Introduction
Gastric cancer is one of the most common and lethal
malignancies, and is the third leading cause of cancer-associated
mortality worldwide (1). The
incidence of gastric cancer in China accounts for >40% of newly
diagnosed patients with gastric cancer in the world (2–4). At
present, the pathogenesis of gastric cancer remains unclear;
however, it is hypothesised that several factors, multiple steps
and multiple genes serve important roles in gastric cancer
pathogenesis (5,6). The International Agency for Research
on Cancer has classified Helicobacter pylori (H.
pylori) as a class I carcinogen (7). H. pylori can exist in the
acidic environment of the stomach for a long time and destroy the
gastric mucosa, generating changes in the release of gastric
mucosal hormones, thus affecting the physiological state of the
stomach; therefore, it represents the most significant risk factor
for malignant gastric tumours (8,9).
Approximately 50% of the world's population is infected with H.
pylori (10,11). The infection rate in China may be
as high as 73.3% (12),
particularly in the Beijing region, where the infection rate may
reach up to 83.4% (13). Although
an increasing number of studies have focussed on H. pylori
infection resulting in gastric cancer, the underlying mechanism
remains unknown.
Heparanase (HPA) is an endoglycosidase capable of
degrading heparan sulfate in the extracellular matrix and basement
membrane (14,15), leading to the release numerous
types of biological mediators, including fibroblast growth factor,
hepatocyte growth factor and vascular endothelial growth factor, in
response to local or systemic signals (16,17).
Thus, HPA is involved in tissue remodelling and cell migration,
leading to inflammation, angiogenesis and tumour metastasis
(18–21). Several studies have demonstrated
that HPA is widely expressed in a number of tumours (22–24),
including stomach, pancreas, colon and bladder tumours. In addition
to its enzymatic activity, recent studies (25–27)
revealed that the non-enzymatic activity of HPA promotes the
aggregation of heparan sulfate proteoglycans, causing a cascade of
intracellular signal amplification that results in the activation
of protein kinase C, Src and Rac. HPA also acts on HPA receptors
located on the cell surface, including mannose-6-phosphate receptor
(MPR), cation-independent MPR and low density lipoprotein
receptor-related protein, which causes signalling cascades. In
addition, HPA serves an important role in inflammation and
autoimmune diseases, including colitis, arthritis, psoriasis and
sepsis (28,29).
A number of studies have demonstrated that H.
pylori infection leads to the development of gastric cancer by
activating nuclear factor (NF)-κB (30,31).
Studies have also revealed that NF-κB upregulates the expression of
HPA in numerous tumours (32–34).
Furthermore, it has been demonstrated that H. pylori
infection causes the development of gastric adenocarcinoma via the
activation of mitogen-activated protein kinase (MAPK) (35,36).
MAPK is activated and translocated to the nucleus, leading to the
activation of transcription factors, such as NF-κB (37,38).
Another study also demonstrated that the activation of the MAPK
signalling pathway is closely associated with the expression of HPA
(39). However, it is not clear
whether MAPK is involved in the regulation of HPA expression
following a H. pylori infection, leading to gastric
cancer.
The present study aimed to investigate whether H.
pylori infection causes the proliferation, invasion and
metastasis of gastric cancer by affecting the expression and
mechanisms of HPA. It was confirmed that H. pylori increased
HPA expression via the MAPK and NF-κB signalling pathways in MKN-45
cells.
Materials and methods
Human cell and bacterial culture
Human gastric cancer MKN-45 cells were obtained from
the Chinese Academy of Sciences (Shanghai, China) and cultivated in
RPMI-1640 medium supplemented with 10% foetal bovine serum (FBS;
both purchased from Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA), penicillin and streptomycin (both from North China
Pharmaceutical Co., Inc., Shijiazhuang, China) in a humidified
atmosphere containing 5% CO2 at 37°C.
H. pylori NCTC11637 bacteria were provided by
the Key Laboratory of Digestive System Tumors of Gansu Province
(Lanzhou, China) and cultured on Columbia agar plates containing 7%
defibrinated goat blood (Solarbio Science and Technology Co., Ltd.,
Shanghai, China) in an anaerobic tank. Next, the bacteria were
acquired and resuspended in RPMI-1640 medium without antibiotics,
but containing 10% FBS. The optical density (OD) at 600 nm was used
to measure the density of the bacteria (one unit of
OD600 was equal to 1×108 colony-forming
units/ml).
Co-culture of cells and bacteria
Following digestion, the MKN-45 cells were seeded
into three culture dishes and cultured in physiological conditions
until they reached the logarithmic growth phase. Next, the old
medium was discarded and replaced with RPMI 1640 medium without
antibiotics supplemented with FBS. H. pylori were then added
to the MKN-45 cells in a bacterium to cell ratio of 100:1. The
bacteria and cells were co-cultured for 6 h at 37°C in an
atmosphere with 5% CO2 and saturated humidity, at which
point HPA expression was measured by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting.
Transfection and reagents
A small interfering RNA (siRNA) against HPA and
scrambled siRNA were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). The siRNA sense strand for HPA was
5′-AGUCCGUCCAUUCAAAUAGUAGUGA-3′, and the antisense strand was
5′-UCACUACUAUUUGAAUGGACGGACU-3′. The scrambled siRNA sense strand
was 5′-UCUAAGCGAGAAUUCGUACGAUAUC-3′, and the antisense strand was
5′-ACGUGACACGUUCGGAUAAUUAUCU-3′. Lipofectamine™ 2000
(Santa Cruz Biotechnology, Inc.) was used to transfect the siRNA
sequences, according to the manufacturer's protocol. SB203580, a
p38 MAPK inhibitor, was purchased from Selleck Chemicals (Houston,
TX, USA). SB203580 (20 µM) was added to MKN-45 cells for 2 h prior
to the co-culture with H. pylori to confirm that the MAPK
signalling pathway was involved in H. pylori-induced HPA
expression in gastric cancer cells. Cells were then used in
RT-qPCR, western blotting, Cell Counting kit-8 (CCK-8), the
Transwell method and the Scratch test.
RT-qPCR assay
Total RNA was extracted from the cells using TRIzol
reagent (Takara Biotechnology Co., Ltd., Dalian, China)
supplemented with trichloromethane, isopropanol and ethyl alcohol
(all obtained from Lianchuang Biotechnology Co., Ltd., Lanzhou,
China). RNA quality was analysed using spectrophotometry (Nanodrop
2000). Next, total RNA was reverse transcribed into cDNA using the
PrimeScript™ RT Reagent kit (Takara Biotechnology Co.,
Ltd.) in accordance with the manufacturer's protocols. The RT
reaction was performed for 5 sec at 85°C and 15 min at 37°C. The
primers used in qPCR were designed by the Key Laboratory of
Digestive System Tumors of Gansu Province and synthesised by
Lianchuang Biotechnology Co., Ltd. (GenBank accession no.
NM002046). The primer sequences were as follows: HPA sense,
5′-CCTCATCCTCCTGGGTTCTC-3′ and antisense,
5′-TATCCTGGTTGACTTGAGATTGC-3′; and GAPDH sense,
5′-AAGGCTGGGGCTCATTTG-3′ and antisense, 5′-AGGAGGCATTGCTGATGATC-3′.
The qPCR amplifications were performed using 7500/7500 Fast
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and SYBR® Premix Ex
Taq™ II (Takara Biotechnology Co., Ltd.). According to
the manufacturer's protocol, a 20 µl reaction volume was used, and
the thermal cycling conditions were as follows: 95°C for 30 sec,
followed by 40 cycles of 95°C for 5 sec and 60°C for 34 sec. HPA
mRNA levels were quantified using ABI Prism 7900HT and the
2−ΔΔCq method (40) was
used to analyze the results.
Western blot analysis
The treated cells were washed with ice-cold PBS, and
total protein was extracted from the cells using
radioimmunoprecipitation assay lysis buffer, phenylmethane sulfonyl
fluoride (both from Beyotime Institute of Biotechnology, Haimen,
China) and protein phosphatase inhibitor (Solarbio Science and
Technology Co., Ltd.). The protein concentration was measured using
a BCA protein assay (Beyotime Institute of Biotechnology)
subsequent to centrifugation at 13,000 × g for 30 min at 4°C. Next,
the protein was separated on a 10% gel by SDS-PAGE and then
transferred onto polyvinylidene fluoride membranes (Solarbio
Science and Technology Co., Ltd.), which were subsequently blocked
with 5% non-fat milk for 2 h at 4°C. Following the blocking step,
the membranes were incubated with the following primary antibodies:
anti-HPA1 (1:1,000; cat. no. ab128931), anti-phosphorylated (p)-p38
MAPK (1:1,000; cat. no. ab195049), anti-p38 MAPK (1:1,000; cat. no.
ab170099), anti-p-p65 NF-κB (1:1,000; cat. no. ab76302), anti-p65
NF-κB (1:1,000; no. ab32536; all Abcam, Cambridge, UK) and
anti-β-actin (1:2,000; cat. no. TA-09; OriGene Technologies, Inc.,
Beijing, China). Following overnight incubation at 4°C, the
membranes were washed three times with Tris-buffered
saline/Tween-20 (Solarbio Science and Technology Co., Ltd.) and
then incubated with horseradish peroxidase-conjugated secondary
antibodies (1:10,000; cat. no. ZA-2301; OriGene Technologies, Inc.)
at room temperature for 1 h. The SuperSignal West Pico
Chemiluminescent Substrate (Thermo Fisher Scientific, Inc.) was
used to visualise the protein bands, which were imaged using the
VersaDoc Imaging System (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Densitometry measurements were performed to analyze protein
expression using Bio-Rad Quantity One Software, version 4.62
(Bio-Rad Laboratories, Inc.).
CCK-8 assay
CCK-8 assay is designed to detect cell
proliferation. Briefly, cells were seeded in 96-well plates at a
density of 1×104 cells per well. SB203580 (20 µM) was
added to MKN-45 cells for 2 h prior to the co-culture with H.
pylori, and then 100 µl complete medium and 10 µl CCK-8 reagent
was added to each well. After the cells were incubated at 37°C in
an atmosphere with 5% CO2 and saturated humidity for 4
h, the absorbance was detected at a wavelength of 450 nm using a
multifunctional enzyme marking instrument.
Colony formation assay
The different groups of cells were cultured for 72
h. Next, cells were seeded into 6-well culture plates at a density
of 300 cells per well, and cell culture medium was added to the
wells. Following a 14-day incubation, the cells were washed twice
with PBS, fixed at room temperature for 15 min using 99% methanol
and stained with crystal violet solution for 20 min. Images were
captured and the number of clones were counted using a
microscope.
Transwell assay
A Transwell assay was used to detect the invasion
capability of the tumour cells. Briefly, a 1:8 mixture of Matrigel
gel to RPMI-1640 medium (100 µl) was added to the lower chamber,
which was placed in 24-well plates at 37°C for 24 h. A serum-free
cell suspension (200 µl) at a concentration of 2.5×105
cells/ml was added to the upper chamber, while serum-containing
medium was added to the lower chamber. Subsequently, the cells were
cultured at 37°C in an atmosphere with 5% CO2 and
saturated humidity for 24 h. Following incubation, the cells
remaining in the upper chamber were carefully wiped off, and cells
in the lower chamber was rinsed twice with PBS and fixed in
methanol for 15 min. The cells were stained with crystal violet
solution in methanol for 30 min, and then the excess crystal violet
was washed off. Finally, the cells were observed and images were
obtained using a microscope. The number of invading cells was
counted in several fields of view.
Scratch test
The scratch test was used to detect the migration
ability of tumour cells. Briefly, horizontal lines were drawn every
0.5–1 cm on the back of a 6-well plate, and 5×105 cells
were added to each well. The cells were incubated overnight until
100% confluence was reached, and then a scratch was made across the
dish with a 200 ml pipette. Subsequently, the cells were rinsed
twice with PBS, serum-free medium was added, and the cells were
incubated at 37°C in an atmosphere with 5% CO2.
Following the incubation, images of the cells were obtained, and
the migration distance was measured.
Statistical analysis
IBM SPSS software (version 22.0; IBM Corp., Armonk,
NY, USA) was used to analyse all data. The experimental data are
expressed as the mean ± standard deviation. SigmaPlot (version
13.0; Systat Software, Inc., Chicago, IL, USA) was used to
construct the graphs. P<0.05 indicated that the difference
between groups was statistically significant.
Results
H. pylori infection induces changes in
HPA expression in MKN-45 cells in a time-dependent manner
According to previously published studies, the ratio
of bacteria to gastric cancer cells is ~100:1 (41,42);
therefore, this ratio was used in the experiments conducted in the
current study. To detect the effects of H. pylori infection
on HPA expression in gastric cancer cells, H. pylori and
MKN-45 cells were co-cultured at the aforementioned ratio for 0, 6,
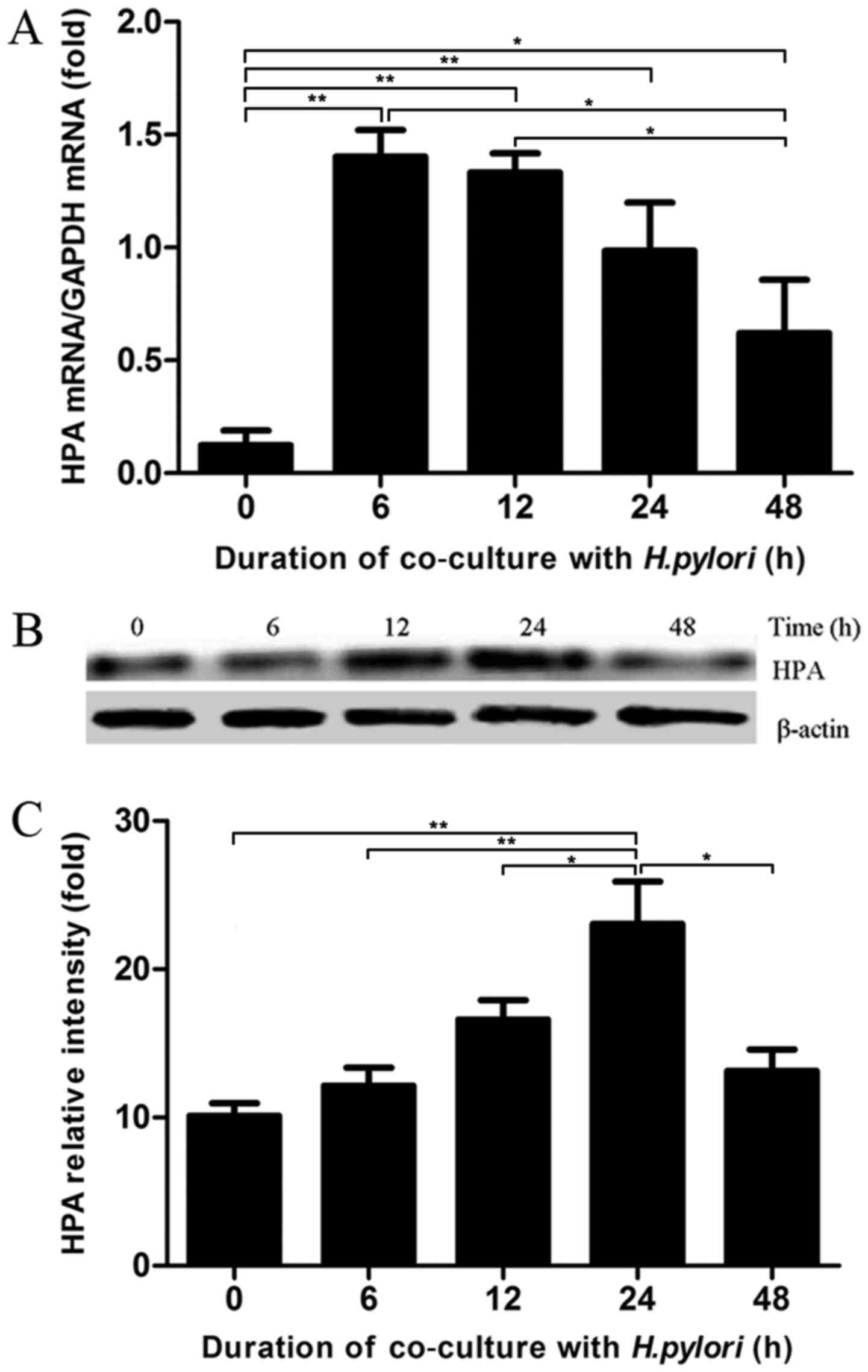
12, 24 and 48 h. The mRNA expression levels of HPA in MKN-45 cells
were analysed by RT-qPCR assays. The co-culture of H. pylori
and MKN-45 cells induced a significant increase in the mRNA
expression level of HPA, which reached a peak level at 6 h and then
decreased (Fig. 1A). The findings
of western blot analysis supported the RT-qPCR results,
demonstrating that HPA expression was also enhanced at the protein
level (Fig. 1B and C). The HPA
protein level peaked at 24 h in H. pylori-infected gastric
cancer cells. Taken together, these results indicated that gastric
cancer cells infected with H. pylori had increased HPA, in a
time-dependent manner.
H. pylori infection mediates the
increase of HPA expression in MKN-45 cells via the MAPK signalling
pathway
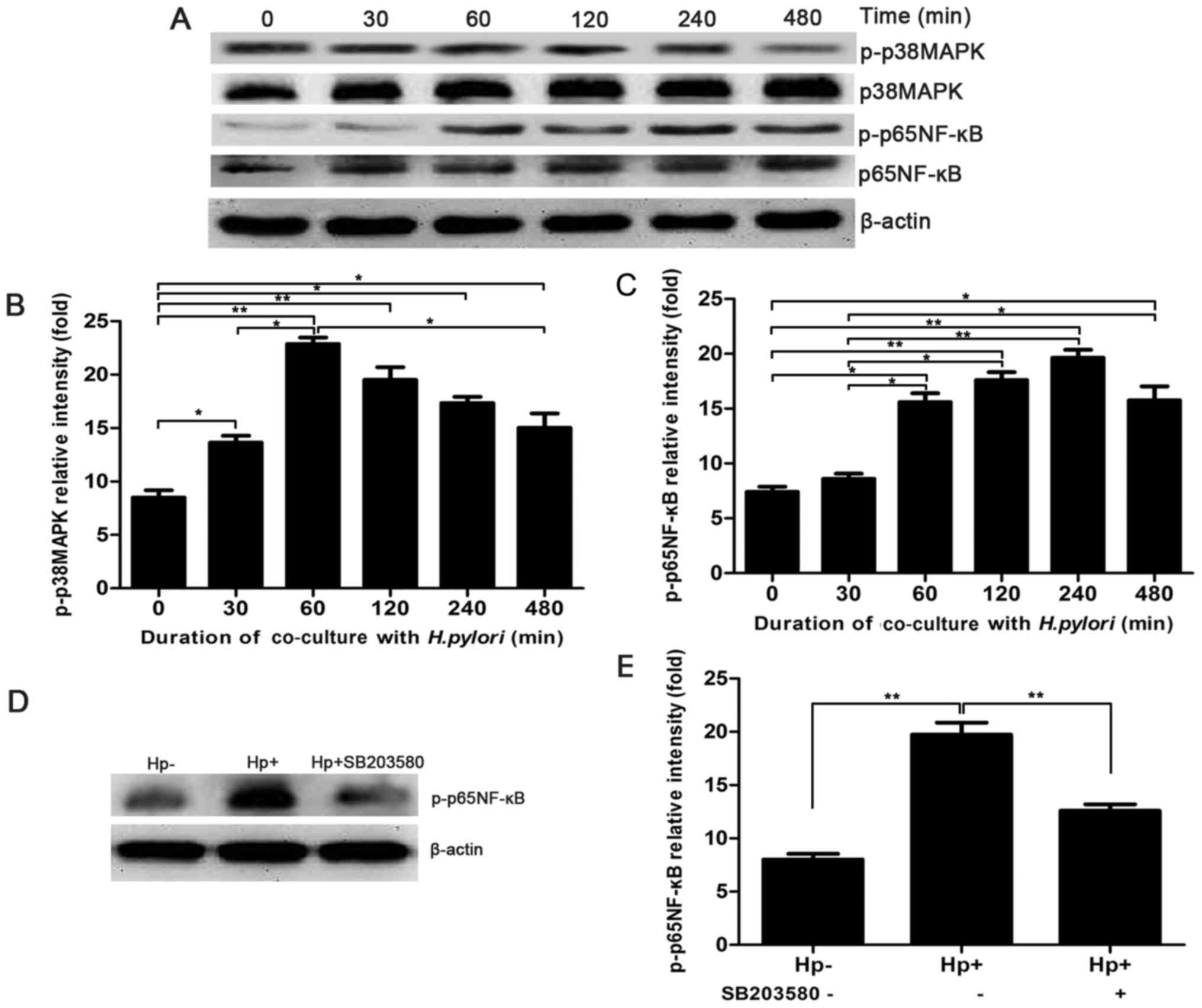
To illustrate whether MAPK signalling is involved in
the H. pylori-induced expression of HPA, the expression of
p-p38 MAPK was detected by western blot analysis after H.
pylori and MKN-45 cells were co-cultured for 0, 30, 60, 120 and
480 min. The expression of p-p38 MAPK was significantly higher at
30 min and peaked at 60 min, whereas the expression of p38 MAPK
remained unchanged (Fig. 2A and
B). To further confirm that H. pylori induces the
activation of the MAPK signalling pathway, leading to the
activation of NF-κB of MKN-45 cells, the expression of p-p65 NF-κB
was also detected by western blot analysis following co-culture of
H. pylori and MKN-45 cells for 0–480 min. The expression of
p-p65 NF-κB gradually increased with the duration of the
co-culture, peaking at 240 min (Fig.
2A and C). Furthermore, MKN-45 cells were pre-treated with a
MAPK inhibitor, SB203580, for 2 h prior to co-culture with H.
pylori. The expression of p65 NF-κB was significantly lower
when MKN-45 cells co-cultured with H. pylori were
pre-treated with SB203580 (Fig. 2D and
E). Therefore, the results revealed that the MAPK/NF-κB
signalling pathway may participate in H. pylori-induced HPA
expression in gastric cancer cells, which requires further
investigation.
Inhibition of MAPK weakens HPA
expression when H. pylori and MKN-45 cells are co-cultured
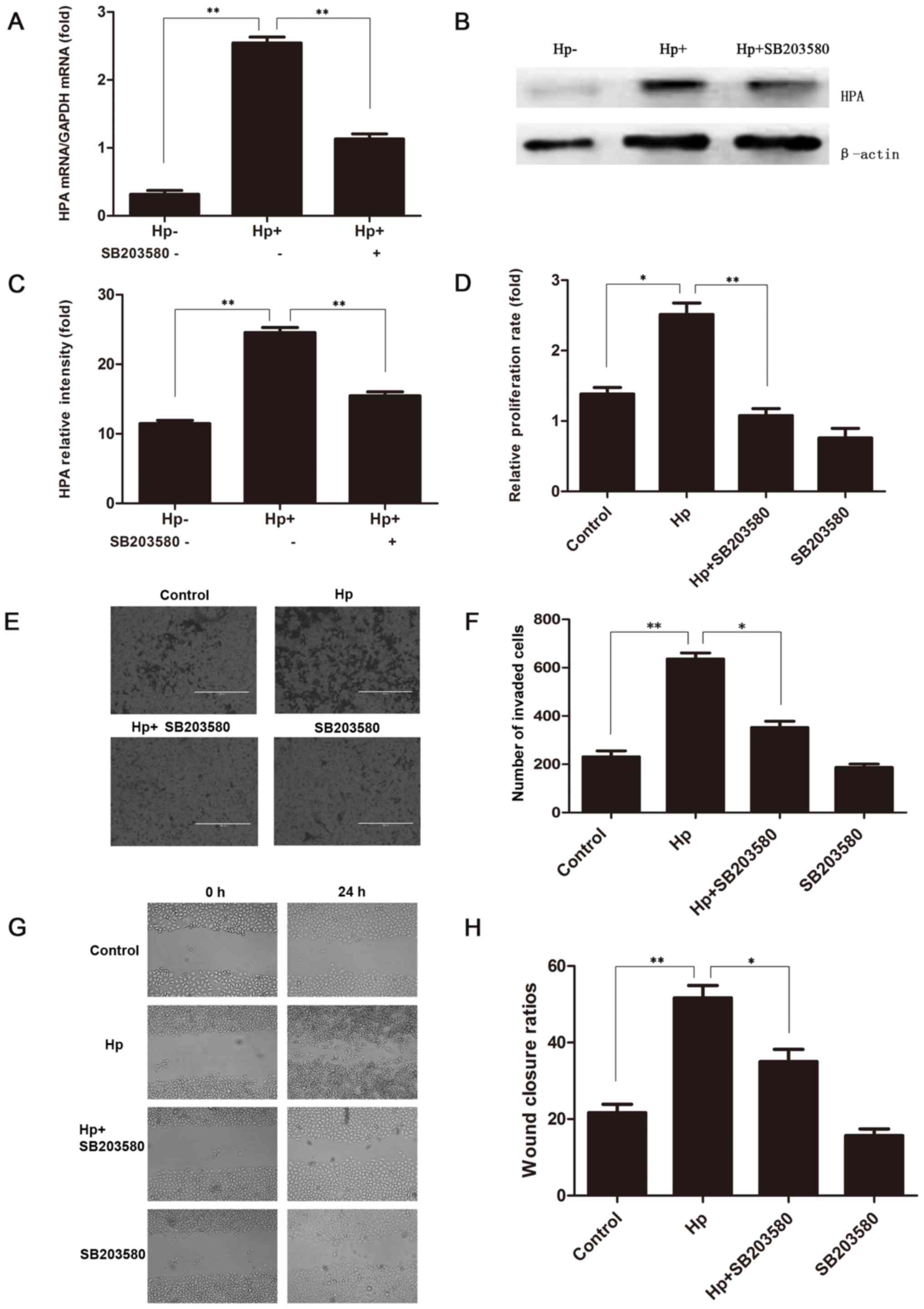
To further illustrate whether the H.
pylori-induced upregulation of HPA was mediated through the
MAPK signalling pathway, 20 µM SB203580 was added to MKN-45 cells
for 2 h prior to the co-culture with H. pylori. The HPA mRNA
expression was significantly higher when H. pylori infected
the MKN-45 cells, but that upregulation was significantly prevented
by SB203580 (Fig. 3A). These
changes were also reflected at the protein level (Fig. 3B and C). Furthermore, the CCK-8
proliferation assay confirmed that the addition of SB203580 to
H. pylori-infected MKN-45 cells significantly reduced the
cell proliferation, as compared with that in untreated H.
pylori-infected MKN-45 cells (Fig.
3D). In addition, the Transwell invasion (Fig. 3E and F) and scratch test migration
(Fig. 3G and H) assays confirmed
that the addition of SB203580 to H. pylori-infected MKN-45
cells markedly decreased the invasion and migration abilities of
MKN-45 cells, respectively. These results indicated that the MAPK
signalling pathway was involved in the H. pylori-induced
upregulation of HPA in gastric cancer cells.
HPA inhibition attenuated the H.
pylori-induced proliferation of MKN-45 cells
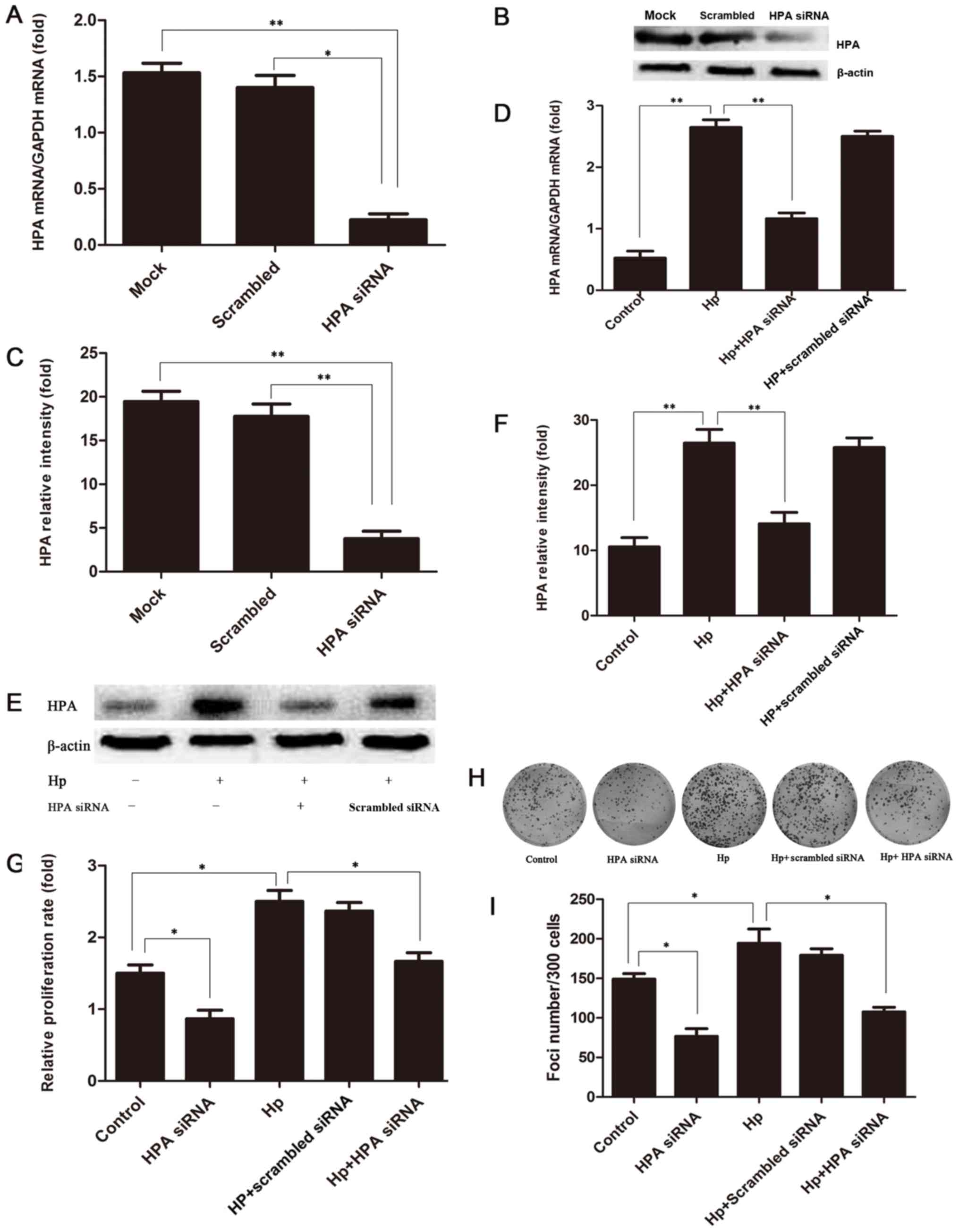
When MKN-45 cells were transfected with siRNA
against HPA, the HPA mRNA and protein expression levels were
significantly decreased (88 and 83%, respectively; Fig. 4A-C). To verify the involvement of
HPA in the proliferation of gastric cancer cells induced by a H.
pylori infection, the expression of HPA in H.
pylori-infected HPA-knockout MKN-45 cells was detected by
RT-qPCR analysis and western blotting. The results indicated that
the expression level of HPA in H. pylori-infected
HPA-knockout MKN-45 cells was significantly lower compared with
that in H. pylori-infected MKN-45 cells at the mRNA and
protein levels (Fig. 4D-F).
Furthermore, it was revealed that proliferation (Fig. 4G) and colony formation (Fig. 4H and I) were significantly
attenuated by HPA knockout in H. pylori-infected MKN-45
cells. These results revealed that HPA may have the potential to
become a target for the treatment of gastric cancer, especially
when H. pylori infection occurs.
Discussion
The present study demonstrated that H. pylori
infection resulted in increased HPA expression in gastric cancer
cells via the MAPK signalling pathway, thus exacerbating the degree
of the proliferation and invasion of gastric cancer cells.
H. pylori infections cause several digestive
diseases, including gastritis, gastric ulcers, gastric cancer,
liver cancer and other diseases (43,44).
H. pylori infection is the most prominent risk factor for
gastric cancer, which is one of the most common type of malignancy
worldwide (45). However, the
exact mechanism by which H. pylori infection causes gastric
cancer has been unclear. HPA has been documented in numerous
primary human tumours (46–48),
such as gastric cancer (49), and
previous studies have demonstrated that H. pylori infection
is vital in the acceleration of tumour growth, angiogenesis and
tumour metastasis (50–52). The current study also confirmed
that HPA was highly expressed in gastric cancer cells, as
previously reported in the literature (53,54),
and that the expression of HPA was greater in H.
pylori-infected gastric cancer cells. Additionally, the present
study demonstrated that the mRNA expression level of HPA was
highest at 6 h, while the protein expression level of HPA was
highest at 24 h in H. pylori-infected gastric cancer cells.
These findings demonstrated that HPA expression is higher in
gastric cancer, and that peak transcription and translation in
tumours occur at 6 and 24 h post-infection, respectively.
Studies have indicated that H. pylori
infection activates MAPK signalling in gastric epithelial cells
(35) and gastric adenocarcinoma
(36). The MAPK signalling pathway
is composed of three major components, including extracellular
signal-regulated kinase, c-Jun N-terminal kinase and p38 MAPK, and
serves an important role in mediating a number of cellular events,
such as genetic transcription, cell adhesion, cell metabolism and
apoptosis (55,56). The physiological function of MAPK
is to produce different reactions to extracellular stimuli, and its
main targets are transcription factors, such as NF-κB (57). When a cell is exposed to external
stimuli, MAPK is phosphorylated, activated and translocated to the
nucleus, where it activates NF-κB, initiating gene expression and
completing the cellular reaction induced by its activation
(58). Multiple studies have
demonstrated that H. pylori infection induces gastric cancer
by activating NF-κB (30,31). It has also been reported that NF-κB
can increase the expression of HPA in multiple tumours (32,34,59,60).
Specifically, studies have revealed that NF-κB was closely
associated with the expression of HPA in gastric cancer cells and
tissues (61,62). Another previous study reported that
the activation of the MAPK signalling pathway increased HPA
expression in cancer (39). The
current study further proved that H. pylori infection
significantly enhanced the expression levels of MAPK and NF-κB in
MKN-45 cells at the mRNA and protein levels. MAPK expression peaked
at 60 min and NF-κB expression peaked at 240 min. Additionally, the
inhibition of MAPK by SB203580 significantly decreased the
expression of NF-κB, indicating that MAPK was activated by H.
pylori and then activated NF-κB in the cell nucleus. Based on
the aforementioned results indicating that HPA transcription and
translation in H. pylori-infected MKN-45 cells peaked at 6
and 24 h, respectively, the authors of the current study speculate
that H. pylori infection in gastric cancer activates
MAPK/NF-κB signalling, thus leading to the activation of HPA.
When the MAPK inhibitor SB203580 was added to H.
pylori-infected gastric cancer cells, the H.
pylori-enhanced expression of HPA was decreased, as determined
by RT-qPCR and western blotting. In addition, cell proliferation,
invasion and migration were all decreased in H.
pylori-infected MKN-45 cells treated with SB203580, as
determined by the CCK-8, Transwell and scratch assays,
respectively. The results of these assays illustrated that H.
pylori infection promoted HPA expression in gastric cancer
cells via the MAPK signalling pathway, thus leading to enhanced
cell proliferation, invasion and migration, which was then
suppressed by the MAPK inhibitor.
Previous studies revealed that H. pylori
infection and HPA cause cell proliferation (43–47).
In the present study, the silencing of HPA significantly lowered
the mRNA and protein expression levels of HPA in H.
pylori-infected MKN-45 cells, and attenuated the proliferation
and colony formation of these cells. These results implied that
H. pylori infection induced the proliferation of gastric
cancer cells through the upregulation of HPA. This mechanism
illustrates that HPA may be a suitable therapeutic target for
gastric cancer, particularly when induced by H. pylori
infection.
In conclusion, the results of the current study
demonstrated that HPA serves a critical role in the development of
gastric cancer in H. pylori-infected cells. This may be an
important mechanism in the induction of gastric cancer by H.
pylori infection. In addition, H. pylori infection may
promote the proliferation, invasion and migration of gastric cancer
cells through the upregulation of HPA expression, and this is may
be mediated via the MAPK/NF-κB signalling pathway. Silencing HPA
with siRNA transfection further confirmed that HPA was involved in
the H. pylori-induced proliferation of MKN-45 cells. These
data suggest that HPA may be used as a therapeutic target in the
treatment of gastric cancer, particularly cancer induced by H.
pylori infection. Additional investigation, including animal
studies, is required to confirm the findings of the present
study.
Acknowledgements
The authors of the present study would like to thank
Dr Zhongtian Bai (The First Hospital of Lanzhou University, Gansu,
China) for a critical reading of this manuscript. The authors
appreciate the technical assistance provided by Professor Wenting
He (The Second Hospital of Lanzhou University, Gansu, China).
Funding
The present study was supported by the Natural
Science Foundation of Gansu Province (grant nos. 1506RJZA255 and
1308RJZA240-01), the Natural Science Foundation of China (grant no.
81572437), the International Science and Technology Cooperation
Program of China (grant no. 2015DFA31650), the Open Topic of the
Key Laboratory of Biological Treatment and Regenerative Medicine in
Gansu Province (grant no. zdsyskfkt-201702) and the fund of
Donggang Branch, The First Hospital of Lanzhou University (grant
no. ldyydgyn-201705).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL and LL designed the experiments. YZ and GF
performed the experiments. LL produced the manuscript. BL and TS
conducted data analysis.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 v1.0, Cancer incidence and mortality
worldwide: IARC cancer base no. 11International agency for research
on cancer. 2013, Lyon, France: http://globocan.iarc.fr
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang XM, Wang Z, Liang JW and Zhou ZX:
Analysis of laparoscopy-assisted gastric cancer operations
performed by inexperienced junior surgeons. Asian Pac J Cancer
Prev. 15:5077–5081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Figueiredo C, Garcia-Gonzalez MA and
Machado JC: Molecular pathogenesis of gastric cancer. Helicobacter.
18 Suppl 1:S28–S33. 2013. View Article : Google Scholar
|
|
6
|
Matsuda K, Tanikawa C and Nakamura Y:
Possible role of genetic factors on reduced risk for gastric cancer
among duodenal ulcer patients. Nihon Rinsho. 71:1491–1496.
2013.PubMed/NCBI
|
|
7
|
Schistosomes, liver flukes and
Helicobacter pylori. IARC Working Group on the evaluation of
carcinogenic risks to humans. Lyon, 7–7 June 1994. IARC Monogr Eval
Carcinog Risks Hum. 61:1–241. 1994.PubMed/NCBI
|
|
8
|
Wang F, Meng W, Wang B and Qiao L:
Helicobacter pylori-induced gastric inflammation and gastric
cancer. Cancer Lett. 345:196–202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang YH, Lv ZF, Zhong Y, Liu DS, Chen SP
and Xie Y: The internalization of Helicobacter pylori plays
a role in the failure of H. pylori eradication.
Helicobacter. 22:2017. View Article : Google Scholar
|
|
10
|
Suerbaum S and Michetti P: Helicobacter
pylori infection. N Engl J Med. 347:1175–1186. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang Y, Wang QL, Cheng DD, Xu WT and Lu
NH: Adhesion and invasion of gastric mucosa epithelial cells by
Helicobacter pylori. Front Cell Infect Microbiol. 6:1592016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Z, Zou D, Ma X, Chen J, Shi X, Gong Y,
Man X, Gao L, Zhao Y, Wang R, et al: Epidemiology of peptic ulcer
disease: Endoscopic results of the systematic investigation of
gastrointestinal disease in China. Am J Gastroenterol.
105:2570–2577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang M, Zhou YZ, Li XY, Tang Z, Zhu HM,
Yang Y and Chhetri JK: Seroepidemiology of Helicobacter
pylori infection in elderly people in the Beijing region,
China. World J Gastroenterol. 20:3635–3639. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vlodavsky I, Singh P, Boyango I,
Gutter-Kapon L, Elkin M, Sanderson RD and Ilan N: Heparanase: From
basic research to therapeutic applications in cancer and
inflammation. Drug Resist Updat. 29:54–75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rivara S, Milazzo FM and Giannini G:
Heparanase: A rainbow pharmacological target associated to multiple
pathologies including rare diseases. Future Med Chem. 8:647–680.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nadir Y and Brenner B: Heparanase multiple
effects in cancer. Thromb Res. 133 Suppl 2:S90–S94. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ilan N, Elkin M and Vlodavsky I:
Regulation, function and clinical significance of heparanase in
cancer metastasis and angiogenesis. Int J Biochem Cell Biol.
38:2018–2039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ni M, Elli S, Naggi A, Guerrini M, Torri G
and Petitou M: Investigating glycol-split-heparin-derived
inhibitors of heparanase: A study of synthetic trisaccharides.
Molecules. 21:E16022016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vlodavsky I, Iozzo RV and Sanderson RD:
Heparanase: Multiple functions in inflammation, diabetes and
atherosclerosis. Matrix Biol. 32:220–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilson JC, Laloo AE, Singh S and Ferro V:
1H NMR spectroscopic studies establish that heparanase is a
retaining glycosidase. Biochem Biophys Res Commun. 443:185–188.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin H and Zhou S: The functions of
heparanase in human diseases. Mini Rev Med Chem. 17:541–548. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yingying X, Yong Z, Zhenning W, Xue Z, Li
J, Yang L and Huimian X: Role ofheparanase-1 in gastric carcinoma
invasion. Asian Pac J Cancer Prev. 10:151–154. 2009.PubMed/NCBI
|
|
23
|
Meirovitz A, Hermano E, Lerner I, Zcharia
E, Pisano C, Peretz T and Elkin M: Role of heparanase in
radiation-enhanced invasiveness of pancreatic carcinoma. Cancer
Res. 71:2772–2780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shafat I, Pode D, Peretz T, Ilan N,
Vlodavsky I and Nisman B: Clinical significance of urine heparanase
in bladder cancer progression. Neoplasia. 10:125–130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gingis-Velitski S, Zetser A, Kaplan V,
Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY,
Vlodavsky I and Ilan N: Heparanase uptake is mediated by cell
membrane heparan sulfate proteoglycans. J Biol Chem.
279:44084–44092. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vreys V, Delande N, Zhang Z, Coomans C,
Roebroek A, Dürr J and David G: Cellular uptake of mammalian
heparanase precursor involves low density lipoprotein
receptor-related proteins, mannose 6-phosphate receptors, and
heparan sulfate proteoglycans. J Biol Chem. 280:33141–33148. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ben-Zaken O, Shafat I, Gingis-Velitski S,
Bangio H, Kelson IK, Alergand T, Amor Y, Maya RB, Vlodavsky I and
Ilan N: Low and high affinity receptors mediate cellular uptake of
heparanase. Int J Biochem Cell Biol. 40:530–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hermano E, Lerner I and Elkin M:
Heparanase enzyme in chronic inflammatory bowel disease and colon
cancer. Cell Mol Life Sci. 69:2501–2513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmidt EP, Yang Y, Janssen WJ, Gandjeva
A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX,
et al: The pulmonary endothelial glycocalyx regulates neutrophil
adhesion and lung injury during experimental sepsis. Nat Med.
18:1217–1223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maeda S, Akanuma M, Mitsuno Y, Hirata Y,
Ogura K, Yoshida H, Shiratori Y and Omata M: Distinct mechanism of
Helicobacter pylori-mediated NF-kappa Bactivation between
gastric cancer cells and monocytic cells. J Biol Chem.
276:44856–44864. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim H, Lim JW and Kim KH: Helicobacter
pylori-induced expression of interleukin-8 and cyclooxygenase-2
in AGS gastric epithelial cells: Mediation by nuclear
factor-kappaB. Scand J Gastroenterol. 36:706–716. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang J, Chen Y, Xin XL, Li QN, Li M, Lin
LP, Geng MY and Ding J: Oligomannurarate sulfate blocks tumor
growth by inhibiting NF-kappaB activation. Acta Pharmacol Sin.
31:375–381. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu WJ, Pan CE, Liu QG, Meng KW, Yu HB,
Wang YL and Zhao L: Expression of heparanase and nuclear factor
kappa B in pancreatic adenocarcinoma. Nan Fang Yi Ke Da Xue Xue
Bao. 27:1267–1270. 2007.(In Chinese). PubMed/NCBI
|
|
34
|
Wu W, Pan C, Yu H, Gong H and Wang Y:
Heparanase expression in gallbladder carcinoma and its correlation
to prognosis. J Gastroenterol Hepatol. 23:491–497. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ding SZ, Smith MF Jr and Goldberg JB:
Helicobacter pylori and mitogen-activated protein kinases
regulate the cell cycle, proliferation and apoptosis in gastric
epithelial cells. J Gastroenterol Hepatol. 23:e67–e78. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen YC, Wang Y, Li JY, Xu WR and Zhang
YL: H pylori stimulates proliferation of gastric cancer
cells through activating mitogen-activated protein kinase cascade.
World J Gastroenterol. 12:5972–5977. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xiang Y, Ye W, Huang C, Lou B, Zhang J, Yu
D, Huang X, Chen B and Zhou M: Brusatol inhibits growth and induces
apoptosis in pancreatic cancer cells via
JNK/p38MAPK/NF-κb/Stat3/Bcl-2 signaling pathway. Biochem Biophys
Res Commun. 487:820–826. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chao W, Deng JS, Li PY, Liang YC and Huang
GJ: 3,4-Dihydroxybenzalactone suppresses human non-small cell lung
carcinoma cells metastasis via suppression of epithelial to
mesenchymal transition, ROS-mediated PI3K/AKT/MAPK/MMP and NFκB
signaling pathways. Molecules. 22:E5372017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu X, Fang H, Chen H, Jiang X, Fang D,
Wang Y and Zhu D: An artificial miRNA against HPSE suppresses
melanoma invasion properties, correlating with a down-regulation of
chemokines and MAPK phosphorylation. PLoS One. 7:e386592012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin CJ, Liao WC, Lin HJ, Hsu YM, Lin CL,
Chen YA, Feng CL, Chen CJ, Kao MC, Lai CH and Kao CH: Statins
attenuate Helicobacter pylori CagA translocation and reduce
incidence of gastric cancer: In vitro and population-based
case-control studies. PLoS One. 11:e01464322016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi IJ, Kim JS, Kim JM, Jung HC and Song
IS: Effect of inhibition of extracellular signal-regulated kinase 1
and 2 pathway on apoptosis and bcl-2 expression in Helicobacter
pylori-infected AGS cells. Infect Immun. 71:830–837. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sachs G, Wen Y and Scott DR: Gastric
infection by Helicobacter pylori. Curr Gastroenterol Rep.
11:455–461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ahn HJ and Lee DS: Helicobacter
pylori in gastric carcinogenesis. World J Gastrointest Oncol.
7:455–465. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Goh LY, Leow AH and Goh KL: Observations
on the epidemiology of gastrointestinal and liver cancers in the
Asia-Pacific region. J Dig Dis. 15:463–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu BW, Li DF, Ke ZF, Ma D, Li YJ, Gang D,
Zheng ZG, Zhang KJ and Zhang YH: Expression characteristics of
heparanase in colon carcinoma and its close relationship with
cyclooxygenase-2 and angiogenesis. Hepatogastroenterology.
57:1510–1514. 2010.PubMed/NCBI
|
|
47
|
Zhou Y, Song B, Qin WJ, Zhang G, Zhang R,
Luan Q, Pan TJ, Yang AG and Wang H: Heparanase promotes bone
destruction and invasiveness in prostate cancer. Cancer Lett.
268:252–259. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Varchalama E, Rodolakis A, Strati A,
Papageorgiou T, Valavanis C, Vorgias G, Lianidou E and Antsaklis A:
Quantitative analysis of heparanase gene expression innormal
cervical, cervical intraepithelial neoplastic, and cervical
carcinoma tissues. Int J Gynecol Cancer. 19:1614–1619. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma XM, Shen ZH, Liu ZY, Wang F, Hai L, Gao
LT and Wang HS: Heparanase promotes human gastric cancer cells
migration and invasion by increasing Src and p38phosphorylation
expression. Int J Clin Exp Pathol. 7:5609–5621. 2014.PubMed/NCBI
|
|
50
|
Vlodavsky I, Ilan N, Naggi A and Casu B:
Heparanase: Structure, biological functions, and inhibition by
heparin-derived mimetics of heparan sulfate. Curr Pharm Des.
13:2057–2073. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vlodavsky I, Friedmann Y, Elkin M, Aingorn
H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal
I, et al: Mammalian heparanase: Gene cloning, expressionand
function in tumor progression and metastasis. Nat Med. 5:793–802.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mogler C, Herold-Mende C, Dyckhoff G,
Jenetzky E, Beckhove P and Helmke BM: Heparanase expression in head
and neck squamous cell carcinomas is associated with reduced
proliferation and improved survival. Histopathology. 58:944–952.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zheng L, Jiang G, Mei H, Pu J, Dong J, Hou
X and Tong Q: Small RNA interference-mediated gene silencing of
heparanase abolishes the invasion, metastasis and angiogenesis of
gastric cancer cells. BMC Cancer. 10:332010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tang W, Nakamura Y, Tsujimoto M, Sato M,
Wang X, Kurozumi K, Nakahara M, Nakao K, Nakamura M, Mori I and
Kakudo K: Heparanase: A key enzyme in invasion and metastasis of
gastric carcinoma. Mod Pathol. 15:593–598. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen Y, Li H, Li M, Niu S, Wang J, Shao H,
Li T and Wang H: Salvia miltiorrhizapolysaccharide activates T
lymphocytes of cancer patients through activation of TLRs
mediated-MAPK and -NF-κB signaling pathways. J Ethnopharmacol.
200:165–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Huang FT, Peng JF, Cheng WJ, Zhuang YY,
Wang LY, Li CQ, Tang J, Chen WY, Li YH and Zhang SN: MiR-143
targeting TAK1 attenuates pancreatic ductal adenocarcinoma
progression via MAPK and NF-κB pathway in vitro. Dig Dis Sci.
62:944–957. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Thakkar N, Yadavalli T, Jaishankar D and
Shukla D: Emerging roles of heparanase in viral pathogenesis.
Pathogens. 6:E432017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ramani VC, Vlodavsky I, Ng M, Zhang Y,
Barbieri P, Noseda A and Sanderson RD: Chemotherapy induces
expression and release of heparanase leading to changes associated
with an aggressive tumor phenotype. Matrix Biol. 55:22–34. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hao NB, Tang B, Wang GZ, Xie R, Hu CJ,
Wang SM, Wu YY, Liu E, Xie X and Yang SM: Hepatocyte growth factor
(HGF) upregulates heparanase expression via PI3K/Akt/NF-κB
signaling pathway for gastric cancer metastasis. Cancer Lett.
361:57–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cao HJ, Fang Y, Zhang X, Chen WJ, Zhou WP,
Wang H, Wang LB and Wu JM: Tumor metastasis and the reciprocal
regulation of heparanase gene expression by nuclear factor kappa B
in human gastric carcinoma tissue. World J Gastroenterol.
11:903–907. 2005. View Article : Google Scholar : PubMed/NCBI
|