnormal movements during sleep with
hyperpigmentation on the back. Laboratory investigations revealed
high blood pressure reaching 140/90 mm/Hg with anemia, and highly
elevated urea and creatinine levels in blood with imbalanced
electrolytes. The patient remained admitted to the hospital due to
chronic kidney disease and renal failure. Ultrasound scan of the
kidneys revealed an echogenic cortex with poor cortico-medullary
differentiation and no back pressure. A renal biopsy and transplant
was also performed for IV:5 and the patient is 16 years old at
present. Histology revealed mesangial matrix expansion,
hypertrophic podocytes and thickened basement membranes (data not
shown; Madinah Maternity and Children Hospital).
Three affected individuals (IV:1, IV:3 and IV:7) had
previously succumbed to mortality. Clinical reports show similar
phenotypic manifestations as found in IV:2 and IV:5. Interviews
with the elder members of the family confirmed that the deceased
individuals had similar clinical manifestations to the affected
living individuals (IV:2 and IV:5). Renal transplantation was not
performed for these deceased individuals. Blood and urine analysis
of the heterozygous carriers (parents) revealed a normal
proteinuria and protein:creatinine ratio range. As symptoms were
present from birth, patients under investigation were most likely
suffering from congenital NS.
Novel variant identification by WES
analysis
The resulting variant call format files obtained
from the fastq files contained ~80,000 variants. Different filters
were applied to these genomic variants, whilst considering any
previous links with the disease phenotype, potential effects on the
protein, genome frequency, quality and pathogenicity. Rare and
potentially harmful variants present in the homozygous state were
of primary interest given the reported positive consanguinity in
this family. Searching for rare homo/hemizygous variants within the
protein coding regions of all genes that have previously been
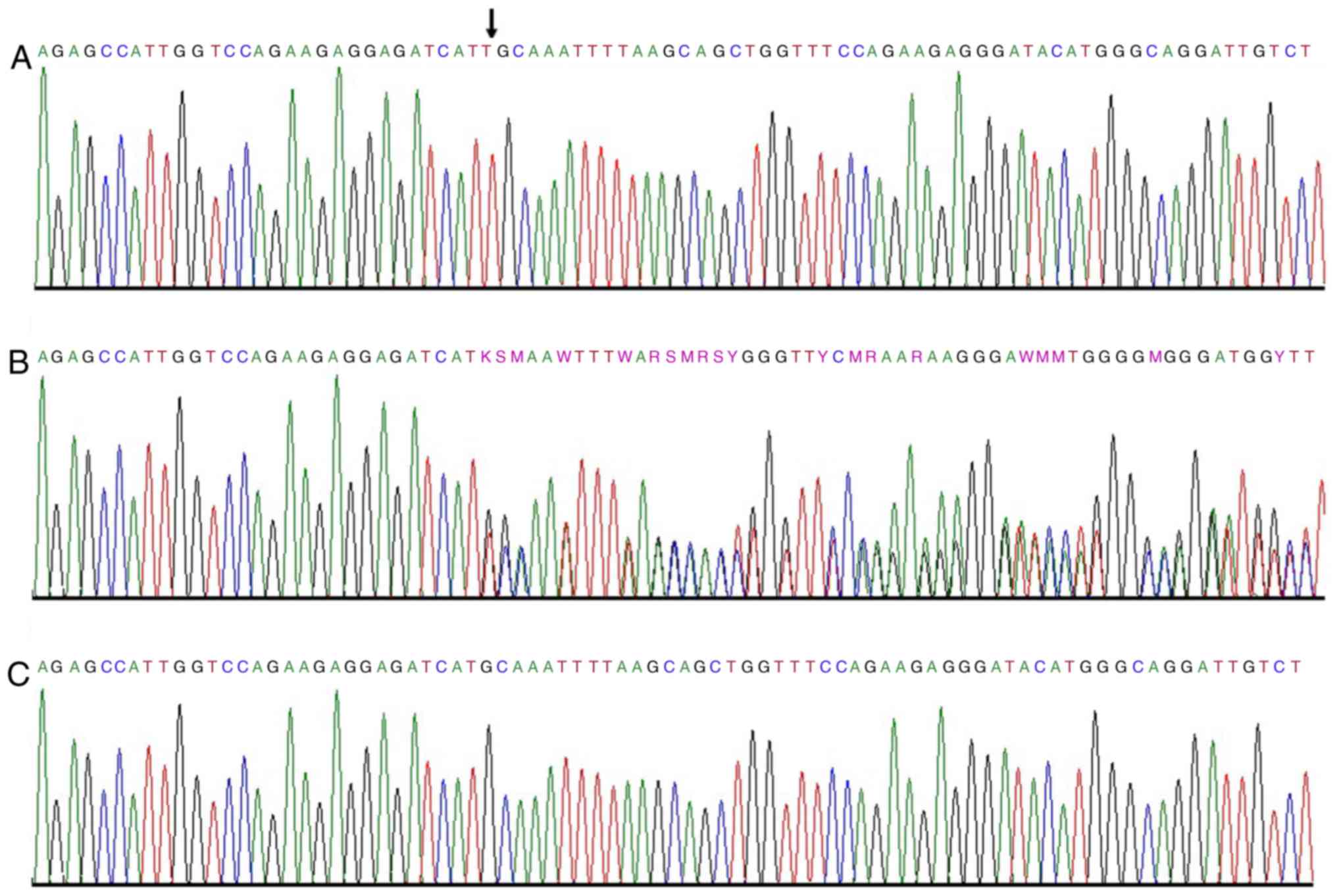
associated with NS yielded a reasonable candidate variant. A novel
homozygous frameshift variant (c.6272_6273insT;
p.2090Met_2091GlnPheSer) in exon 29 of the PLCE1 gene was detected
in the two affected individuals (Fig.
2). Recessive mutations in this gene cause early-onset
nephrotic syndrome (13,27). Taken together, this variant is
likely to contribute to the phenotype of these patients based on
its novelty, predicted deleterious effect and good phenotype
match.
c.6272_6273insT is inherited in an
autosomal recessive manner
The identified variant (c.6272_6273insT) in
PLCE1 gene was further validated through Sanger sequencing.
All members of the family were screened and it was determined that
the variant segregated in the family in an autosomal recessive
manner (Fig. 3). Furthermore, 30
unrelated, normal control individuals were screened from the same
population. Variant (c.6272_6273insT) was not found in the DNA
isolated from any of the control individuals.
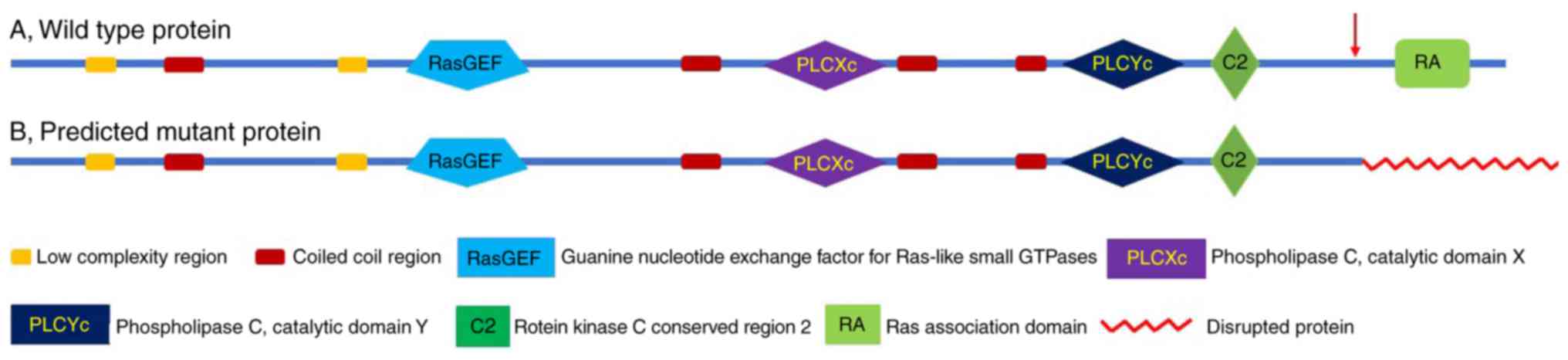
Mutant PCLE1 lacks a complete
Ras-association (RA) domain
SMART was used to predict the effect of the
identified single base pair insertion (c.6272_6273insT) on the
PLCE1 enzyme product. The insertion was determined to result in a
frameshift of the protein from position 2090Met_2091GlnPheSer, just
prior to the RA domain. The RA domain consists of 104 amino acids
and the mutant protein lacked the complete RA domain (Fig. 4).
Discussion
A novel homozygous insertion mutation was identified
in exon 29 of the PLCE1 gene in two siblings with SRNS using the
WES method. The resultant genetic variation results in a frameshift
of the genetic code from position 2090Met_2091GlnPheSer. PLCE1 is
one of the genes implicated in early onset NS.
The frameshift mutation identified in the present
study was predicted to cause inactivation of the RA domain that
subsequently leads to PLCE1 protein loss of function, resulting in
the aforementioned patient phenotype. Ras proteins function as
molecular switches, transmitting a signal in the active guanosine
5′-triphosphate (GTP)-bound state and reverting to an inactive
state when the bound GTP is hydrolyzed to guanosine 5′-diphosphate.
Ras interacts with the RA domain of PLCE1 to initiate downstream
signaling pathways. The congenital NS, in the present case, was
steroid resistant, as the patients did not respond to the
standardized steroid therapy.
NS is a heterogeneous genetic disorder. To date,
>39 different disease-causing genes have been identified in
individuals with syndromic and non-syndromic forms of NS (11–14,28).
When mutated, these genes lead to dysfunctional glomeruli and
subsequently, focal segmental glomerulosclerosis (29). The proteins encoded by these genes
are involved in either the development and/or function of the
glomerular filtration barrier.
NGS has provided a cost-effective platform for the
accurate analysis of a large number of genes. WES has broadened the
genetic heterogeneity and mutation continuum of the disease by
identifying novel candidate genes and novel mutations responsible
for NS (30).
The PLCE1 gene spans over 334.4 kb on chr10q23.33
and has 34 exons. The protein encoded by PLCE1 gene belongs to the
phospholipase family of proteins. In 2006, positional cloning was
used to identify mutations in PLCE1 responsible for a novel cause
of recessive NS type 3 (31).
Mutations in PLCE1 are associated with early onset NS (32). At present, 17 different mutations
have been identified in PLCE1.
PLCE1 protein is essential for the functional
development of the glomeruli at the capillary loop stage. The
identification of a novel pathogenic mutation in PLCE1 gene using
WES in a Saudi family with congenital nephrotic syndrome expands
the existing mutation spectrum of PLCE1 gene within the Saudi
population and worldwide. Besides its potential role in diagnosis
and disease pathogenicity, this discovery also highlighted the
importance of WES as a tool for the molecular diagnosis of NS.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Strategic
Technology Program of the National Plan for Science, Technology and
Innovation (Riyadh, Saudi Arabia; grant no. 13-MED2088-05).
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JAH performed variant validation using the Sanger
approach, and wrote the initial draft of the manuscript. RAS
recruited the family included in the present study and performed
clinical diagnosis. SA performed polymerase chain reactions using
the control samples. FA designed the primer sequences. AMA
extracted and quantified the genomic DNA, and performed exome
sequencing. ZI performed experiments investigating variant
identification. SB designed the study and analyzed exome data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in the present study were
approved by The Ethical Review Committee of Taibah University
(Medina, Saudi Arabia; approval no. TU-REC-2016018) and were also
in accordance with the Declaration of Helsinki. Written informed
consent was obtained from all individual participants or the
guardians of underage participants included in the study.
Patient consent for publication
All patients and unaffected participants have
provided consent for publication of genetic data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Franceschini N, North KE, Kopp JB,
McKenzie L and Winkler C: NPHS2 gene, nephrotic syndrome and focal
segmental glomerulosclerosis: A HuGE review. Genet Med. 8:63–75.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wong W: Idiopathic nephrotic syndrome in
New Zealand children, demographic, clinical features, initial
management and outcome after twelve-month follow-up: Results of a
three-year national surveillance study. J Paediatr Child Health.
43:337–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schlesinger ER, Sultz HA, Mosher WE and
Feldman JG: The nephrotic syndrome. Its incidence and implications
for the community. Am J Dis Child. 116:623–632. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cil O, Besbas N, Duzova A, Topaloglu R,
Peco-Antić A, Korkmaz E and Ozaltin F: Genetic abnormalities and
prognosis in patients with congenital and infantile nephrotic
syndrome. Pediatr Nephrol. 30:1279–1287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Niaudet P and Boyer O: Pediatric
Nephrology. 6th. Avner E..Harmon W..Niaudet P..Yoshikawa N.:
Springer; New York, NY, USA: pp. 667–702. 1968
|
|
6
|
The primary nephrotic syndrome in
children. Identification of patients with minimal change nephrotic
syndrome from initial response to prednisone. A report of the
International Study of Kidney Disease in Children. J Pediatr.
98:561–564. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Habib R, Lévy M and Gubler MC:
Clinicopathologic correlations in the nephrotic syndrome.
Paediatrician. 8:325–348. 1979.PubMed/NCBI
|
|
8
|
Rood IM, Deegens JK and Wetzels JF:
Genetic causes of focal segmental glomerulosclerosis: Implications
for clinical practice. Nephrol Dial Transplant. 27:882–890. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tarshish P, Tobin JN, Bernstein J and
Edelmann CM Jr: Prognostic significance of the early course of
minimal change nephrotic syndrome: Report of the International
Study of Kidney Disease in Children. J Am Soc Nephrol. 8:769–776.
1997.PubMed/NCBI
|
|
10
|
Kestilä M, Lenkkeri U, Männikkö M,
Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T,
Nissinen M, Herva R, et al: Positionally cloned gene for a novel
glomerular protein-nephrin-is mutated in congenital nephrotic
syndrome. Mol Cell. 1:575–582. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lovric S, Ashraf S, Tan W and Hildebrandt
F: Genetic testing in steroid-resistant nephrotic syndrome: When
and how? Nephrol Dial Transplant. 31:1802–1813. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gbadegesin R, Lavin P, Foreman J and Winn
M: Pathogenesis and therapy of focal segmental glomerulosclerosis:
An update. Pediatr Nephrol. 26:1001–1015. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sadowski CE, Lovric S, Ashraf S, Pabst WL,
Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J,
et al: A single-gene cause in 29.5% of cases of steroid-resistant
nephrotic syndrome. J Am Soc Nephrol. 26:1279–1289. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saleem MA: New developments in
steroid-resistant nephrotic syndrome. Pediatr Nephrol. 28:699–709.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dyment DA, Sawyer SL, Chardon JW and
Boycott KM: Recent advances in the genetic etiology of brain
malformations. Curr Neurol Neurosci Rep. 13:3642013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rabbani B, Mahdieh N, Hosomichi K, Nakaoka
H and Inoue I: Next-generation sequencing: Impact of exome
sequencing in characterizing Mendelian disorders. J Hum Genet.
57:621–632. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dixon-Salazar TJ, Silhavy JL, Udpa N,
Schroth J, Bielas S, Schaffer AE, Olvera J, Bafna V, Zaki MS,
Abdel-Salam GH, et al: Exome sequencing can improve diagnosis and
alter patient management. Sci Transl Med. 4:138ra782012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Basit S, Al-Harbi KM, Alhijji SA, Albalawi
AM, Alharby E, Eldardear A and Samman MI: CIT, a gene involved in
neurogenic cytokinesis, is mutated in human primary microcephaly.
Hum Genet. 135:1199–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alharby E, Albalawi AM, Nasir A, Alhijji
SA, Mahmood A, Ramzan K, Abdusamad F, Aljohani A, Abdelsalam O,
Eldardear A and Basit S: A homozygous potentially pathogenic
variant in the PAXBP1 gene in a large family with global
developmental delay and myopathic hypotonia. Clin Genet.
92:579–586. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Hamed M, Sayer JA, Al-Hassoun I,
Aldahmesh MA and Meyer B: A novel mutation in NPHS2 causing
nephrotic syndrome in a Saudi Arabian family. NDT Plus. 3:545–548.
2010.PubMed/NCBI
|
|
21
|
Al-Hamed MH, Al-Sabban E, Al-Mojalli H,
Al-Harbi N, Faqeih E, Al Shaya H, Alhasan K, Al-Hissi S, Rajab M,
Edwards N, et al: A molecular genetic analysis of childhood
nephrotic syndrome in a cohort of Saudi Arabian families. J Hum
Genet. 58:480–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hashmi JA, Al Harbi KM, Ramzan K, Albalwi
AM, Mehmood A, Samman MI and Basit S: A novel splice-site mutation
in the ASPM gene underlies autosomal recessive primary
microcephaly. Ann Saudi Med. 36:391–396. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. arXiv.
1303:39972013.
|
|
24
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high-confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 11:11.10.1. 11.10.33. 2013. View Article : Google Scholar :
|
|
25
|
Basit S, Malibari O, Al Balwi AM,
Abdusamad F and Ismail Abu F: A founder splice site mutation
underlies glycogen storage disease type 3 in consanguineous Saudi
families. Ann Saudi Med. 34:390–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Letunic I, Doerks T and Bork P: SMART:
Recent updates, new developments and status in 2015. Nucleic Acids
Res. 43:(Database Issue). D257–D260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al-Hamed MH, Al-Sabban E, Al-Mojalli H,
Al-Harbi N, Faqeih E, Al Shaya H, Alhasan K, Al-Hissi S, Rajab M,
Edwards N, et al: A molecular genetic analysis of childhood
nephrotic syndrome in a cohort of Saudi Arabian families. J Hum
Genet. 58:480–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tadano M, Edamatsu H, Minamisawa S,
Yokoyama U, Ishikawa Y, Suzuki N, Saito H, Wu D, Masago-Toda M,
Yamawaki-Kataoka Y, et al: Congenital semilunar valvulogenesis
defect in mice deficient in phospholipase C epsilon. Mol Cell Biol.
25:2191–2199. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogino D, Hashimoto T, Hattori M, Sugawara
N, Akioka Y, Tamiya G, Makino S, Toyota K, Mitsui T and Hayasaka K:
Analysis of the genes responsible for steroid-resistant nephrotic
syndrome and/or focal segmental glomerulosclerosis in Japanese
patients by whole-exome sequencing analysis. J Hum Genet.
61:137–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bullich G, Trujillano D, Santín S,
Ossowski S, Mendizábal S, Fraga G, Madrid Á, Ariceta G, Ballarín J,
Torra R, et al: Targeted next-generation sequencing in
steroid-resistant nephrotic syndrome: Mutations in multiple
glomerular genes may influence disease severity. Eur J Hum Genet.
23:1192–1199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hinkes B, Wiggins RC, Gbadegesin R,
Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins
BE, et al: Positional cloning uncovers mutations in PLCE1
responsible for a nephrotic syndrome variant that may be
reversible. Nat Genet. 38:1397–1405. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boyer O, Benoit G, Gribouval O, Nevo F,
Pawtowski A, Bilge I, Bircan Z, Deschênes G, Guay-Woodford LM, Hall
M, et al: Mutational analysis of the PLCE1 gene in steroid
resistant nephrotic syndrome. J Med Genet. 47:445–452. 2010.
View Article : Google Scholar : PubMed/NCBI
|