Introduction
Hepatic ischemia/reperfusion injury (HIRI), a major
cause of mortality post-liver surgery, occurs in a variety of
circumstances when liver blood flow is interrupted, including
during liver transplantation, liver resection and shock, and is
compounded by multiple factors including ATP depletion, activation
of neutrophils/macrophages, formation and release of cytokines and
DNA damage (1). However, the
specific mechanisms and mediators involved in postoperative HIRI
remain largely unknown (1).
Previous evidence has suggested that mitochondrial autophagy may
act as a protective factor in canine hepatic injury (2). Protective effects have additionally
been observed in porcine livers, and murine liver injury is more
severe in autophagy-deficient rats compared with wild type
counterparts, while inhibition of autophagy resulted in more severe
injury of hepatocytes and endothelial cells (3). Using an adenoviral vector, an
additional previous study demonstrated that overexpressed autophagy
resulted in sustained protection of hepatocytes from hepatocyte
injury (4). This previous evidence
demonstrates that mitochondrial autophagy has a negative
correlation with HIRI.
The specific regulatory factors of mitochondrial
autophagy have not been completely elucidated (5). However, increasing numbers of studies
have linked mitochondrial autophagy function to Parkin (6–8). In
patients with Parkin mutations, the activity of the mitochondrial
complex has been demonstrated to be reduced in leucocytes; rat
Parkin knockout models exhibit reduced mitochondrial proteins and
certain mitochondrial deficits, and an increased vulnerability
towards autophagy function was observed in a Parkin deletion model
(9). In addition, an association
of Parkin with the stimulation of mitochondrial biogenesis has been
observed in cells which overexpress Parkin, suggesting that Parkin
serves an important and phylogenetically-conserved mitochondrial
function (10). However, it
remains unclear how Parkin mediates these effects on mitochondrial
autophagy.
In addition to mitochondrial autophagy, hepatocyte
apoptosis, the biogenetic and metabolic process which serves a role
in the initiation of cell death (11), contributes to HIRI as a key
regulator, consistent with a previous study which demonstrated that
short hairpin (sh)RNA-infected livers exhibited increased apoptosis
compared with controls following HIRI (12). In a pig model, Parkin was able to
protect hepatocytes from apoptosis and necrosis to improve survival
during HIRI (13). Similar effects
on apoptosis have been observed in the brain and intestinal tract
(10). Previous experiments have
identified that certain pro-apoptotic genes, including cellular
tumor antigen p53, are upregulated in Parkin knockdown rats, and
expression of the anti-apoptotic gene apoptosis regulator Bcl-2
(Bcl-2) is altered. These previous data suggest that apoptosis is
simultaneously regulated by Parkin (5). However, it remains unclear how Parkin
deficiency triggers apoptosis. The regulatory effect of Parkin on
mitochondrial autophagy and apoptosis may underlie the association
between autophagy and apoptosis post-HIRI.
As important regulatory factors of cell death, DNA
damage repair and cell cycle arrest are associated with liver
injury (14). The initiation of
DNA damage and unrepaired damage in the liver post-HIRI may be due
to DNA double strand breaks (DSBs), which may directly lead to
apoptosis, cause growth arrest and induce liver injury (15). In addition to the effects of DSB
and G2/M cell cycle arrest, the upregulation of apoptotic genes
contributes to the promotion of apoptosis and cell dysfunction
during HIRI (16). The mechanisms
involved in Parkin-mediated DNA damage repair and cell cycle arrest
remain unclear.
The objective of the present study was to
investigate the role of Parkin in HIRI and cell survival,
particularly its influence on autophagy, apoptosis, DNA damage
repair and cell cycle arrest. In an inducible under-expression
system, the involvement of Parkin in the regulation of
mitochondrial autophagy, apoptosis gene expression and the
maintenance of the DSB repair protein was observed. It is
hypothesized that these functions of Parkin contribute to the
survival of normal hepatocytes post HIRI.
Materials and methods
Animals and ethics statement
Ninety adult male Sprague Dawley rats of 15–16 weeks
of age (weighing 250–300 g) were obtained from the Animal
Bio-Safety Level-III (ABSL-III) laboratory of the Wuhan University
School of Medicine and were housed in accordance with the
regulations of the National Institutes of Health (Bethesda, MA,
USA). The animals were individually housed in stainless steel
wire-bottomed cages and allowed access to standard chow diet and
water, under a standard 12-h light/dark cycle. All surgical
procedures were performed under sterile conditions. Care was taken
to minimize suffering and pain. All of the animal experiments were
approved by the Institutional Animal Care and Use Committee of
Wuhan University (Wuhan, China).
Parkin knockdown vector
construction
The shRNA was designed according to the Parkin
sequence in the NCBI database (www.ncbi.nlm.nih.gov/). The fitted target sequence was
selected in the coding region and exhibited no homology to any
other gene, as observed using the basic local alignment search
tool. The shRNA contained a unique 19-nucleotide double-stranded
human Parkin sequence, which was presented as an inverted
complementary repeat loop with a 9-nucleotide spacer and the
concrete sequence GCCCCGATATTTAAGCAAA and scramble sequence
GACTCTCCGAACGTGTCAC. The shRNA was cloned into plasmid pGenesil-1
(Genesil, Wuhan, China; www.genesil.com/)to generate the novel vector
pshRNA-Parkin.
HIRI model and transfection
The HIRI model was constructed according to the
method of Ohmori et al (17). A 5-cm midline incision was made on
the abdominal wall of rats under brief light diethyl ether
anesthesia (20 mg/l) according to the Ohmori et al (17). The portal pedicle, portal vein, and
common hepatic artery were clamped for 20 min using a microclip.
For the Parkin-knockdown experiments, the plasmids and the
transfection agent Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) were mixed with
Opti-MEM (Invitrogen; Thermo Fisher Scientific, Inc.) and incubated
at room temperature for 5 min, according to the manufacturer's
protocol. The cultured SD rat hepatocytes were obtained according
to the method described by Ohmori et al (17) were seeded following intraperitoneal
injection with mixed shRNA plasmids 48 h prior to surgery. As a
control, negative control plasmids were transfected into a subset
of hepatocytes, and intraperitoneal injection was performed for the
scramble group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
All of the hepatocytes digested by pancreatin
treatment (25°C, 15 min) were harvested in order to extract mRNA
using TRIZOL reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. After a pre denaturation
at 95°C for 30 sec, qPCR was performed using 35 cycles of
denaturation at 95°C for 5 sec, annealing at 62°C for 20 sec and
elongation at 72°C for 30 sec. First strand cDNA synthesis was
performed using a PrimeScript RT regent kit (cat. no. RR820A;
Takara Bio, Inc., Otsu, Japan) and RT-qPCR analysis for Parkin was
performed using the TaqMan kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The Parkin primer sequences were as follows:
5′-CCGAGTGACACTGATAGTGTTTGT-3′ (forward) and
5′-ATCGTCTGCGGATTGGCTGTAGTT-3′ (reverse); with the internal control
gene GAPDH primer as follows: 5′-TCCTCTGACTTCAACAGCGACAC-3′
(forward) and 5′-TCTCTCTTCCTCTTGTGCTCTTGG-3′ (reverse). The
relative expression was calculated using the 2−ΔΔCq
method (18).
Immunoblotting
The hepatic tissues were washed twice with PBS and
mixed with pancreatic treatment buffer. When the cells were
homogenized, they were centrifuged at 13,400 × g at 4°C for 5 min.
Total protein was extracted with Cell Lysis reagent (Pierce; Thermo
Fisher Scientific, Inc.), quantified by the bicinchoninic acid
method (Pierce; Thermo Fisher Scientific, Inc.), was incubated in
loading buffer, and was boiled for 5 min. Equal amounts of protein
extracts (20 ul) were electrophoresed using SDS-PAGE on a 10% gel,
and all the proteins were transferred to a polyvinylidene fluoride
membrane and processed for immunoblotting. The membrane was blocked
in 5% non-fat milk in TBS/Tween-20 for 2 h at 25°C and incubated
with anti-Parkin antibody (cat. no. ab77924, 1:2,500; Abcam,
Cambridge, UK), anti-microtubule associated protein 1 light chain 3
beta (LC3B) antibody (cat. no. 48397, 1:2,500; Abcam), anti BCl-2
antibody (cat. no. 59348, 1:2,500; Abcam), anti Ku-70 antibody
(cat. no. 201963, 1:2,500; Abcam), anti Chk1 antibody (cat. no.
47574, 1:2,500; Abcam) or anti-beta actin antibody (cat. no.
ab8226, 1:2,500; Abcam) overnight at 4°C. Membranes were further
incubated with horseradish peroxidase-conjugated (HRP) secondary
antibody diluted (cat. no. ab6789, 1:5,000; Abcam) for 60 min at
room temperature and bands were visualized using an enhanced
chemiluminescence kit (Beyotime Institute of Biotechnology, Haimen,
China). The autoradiographs were exposed onto X-Omat AR film
(Kodak, Rochester, NY, USA). The density of bands in the films was
quantified using Image J software (Version 2.1.4.7, National
Institutes of Health).
Apoptosis and cell cycle assays
All of the hepatocytes digested by pancreatin
treatment were harvested 48 h post-transfection, for apoptosis and
cell cycle assays, and stained with 5 ml annexin V-fluorescein
isothiocyanate (FITC) and 5 ml propidium iodide using the Annexin
V-FITC Apoptosis Detection kit, with cell cycle distribution
detected using the Cell Cycle Detection kit (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. The
percentage of apoptotic cells or cell cycle distribution was
measured using flow cytometry with analyzing software (CytoDiff CXP
2.0; Beckman Coulter, Inc., Brea, CA, USA). All analyses were
performed in triplicate.
Statistical analysis
All of the experiments were conducted in triplicate
and data are expressed as the mean ± standard deviation.
Statistical analyses of data were performed using one-way analysis
of variance with SPSS software (version 13.0; SPSS, Inc., Chicago,
IL, USA) and GraphPad Prism software (version 6.0; GraphPad
Software, Inc., La Jolla, CA, USA). P-values are based on two-sided
hypothesis testing. P<0.05 was considered to indicate a
statistically significant difference.
Results
Parkin expression and mitochondrial
autophagy are dramatically up-regulated following HIRI
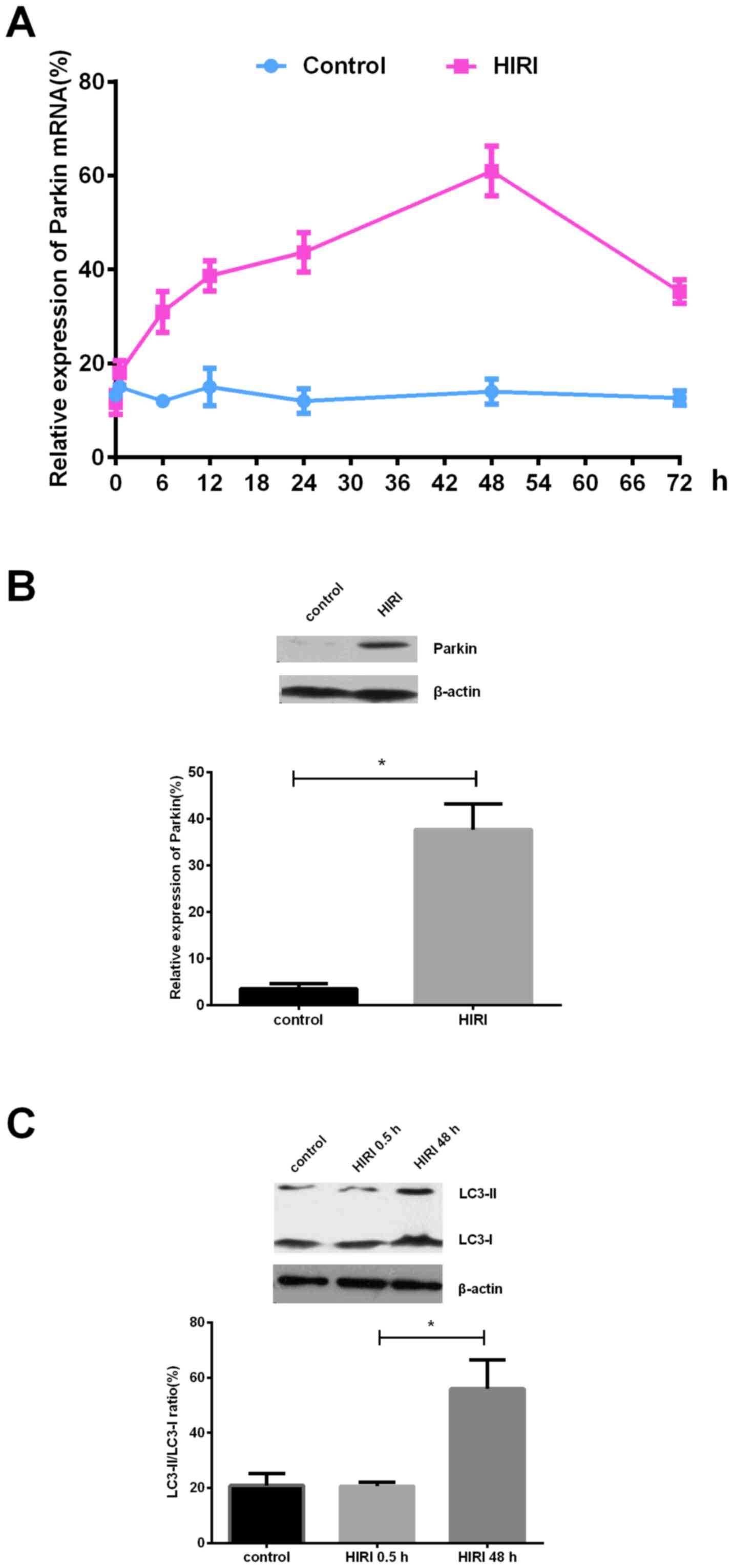
The results of the present study demonstrated that
the expression of Parkin mRNA was markedly upregulated following
HIRI in a time dependent manner, with the most abundant expression
~48 h post-HIRI (Fig. 1A). Similar
to mRNA, Parkin protein expression was upregulated post-HIRI
compared with the scramble control (Fig. 1B). The expression ratio of
autophagy-associated proteins LC3-II/LC3-I, used as an indicator of
mitochondrial autophagy, was upregulated in a time dependent manner
post-HIRI (Fig. 1C), indicating an
increased internal mitochondrial autophagy level post-HIRI.
Parkin knockdown suppresses the levels
of autophagy during HIRI
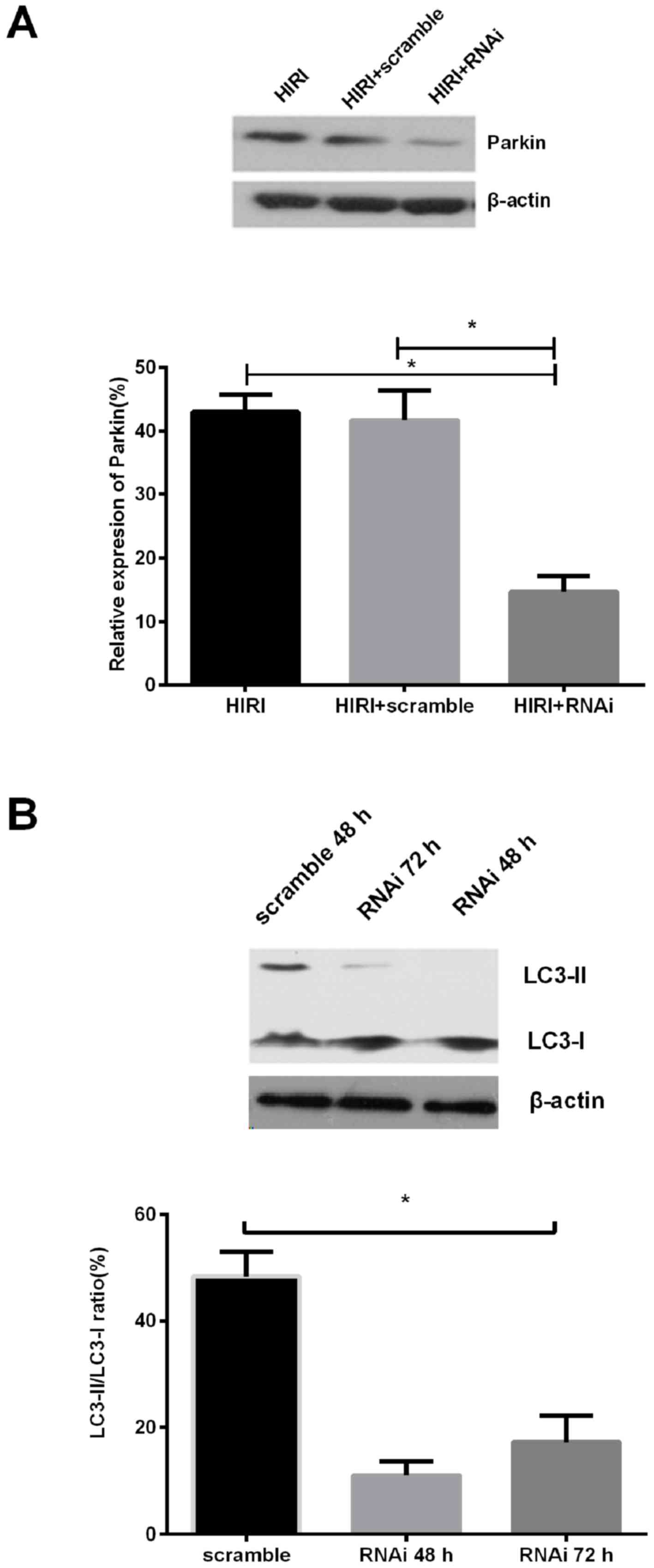
The Parkin-knockdown experiment demonstrated that
the quantity of Parkin protein in the Parkin knockdown group was
markedly reduced by >60% compared with the negative control
group (Fig. 2A). The expression
ratio of LC3-II/LC3-I protein was reduced compared with the
scramble control, in a time dependent manner (Fig. 2B). The results of the present study
indicated that Parkin knockdown reversed the level of mitochondrial
autophagy during HIRI.
Parkin knockdown promotes hepatocyte
apoptosis by suppressing Bcl-2
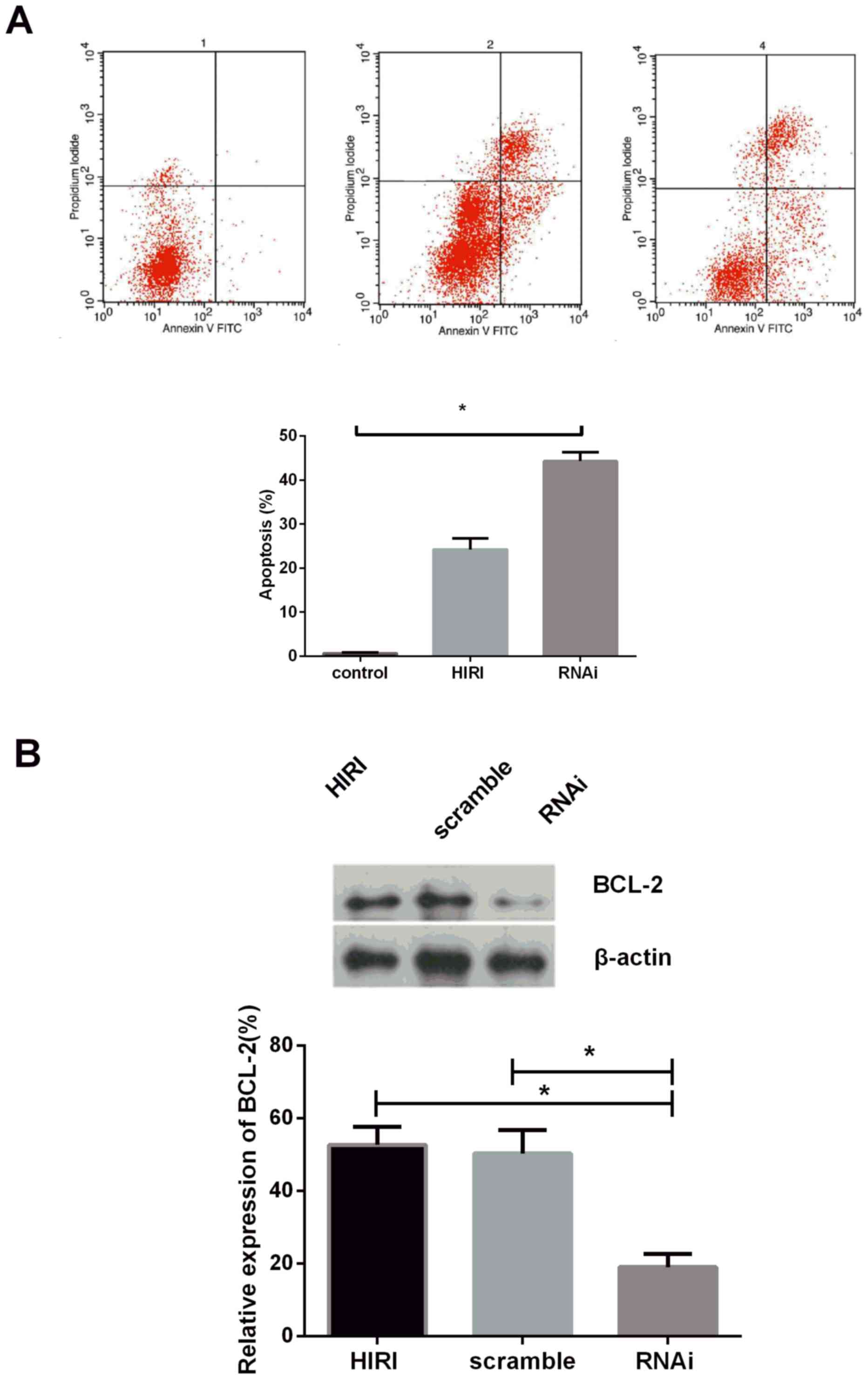
In order to investigate the effect of Parkin on
apoptosis during HIRI, Parkin protein was suppressed by
shRNA-Parkin, the results of which indicated that the rate of
apoptosis was markedly increased following Parkin knockdown
(Fig. 3A). Bcl-2 expression was
determined in rat hepatocytes transfected with shRNA-Parkin plasmid
or scramble control using immunoblotting analysis, and the results
demonstrated that Bcl-2 expression was markedly downregulated in
the knockdown group compared with the scramble group (Fig. 3B). The results of the present study
suggested that Parkin deficiency may promote apoptosis by
suppressing Bcl-2 expression in rat hepatocytes.
Parkin knockdown alters cell cycle
distribution in HIRI
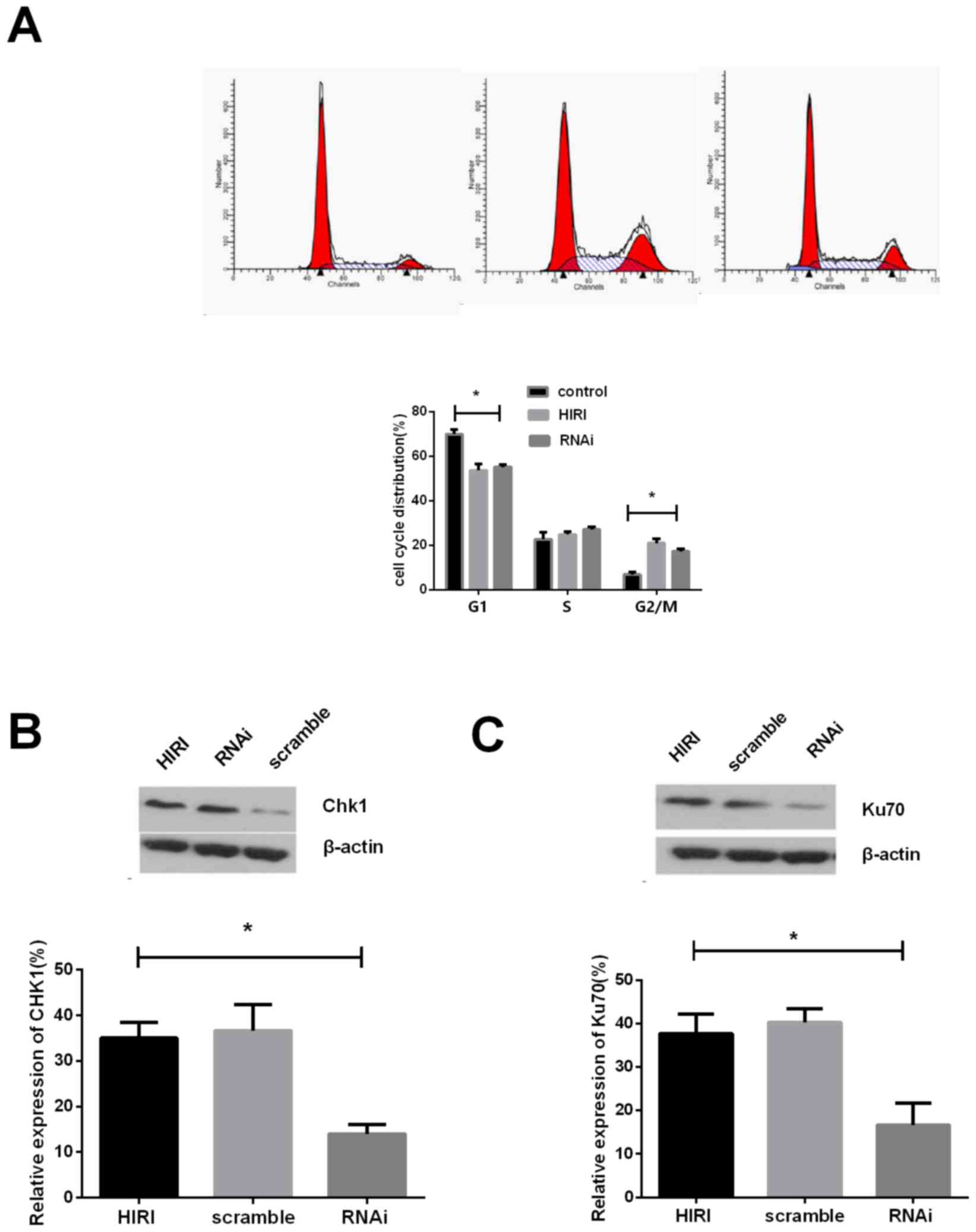
Following knockdown of Parkin expression, the
proportion of cells in the G2/M phase decreased to 8.05, suggesting
a decrease in G2/M arrest (Fig.
4A). The expression of serine/threonine protein kinase chk1
(Chk1) post-HIRI was detected and the results demonstrated that,
compared with the scramble control group, Chk1 expression was
markedly reduced following Parkin knockdown (Fig. 4B). The results of the present study
demonstrated that Parkin knockdown alters cell cycle
distribution.
Parkin knockdown impairs process of
DNA damage repair induced by HIRI
In order to investigate the effect of Parkin on the
DNA damage repair response, expression of ATP-dependent DNA
helicase II subunit 1 (Ku70), a typical DNA damage repair protein
involved in the DSB repair mechanism, was determined in order to
measure the level of DSB repair. The results of the present study
demonstrate that the quantity of Ku-70 in the Parkin knockdown
group was decreased compared with the HIRI samples (Fig. 4C). Therefore, Parkin deficiency may
impair the DNA repair process in rat hepatocytes following
HIRI.
Discussion
Parkin, exhibiting E3-ligase activity and mediating
the covalent attachment of activated ubiquitin to target proteins,
was initially hypothesized to function in a variety of cellular
metabolic processes and numerous substitutes for Parkin have been
identified, several of which are involved in HIRI (19). However, the definitive association
of Parkin with proteasome-independent activities, which serve a
role in mitochondrial autophagy, apoptosis, DNA damage response and
HIRI, remains unclear (16).
Previous studies have investigated HIRI-induced
liver damage with insufficient energy supply, which may be
exacerbated by the degradation of mitochondrial autophagy (20). It is well established that
mitochondrial autophagy and its derivatives potently and reversibly
decrease mitochondrial transmembrane potentials and inhibit cell
death in a concentration-dependent manner. The results of the
present study demonstrate that mitochondrial autophagy may decrease
cell death and liver injury post-HIRI; decreased levels of
autophagy proteins and increased cell death following Parkin
knockdown were observed post-HIRI. The results of the present study
indicate that Parkin may function in the HIRI process as a
protective factor, partly by promoting mitochondrial autophagy.
It has been previously demonstrated that there is an
association between apoptosis and sensitivity to HIRI in mammals
(4). Livers without injury have
been demonstrated to exhibit decreased apoptosis compared with
their counterparts, following HIRI and a direct effect of apoptosis
on tissues is the suppression of energy production by mitochondria
(21). At the gene expression
level, certain studies (20) have
reported that Bcl-2 secured correct rejoining of the DNA broken end
to protect DNA integrity as DNA damage response post-HIRI,
indicating that Bcl-2-associated regulatory mechanisms serve an
important role in HIRI; this may explain why Bcl-2 downregulation
results in increased sensitivity to HIRI. These previous studies
suggest that the downregulation of anti-apoptotic genes and a
subsequent increase in apoptosis may impair cell survival post HIRI
(12). In the present study,
Parkin inhibition induced Bcl-2 downregulation and an increase in
apoptosis, which provides evidence that Parkin may be involved in
Bcl-2 regulation and apoptosis control. Therefore, Parkin
deficiency may cause a decreased efficiency in apoptosis
suppression and increased cell death.
In addition to autophagy and apoptosis, certain
studies have demonstrated that the G2/M cell cycle distribution may
represent a novel mechanism of HIRI associated injury, wherein cell
cycle arrest may provide sufficient time for cell damage to be
repaired (16). Removal of Chk1, a
specific marker of G2/M cell cycle arrest, or other cell checkpoint
control proteins, may also contribute to injury (14). In the present study, following
ischemia-reperfusion expression of Chk1 was measured; the results
demonstrate that, compared with the control group, Chk1 expression
was markedly reduced following Parkin knockdown, suggesting that
Parkin is able to maintain the G2/M arrest, in part, by regulating
Chk1. However, the detailed mechanisms of Parkin function in cell
cycle control remain unknown (22).
DNA damage and repair has been demonstrated to
impact upon the outcome of HIRI (23). The repair of DSB, and the residual
damage, appear to be important factors affecting HIRI. In the
present study, DNA damage repair responses were observed in rat
hepatocytes post-HIRI, following Parkin knockdown. The results of
the present study demonstrated that there may be a negative
association between DSB repair and Parkin, suggesting that Parkin
may serve an important role in DSB recovery. However, the detailed
mechanisms underlying the association between Parkin and DNA damage
repair proteins remain unclear. The results of the present study
provide a novel hypothesis that Parkin may function to protect cell
survival by enhancing DSB repair and minimizing the residual DNA
damage.
In conclusion, the results of the present study
demonstrate physical evidence that Parkin deficiency may decrease
the survival of hepatic cells, in addition to mitochondrial
autophagy suppression, apoptosis promotion and DNA damage
elevation, during the initiation and development of HIRI. The
results of the present study demonstrate that Parkin serves a role
in the induction of mitochondrial autophagy, the suppression of
apoptosis, cell cycle distribution and DNA repair promotion during
HIRI. The present data provide novel evidence that Parkin may act
as a protective effector to protect hepatic cells from injury in
the rat liver following HIRI.
Acknowledgements
The present study was supported by the Xinjiang
Joint Funds of the National Natural Science Foundation of China
(grant no. U1403222).
References
|
1
|
Teoh NC and Farrell GC: Hepatic ischemia
reperfusion injury: Pathogenic mechanisms and basis for
hepatoprotection. J Gastroenterol Hepatol. 18:891–902. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kupiec-Weglinski JW and Busuttil RW:
Ischemia and reperfusion injury in liver transplantation.
Transplant Proc. 37:1653–1656. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kao SY: DNA damage induces nuclear
translocation of parkin. J Biomed Sci. 16:672009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Czaja MJ, Ding WX, Donohue TM Jr, Friedman
SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et
al: Functions of autophagy in normal and diseased liver. Autophagy.
9:1131–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomas RL and Gustafsson AB: Mitochondrial
autophagy-an essential quality control mechanism for myocardial
homeostasis. Circ J. 77:2449–2454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scarffe LA, Stevens DA, Dawson VL and
Dawson TM: Parkin and PINK1: Much more than mitophagy. Trends
Neurosci. 37:315–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Checler F, Goiran T and da Costa Alves C:
Presenilins at the crossroad of a functional interplay between
PARK2/PARKIN and PINK1 to control mitophagy: Implication for
neurodegenerative diseases. Autophagy. 1–2. 2017.
|
|
8
|
Drapalo K and Jozwiak J: Parkin, PINK1 and
DJ1 as possible modulators of mTOR pathway in ganglioglioma. Int J
Neurosci. 28:102–104. 2017.
|
|
9
|
Miklya I, Göltl P, Hafenscher F and Pencz
N: The role of parkin in Parkinson's disease. Neuropsychopharmacol
Hung. 16:67–76. 2014.PubMed/NCBI
|
|
10
|
Siriussawakul A, Zaky A and Lang JD: Role
of nitric oxide in hepatic ischemia-reperfusion injury. World J
Gastroenterol. 16:6079–6086. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salminen A, Kaarniranta K, Kauppinen A,
Ojala J, Haapasalo A, Soininen H and Hiltunen M: Impaired autophagy
and APP processing in Alzheimer's disease: The potential role of
Beclin 1 interactome. Prog Neurobiol. 106–107:33–54. 2013.
View Article : Google Scholar
|
|
12
|
Sasaki H, Matsuno T, Nakagawa K and Tanaka
N: Induction of apoptosis during the early phase of reperfusion
after rat liver ischemia. Acta Med Okayama. 51:305–312.
1997.PubMed/NCBI
|
|
13
|
Suzuki T, Yoshidome H, Kimura F, Shimizu
H, Ohtsuka M, Takeuchi D, Kato A, Furukawa K, Yoshitomi H, Iida A,
et al: Hepatocyte apoptosis is enhanced after ischemia/reperfusion
in the steatotic liver. J Clin Biochem Nutr. 48:142–148. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brenner C, Galluzzi L, Kepp O and Kroemer
G: Decoding cell death signals in liver inflammation. J Hepatology.
59:583–594. 2013. View Article : Google Scholar
|
|
15
|
Zhang Y and Hunter T: Roles of Chk1 in
cell biology and cancer therapy. Int J Cancer. 134:1013–1023. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kao SY: Regulation of DNA repair by
parkin. Biochem Biophys Res Commun. 382:321–325. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ohmori M, Miyashita F, Uchida H, Kitoh Y,
Tsuruoka S, Harada K, Sugimoto K, Fujimura A and Kobayashi E:
Effect of erythromycin on ischemia-reperfusion injury of liver in
rats. Transplant Proc. 32:811–814. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du H, Yang W, Chen L, Shen B, Peng C, Li
H, Ann DK, Yen Y and Qiu W: Emerging role of autophagy during
ischemia-hypoxia and reperfusion in hepatocellular carcinoma. Int J
Oncol. 40:2049–2057. 2012.PubMed/NCBI
|
|
20
|
Hashmi SK, Baranov E, Gonzalez A, Olthoff
K and Shaked A: Genomics of liver transplant injury and
regeneration. Transplant Rev (Orlando). 29:23–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guicciardi ME, Malhi H, Mott JL and Gores
GJ: Apoptosis and necrosis in the liver. Compr Physiol. 3:977–1010.
2013.PubMed/NCBI
|
|
22
|
Dawson TM and Dawson VL: Parkin plays a
role in sporadic Parkinson's disease. Neurodegener Dis. 13:69–71.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cuervo AM and Wong E: Chaperone-mediated
autophagy: Roles in disease and aging. Cell Res. 24:92–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|