Introduction
Retinitis pigmentosa (RP) is an inherited retinal
dystrophy that causes adult blindness, and is characterized by the
degeneration of rod and cone photoreceptors. Typical symptoms
include night blindness at an early age and progressive
constriction of the mid-peripheral visual field. Patients with RP
eventually become completely blind. There are three major modes of
inheritance: Autosomal dominant (ADRP), autosomal recessive (ARRP)
and X-linked (XLRP) (1). In
addition, other modes of inheritance, including digenic or
mitochondrial inheritance, have been reported (2,3).
RP can also occur as a syndrome accompanying other
diseases. Usher syndrome is the most frequent form of syndromic RP,
characterized by RP accompanied by sensorineural hearing loss.
Usher syndrome can be divided into three types based on the
assessment of clinical features.
Given the complexity and perniciousness of RP,
molecular diagnosis is a potentially effective method of analysis
that can be used to diagnose RP and assess disease prognosis.
Although the traditional method, Sanger sequencing, can be a valid
strategy used to detect genetic abnormities, analyzing a large
number of candidate genes with this method is expensive (4). With the advance of whole-exome
sequencing technology, determining full-coding regions has become
faster (5). Whole-exome sequencing
is particularly useful in the detection of novel disease-causing
genes (6).
In the present study, one Chinese pedigree with ARRP
and one Chinese pedigree with XLRP were investigated. Whole-exome
sequencing was performed on the affected patients, identifying a
compound heterozygous mutation in usherin (USH2A) and a
heterozygous X-linked mutation in ARL3 GTPase-activating protein
(RP2). The mutations co-segregated with the diseases in the
pedigrees and, to the best of our knowledge, the pathogenic
mutation (c.6,485+5G>A) in USH2A has not been reported
previously among Chinese patients.
Materials and methods
Subjects and clinical assessment
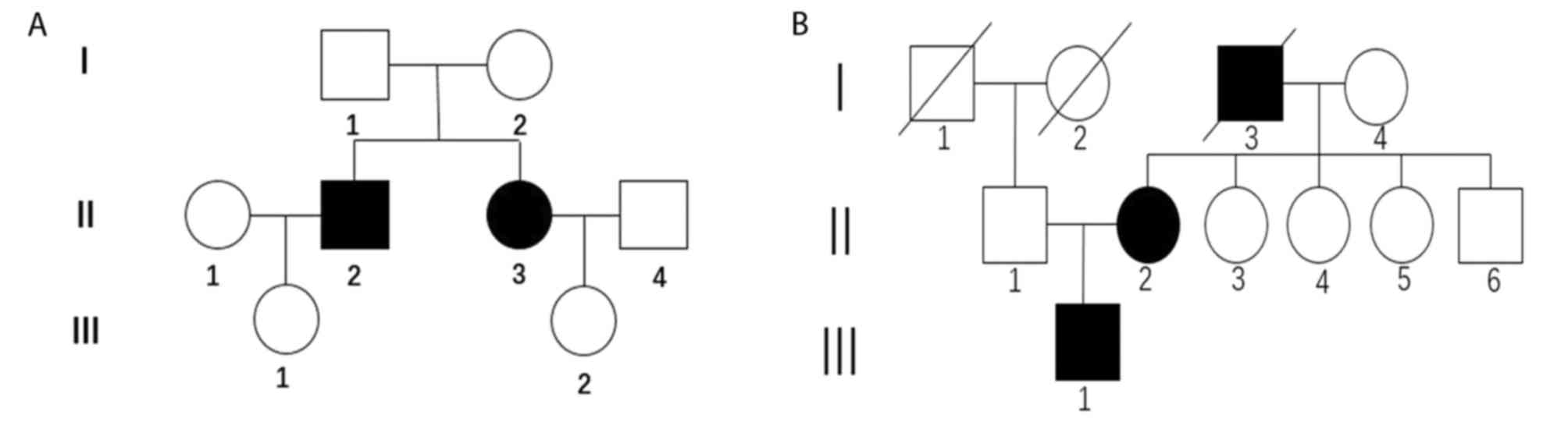
The present study involved seven individuals from
two Chinese families with typical clinical features of RP. The two
pedigrees were labeled Family D and Family H and the pedigrees of
the two families are shown in Fig. 1A
and B. The present study was performed in accordance with the
tenets of the Declaration of Helsinki and the ethical guidelines of
the Shanghai Renji Hospital, Shanghai Jiaotong University School of
Medicine (Shanghai, China). Written informed consent was obtained
from all participants. The clinical examinations performed to
diagnose RP predominantly included assessing the best-corrected
visual acuity (BCVA) using a Snellen chart, capturing images of
each fundus, assessing computerized test of central and peripheral
visual fields, and optical coherence tomography (OCT).
DNA extraction
Peripheral blood samples were obtained from all
subjects, and used for DNA extraction using a kit (Qiagen,
Shanghai) according to the manufacturer's protocols. DNA yield was
quantified with NanoDrop One (Thermo Fisher Scientific, Inc.,
Wilmington, DE, USA) and 1.5 µg of DNA was used for the following
assay. The DNA samples were stored at −80°C until use.
Exome capture and sequencing
Whole-exome sequencing was performed by Beijing
Youle Fusheng Technology Co., Ltd. (Beijing, China). Sample
libraries of gDNA were prepared using the TruSeq DNA Sample
Preparation kit (Illumina, Inc., San Diego, CA, USA) and the exomic
regions were enriched using the Roche Nimblegen SeqCap EZ v5.1 kit
(Roche Applied Science, Madison, WI, USA) according to
manufacturers' protocols. Paired-end short read sequencing was
performed on an Illumina platform with a PE150 read length
(Illumina, Inc.).
Identification of pathogenic mutations
from exome data
Bioinformatics analyses were performed using an
in-house pipeline. Briefly, sequencing reads that passed quality
filtering were aligned with the human reference genome, hg19, using
the Burrows-Wheeler Aligner program (BWA version 0.7.15) (7). Variants were identified using a
combination of the FreeBayes (version 0.9.21) (8), Genome Analysis Toolkit (GATK version
3.8) (9–11), CNVnator (version 0.2.7) (12) and in-house software programs.
The in-house software program, Langya, was used for
variant annotation and filtering. The variants were prioritized
based on variant frequency, pathogenicity, inheritance pattern and
how closely a gene associated with the given phenotype. The variant
frequency was obtained from the 1,000 Genomes Project, Single
Nucleotide Polymorphism database (http://www.1000genomes.org) and Exome Aggregation
Consortium databases (http://exac.broadinstitute.org). Predicted
pathogenicity data were obtained using the SNPEff (http://snpeff.sourceforge.net), Ensembl Variant Effect
Predictor (http://asia.ensembl.org/info/docs/tools/vep/index.html),
Sorting Intolerant From Tolerant, Polymorphism Phenotyping version
2 (http://genetics.bwh.harvard.edu/pph2/) and dbscSNV
databases (https://sites.google.com/site/jpopgen/dbNSFP).
Gene-phenotype association was characterized from structured
resources including the online Mendelian Inheritance in Man
database (https://www.omim.org), and unstructured
resources, including literature, utilizing natural language
processing techniques. The average sequencing depth in Family D was
>100X, whereas it was ~85X in Family H. The average coverage of
the two families was ~99.8%.
Sanger sequencing
The candidate variants in USH2A and RP2 identified
in the exome data were validated by Sanger sequencing.
Results
Clinical assessment of families with
RP
The clinical features of the affected patients in
the two pedigrees are listed in Table
I. The male patient D-II-2 suffered from decreased visual
acuity and night blindness at the age of 14 years. The
best-corrected visual acuity (BCVA) of D-II-2 decreased to light
perception as the patient aged. Although the central vision of the
affected female individual, D-II-3, was 20/20, the visual field of
the eyes of D-II-3 exhibited severe mid-peripheral scotomata due to
a coalescence of scotomata. All of the clinical features fit with
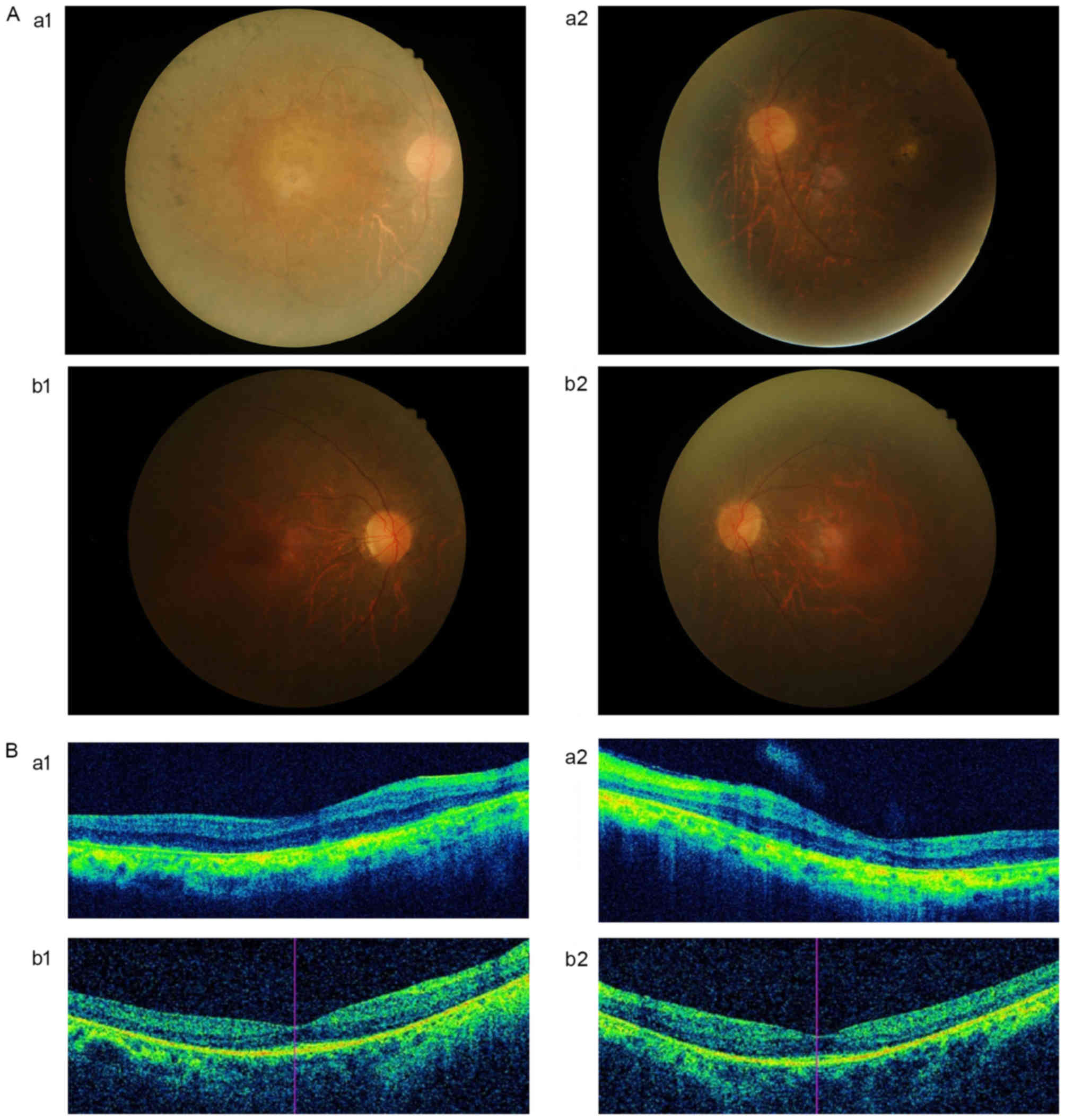
the diagnostic criteria of RP. The results of fundus examinations
revealed that the patients exhibited a typical RP appearance: Pale
optic discs, attenuated retinal arterioles and ‘bone spicule’
pigmentation. The OCT indicated that marked thinning and disruption
of the retinal nerve fiber layer, and the retinitis pigment
epithelium was evident (Fig. 2A and
B). The hearing examinations performed on the patients were
normal.
 | Table I.Clinical phenotype of the patients in
Families D and H. |
Table I.
Clinical phenotype of the patients in
Families D and H.
| Family | ID | Age (years) | Gender | Age at onset
(years) | BCVA | Slit lamp | Refraction |
|---|
| D | D-II-2 | 49 | Male | 14 | R: LP/LP | Mild cataract | R:
−2.50DS/−1.75DC*165 |
|
|
|
|
|
| L: LP/LP |
| L:
−2.00DS/−2.00DC*10 |
|
| D-II-3 | 47 | Female | 20 | R: 20/20 | Mild cataract | R:
−7.00DS/−1.00DC*160 |
|
|
|
|
|
| L: 20/20 |
| L:
−7.25DS/−0.75DC*5 |
| H | H-II-2 | 53 | Female | 20 | R: FC/5 cm | Mild cataract | R:
−4.50DS/−0.75DC*175 |
|
|
|
|
|
| L: 12/200 |
| L: +0.50DC*95 |
|
| H-III-1 | 28 | Male | 7 | R: 12/200 | Normal | R: −1.25DS |
|
|
|
|
|
| L: 12/200 |
| L: +1.25DS |
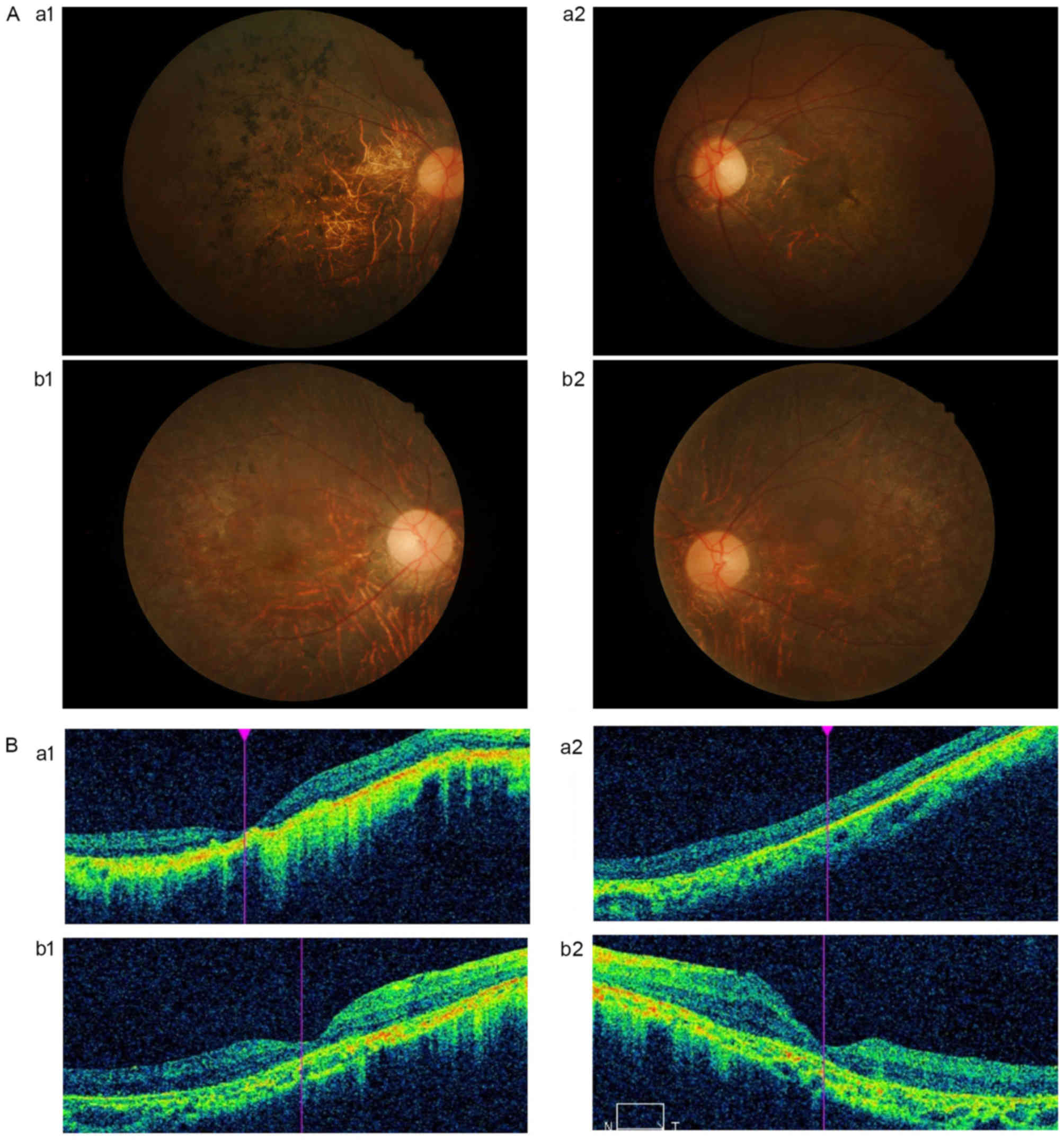
In Family H, a 48-year-old female patient suffered
from visual difficulties at night at the age of 20 years; visual
field loss and decreased visual acuity progressed markedly from the
age of 30 years. In the present study, the BCVA of the patient was
finger count (FC)/5 cm. The visual field of the patient's eyes
demonstrated mid-peripheral scotomata due to a coalescence of
scotomata. The deceased father of the patient had been diagnosed
with RP. The patient's son (H-III-1) was diagnosed with RP at the
age of 7 years due to night blindness. The visual acuity of H-III-1
decreased significantly following diagnosis. The detailed
ophthalmic examination results are shown in the Fig. 3A and B. The fundus examinations
indicated that H-III-1 exhibited a typical RP appearance, including
‘bone spicule’ pigmentary deposits, retinal vessel attenuation and
waxy optic discs. The OCT revealed that the retinal thickness was
decreased and that the foveal and retinal layers were atrophied.
The unaffected individuals exhibited normal phenotypes in all of
the ophthalmological examinations.
Pathogenic mutation detection
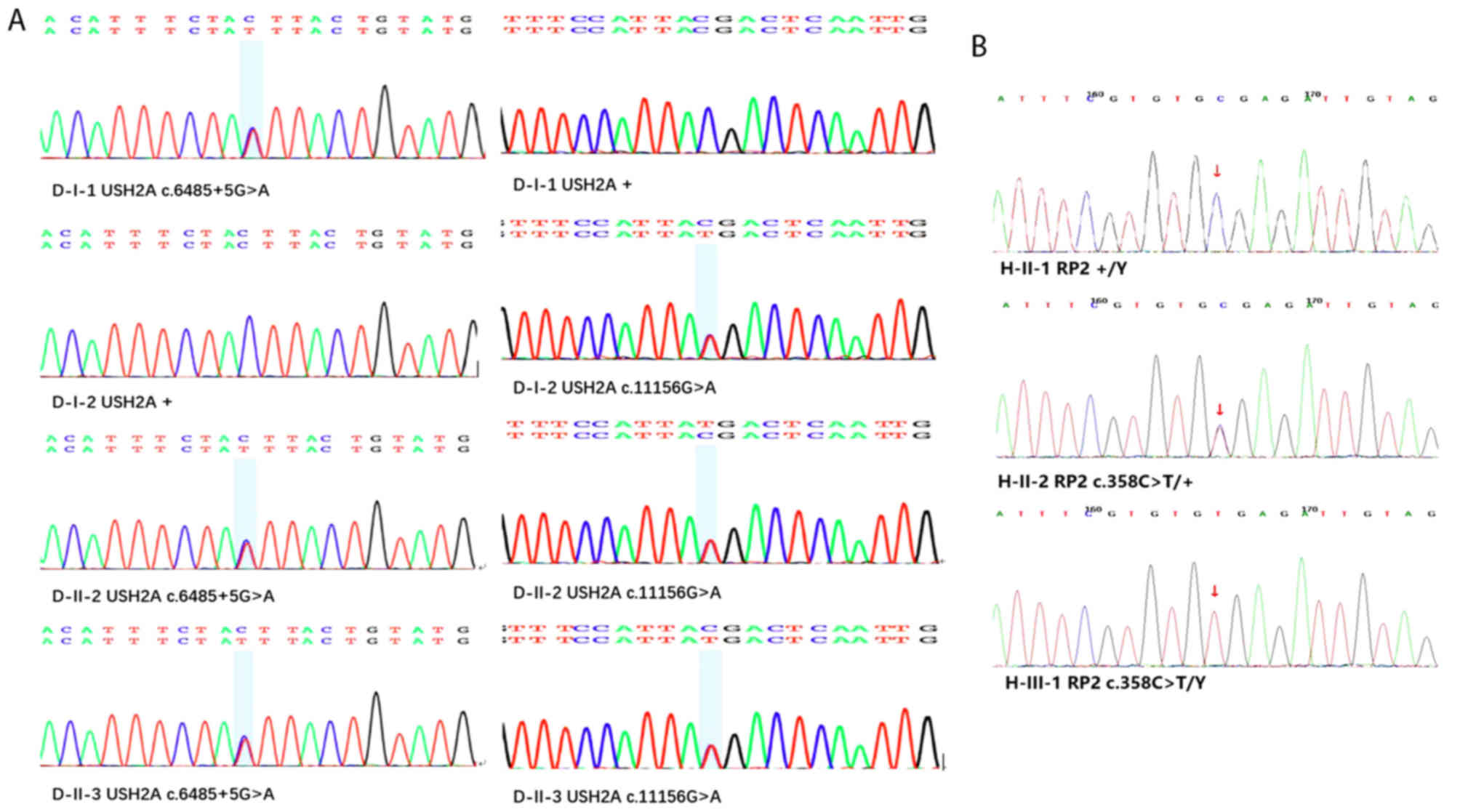
Whole-exome sequencing was performed on samples from
two Chinese families with RP. In Family H, a heterozygous mutation
(c.358C>T) in RP2 was identified as a pathogenic mutation,
co-segregating with the disease. The c.358C>T mutation in RP2
was demonstrated to be involved in pathogenic mutation in XLRP. The
two patients in Family H possessed the heterozygous variants,
whereas the unaffected individual did not. The two patients in
Family D possessed the same compound heterozygous mutation
(c.6,485+5G>A/c.11,156G>A) in the USH2A gene, which
co-segregated with the disease (Table
II). The c.6,485+5G>A mutation is a pathogenic mutation in
ARRP, which has not been reported previously in Chinese patients.
The results of Sanger sequencing of the validated pathogenic
mutations is shown in Fig. 4A and
B. Based on the clinical assessments and mutation analyses, the
present study concluded that the three heterozygous mutations were
the pathogenic variants involved in RP in the two families.
| Table II.Pathogenic mutations of patients in
Families D and H diagnosed with retinitis pigmentosa. |
Table II.
Pathogenic mutations of patients in
Families D and H diagnosed with retinitis pigmentosa.
| Family | Gene | Chromosome
position | cDNA mutation | Type of mutation | Hom/Het | Amino acid
change | Inheritance | Origin |
|---|
| D | USH2A | 1:215933077 | c.6485+5G>A |
| Het |
| Autosomal
recessive | Father |
|
| USH2A | 1:215933077 | c.11156G>A | Missense variant | Het | Arg3719Leu | Autosomal
recessive | Mother |
| H | RP2 | X:46713166 | c.358C>T | Stop-gain | Het | Arg120Ter | X-linked | Mother |
Discussion
In the present study, three mutations associated
with RP were reported. In humans, XLRP is one of the most severe
forms of RP, with six associated genetic loci. It is estimated that
~20% of patients with XLRP have pathogenic mutations in RP2. RP2
has been reported as an activator of small GTPase ADP-ribosylation
factor like-3, and has been implicated in cell signaling and
vesicular transportation by linking the cell membrane with the
cilia and cytoskeleton in photoreceptor cells (13,14).
Patil et al (15) revealed
that RP2 was crucial in photoreceptor development, and distinct
mutations in RP2 caused dysfunctions in cone and rod
photoreceptors, resulting in a wide-spectrum of clinical
phenotypes. In the present study, the c.358C>T mutation in exon
2 resulted in a truncated RP2 protein through a change of the
arginine codon to a nonsense codon. c.358C>T was first reported
by Mears et al (16) in
1999. With the development of sequencing techniques, whole-exome
sequencing has allowed for significant advances to be made in the
identification of novel RP pathogenic genes and screening of
mutations. In Family H, the affected male (H-III-1) exhibited more
severe clinical features compared with the affected female
(H-II-2); H-III-1 had an earlier age of onset, rapid progression of
visual field constriction and loss of visual acuity. RP is
generally determined by random X-chromosome inactivation, the
process responsible for silencing one of the X chromosomes in
females. The X-inactivation center produces a long functional
non-coding RNA, X inactive specific transcript (Xist), which is
directly involved in the mediation of chromosome silencing
(17). However, the mechanism of
Xist silencing requires further investigation.
RP can also occur as a syndrome, which accompanies
other diseases. Usher syndrome is the most frequent form of
syndromic RP associated with hearing loss (18). It can be divided into three types
based on the assessment of clinical features. At present, ~500
coding variants in USH2A have been reported to be pathogenic.
However, Jacobson et al (19) analyzed photoreceptors and the
retinal pigment epithelia of individuals with different mutations
in USH2A using OCT, demonstrating consistent results between the
individuals with the same USH2A genotype. USH2A is located on
chromosome 1q41 and is the most commonly mutated splice variant of
USH2, which consists of two main spliced transcripts. The shorter
splice variant consists of 21 exons and encodes a 170 kDa protein.
The longer splice variant consists of 72 exons and encodes a 600
kDa protein, which contains a membrane-spanning segment followed by
an intracellular PDZ-binding domain at the C-terminus (20). The expression of the longer splice
variant is restricted to photoreceptors and developing cochlear
hair cells (21). Liu et al
(22) disrupted the expression of
USH2A in mice, resulting in the progressive degeneration of
photoreceptors and non-progressive hearing impairment, suggesting
that USH2A may be essential for the maintenance and development of
photoreceptors and cochlear hair cells.
In Family D, D-II-3 and D-II-2 possessed the same
compound heterozygous mutation (c.6,485+5G>A/c.11,156G>A) in
USH2A. Although the two shared the same mutations, D-II-3 had a
central visual field, whereas D-II-2 exhibited almost complete
visual field loss. It has been suggested that the same mutations
can cause a variety of intrafamilial phenotypes. Previously, it was
reported that mutations in USH2A can cause patients to suffer from
RP without hearing loss. The missense mutation in USH2A,
c.11,156G>A, affects the long splice variant of USH2A, resulting
in a substitution of arginine for leucine at protein position 3,719
(23). This change may affect the
insertion of the protein into the membrane, leading to the
degeneration of photoreceptors. As the mRNA expression levels of
the long splice variant in the cochlea are low, the function of
USH2A has been demonstrated to persist in cochlea cells (22). The mutation c.6,485+5G>A in
USH2A was previously reported in a Japanese patient with Usher
syndrome type 2, however, it was only identified in one individual
without families involvement (24). In the present study, the two
patients shared the same compound mutation in family H,
co-segregating with clinical features. In addition, it demonstrated
pleiotropy when patients in family H did not suffer from hearing
impairment.
In conclusion, the present study described two
Chinese families comprising individuals diagnosed from RP. Through
the use of whole-exome sequencing, a compound heterozygous mutation
co-segregating with the disease was identified as the pathogenic
mutation in ARRP in Family D. To the best of our knowledge, this is
the first identification of compound mutation c.6,485+5G>A in
USH2A as a pathogenic mutation among Chinese patients with RP.
However, the pathogenic mechanisms resulting from c.6,485+5G>A
require further investigation. In Family H, the XLRP mutation
(c.358C>T) in RP2 was confirmed to be a pathogenic mutation
caused by random X-chromosome inactivation. Future investigations
aim to analyze more pedigrees with RP, and examine the pathogenic
mechanism of the gene mutation.
Acknowledgements
The authors are grateful to patients and their
family members for the participation.
Funding
The present study was supported by grants from
Shanghai Hospital Development Center (grant no. SHDC12016116) and
Renji Hospital (grant no. PYIII-17-030).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YF contributed to conception and design of the
study, blood sample collection, data acquisition, statistical
analysis, data interpretation and drafting of the manuscript; NC
contributed to data acquisition, data interpretation and critical
revision of the manuscript for scientific and factual content; ZQ
contributed to critical revision of the design of the study; JS
contributed to supervision, conception and design of the study,
data interpretation and critical revision of the manuscript; LL
contributed to supervision, conception and design of the study and
critical revision of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Ren Ji Hospital [reference number (2016) 219 (2)]. Informed consent was obtained from
all the patients involved.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pawlyk BS, Bulgakov OV, Sun X, Adamian M,
Shu X, Smith AJ, Berson EL, Ali RR, Khani S, Wright AF, et al:
Photoreceptor rescue by an abbreviated human RPGR gene in a murine
model of X-linked retinitis pigmentosa. Gene Ther. 23:196–204.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dryja TP, Hahn LB, Kajiwara K and Berson
EL: Dominant and digenic mutations in the peripherin/RDS and ROM1
genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci.
38:1972–1982. 1997.PubMed/NCBI
|
|
3
|
Holt IJ, Harding AE, Petty RK and
Morgan-Hughes JA: A new mitochondrial disease associated with
mitochondrial DNA heteroplasmy. Am J Hum Genet. 46:428–433.
1990.PubMed/NCBI
|
|
4
|
Benjaminy S, Kowal SP, MacDonald IM and
Bubela T: Communicating the promise for ocular gene therapies:
Challenges and recommendations. Am J Ophthalmol. 160:408–415, e2.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Y, Huang L, Sundaresan P, Li S, Kim R,
Saikia Ballav B, Qu C, Zhu X, Zhou Y, Jiang Z, et al: Whole-exome
sequencing analysis identifies mutations in the EYS gene in
retinitis pigmentosa in the Indian population. Sci Rep.
6:194322016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Avila-Fernandez A, Perez-Carro R, Corton
M, Lopez-Molina MI, Campello L, Garanto A, Fernandez-Sanchez L,
Duijkers L, Lopez-Martinez MA, Riveiro-Alvarez R, et al:
Whole-exome sequencing reveals ZNF408 as a new gene associated with
autosomal recessive retinitis pigmentosa with vitreal alterations.
Hum Mol Genet. 24:4037–4048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lan X, Gao H, Wang F, Feng J, Bai J, Zhao
P, Cao L, Gui S, Gong L and Zhang Y: Whole-exome sequencing
identifies variants in invasive pituitary adenomas. Oncol Lett.
12:2319–2328. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1. 332013.
|
|
12
|
Abyzov A, Urban AE, Snyder M and Gerstein
M: CNVnator: An approach to discover, genotype, and characterize
typical and atypical CNVs from family and population genome
sequencing. Genome Res. 21:974–984. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Evans RJ, Chapple JP, Grayson C,
Hardcastle AJ and Cheetham ME: Assay and functional analysis of the
ARL3 effector RP2 involved in X-linked retinitis pigmentosa.
Methods Enzymol. 404:468–480. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuhnel K, Veltel S, Schlichting I and
Wittinghofer A: Crystal structure of the human retinitis pigmentosa
2 protein and its interaction with Arl3. Structure. 14:367–378.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patil SB, Hurd TW, Ghosh AK,
Murga-Zamalloa CA and Khanna H: Functional analysis of retinitis
pigmentosa 2 (RP2) protein reveals variable pathogenic potential of
disease-associated missense variants. PLoS One. 6:e213792011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mears AJ, Gieser L, Yan D, Chen C, Fahrner
S, Hiriyanna S, Fujita R, Jacobson SG, Sieving PA and Swaroop A:
Protein-truncation mutations in the RP2 gene in a North American
cohort of families with X-linked retinitis pigmentosa. Am J Hum
Genet. 64:897–900. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wutz A: Gene silencing in X-chromosome
inactivation: Advances in understanding facultative heterochromatin
formation. Nat Rev Genet. 12:542–553. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fishman GA, Kumar A, Joseph ME, Torok N
and Anderson RJ: Usher's syndrome. Ophthalmic and neuro-otologic
findings suggesting genetic heterogeneity. Arch Ophthalmol.
101:1367–1374. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jacobson SG, Cideciyan AV, Aleman TS,
Sumaroka A, Roman AJ, Gardner LM, Prosser HM, Mishra M, Bech-Hansen
NT, Herrera W, et al: Usher syndromes due to MYO7A, PCDH15, USH2A
or GPR98 mutations share retinal disease mechanism. Hum Mol Genet.
17:2405–2415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Wijk E, Pennings RJ, te Brinke H,
Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW and
Kremer H: Identification of 51 novel exons of the Usher syndrome
type 2A (USH2A) gene that encode multiple conserved functional
domains and that are mutated in patients with Usher syndrome type
II. Am J Hum Genet. 74:738–744. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aller E, Sánchez-Sánchez AV, Chicote JU,
García-García G, Udaondo P, Cavallé L, Piquer-Gil M, García-España
A, Díaz-Llopis M, Millán JM and Mullor JL: Analysis of the Ush2a
gene in medaka fish (Oryzias latipes). PLoS One. 8:e749952013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu X, Bulgakov OV, Darrow KN, Pawlyk B,
Adamian M, Liberman MC and Li T: Usherin is required for
maintenance of retinal photoreceptors and normal development of
cochlear hair cells. Proc Natl Acad Sci USA. 104:4413–4418. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McGee TL, Seyedahmadi BJ, Sweeney MO,
Dryja TP and Berson EL: Novel mutations in the long isoform of the
USH2A gene in patients with Usher syndrome type II or non-syndromic
retinitis pigmentosa. J Med Genet. 47:499–506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakanishi H, Ohtsubo M, Iwasaki S, Hotta
Y, Mizuta K, Mineta H and Minoshima S: Identification of 11 novel
mutations in USH2A among Japanese patients with Usher syndrome type
2. Clin Genet. 76:383–391. 2009. View Article : Google Scholar : PubMed/NCBI
|