Introduction
Head and neck squamous cell carcinoma (HNSCC), a
common malignant tumor of the head and neck region, is primarily
comprised of lip, oral cavity, larynx, nasopharynx and other
pharynx carcinomas. In 2012, the number of new HNSCC cases reported
worldwide was ~686,000, while the HNSCC-associated mortality cases
were 375,000 (1). Currently,
smoking and alcohol consumption are deemed to be risk factors for
HNSCC development; furthermore, human papilloma virus (HPV)
infection is considered to have an important role in the occurrence
and prognosis of HNSCC (2). In
spite of the application of surgery, chemoradiation and multimodal
treatment approaches, the prognosis of HNSCC remains poor due to
local recurrence and metastasis, and the 5-year overall survival is
~50% (1). Furthermore, poor
prognosis is partially attributed to the lack of understanding of
the molecular mechanism underlying the development of this cancer.
The oncogenesis and progression of HNSCC is a complicated process
involving multiple molecules, including microRNA-98 (3), Twist family BHLH transcription factor
1 (4) and Mastermind-like 1
(4). With the application of
immunotherapy, multimodal therapeutic approaches may be improved in
the future (2). Thus, identifying
specific tumor markers and novel molecular targets for the
treatment of HNSCC is important.
Huang et al (5) has recently proposed that lysine
(K)-specific demethylase 5B (KDM5B) is overexpressed in HNSCC
tissues as compared with its levels in adjacent noncancerous
tissues, and may be a significant prognostic biomarker of HNSCC on
account of the association between KDM5B and overall survival
times. A previous study by Trivedi et al (6) suggested an association between the
expression of several tumor markers (including pemphigus vulgaris
antigen, parathyroid hormone-related peptide and tumor-associated
calcium signal transducer 1) and the metastasis of HNSCC to the
lymph nodes. Furthermore, numerous established or emerging
biomarkers associated with HNSCC have previously been explored,
including hypoxia-inducible factor 1, carbonic anhydrase IX,
programmed death ligand-1 and cytotoxic T-lymphocyte antigen 4,
which are involved in immune checkpoints, hypoxia and radiation
sensitivity (7). Tumor markers
vary widely due to the multiple anatomical sites and histological
types of HNSCC; thus, the identification of more valuable tumor
markers of HNSCC is required.
The approach for screening tumor markers and
molecular targets has improved with the application of microarray
and transcriptional sequencing technologies (8,9).
Microarray analysis and high-throughput sequencing technology
provide valuable information and have been successfully used in
marker screening for numerous tumors (10,11).
The transcriptome data containing large amounts of information is
stored in a public database for sharing. Data mining and
bioinformatics analysis allow for transcriptome data to be fully
utilized by researchers and enable more reliable biological
information to be obtained (12–14).
Biological and medical research has improved through the generation
and development of bioinformatics (15). Numerous biological websites and
software are available for use in bioinformatics analysis,
including The Cancer Genome Atlas (TCGA) (16), Gene Expression Omnibus (GEO) 2R
(17), The Database for Annotation
Visualization and Integrated Discovery (DAVID) (18), Gene Expression Profiling
Interactive Analysis (GEPIA) (19). These websites and software have
been successfully applied in numerous biological studies (13,14).
Despite the extensive application of bioinformatics
analysis in biomarker screening, investigations regarding HNSCC are
limited. Demokan et al (20) have demonstrated that guanine
nucleotide-binding protein γ-7 (GNG7) is downregulated in HNSCC
through expression profile screening, and the expression level of
GNG7 was then validated by quantitative methylation-specific
polymerase chain reaction. A recent study also identified
differentially expressed miRNAs and mRNAs in laryngeal squamous
cell carcinoma through data mining and bioinformatics analysis
(21). Therefore, the present
study aimed to identify novel diagnostic, prognostic or predictive
biomarkers for HNSCC, and highlight the core genes associated with
HNSCC through bioinformatics analysis.
In the present study, three mRNA expression profiles
associated with HNSCC were retrieved and filtered from the National
Center for Biotechnology Information (NCBI)/GEO datasets, which are
public and freely available databases (17,22).
The differentially expressed genes (DEGs) in each data series were
analyzed using GEO2R. The intersection of the three sets of DEGs
extracted from the three data series included 19 mRNAs, which were
defined as the hub genes and deemed to be highly associated with
HNSCC. Through bioinformatics analysis, validation and survival
analysis of a large sample size based on TCGA, a total of nine hub
genes were identified as potential tumor markers and important
prognostic indicators for HNSCC.
Materials and methods
Data mining
Transcriptome expression profiles of HNSCC patients
were retrieved from the NCBI-GEO datasets (https://www.ncbi.nlm.nih.gov/gds/) and used for
further screening. Three data series were selected for
investigation in the present study, including GSE6631, GSE58911 and
GSE83519. The screening criteria were set as follows: The mRNA
expression profiles selected were derived from matched-pairs
sample, while data derived from non-paired sample or cell lines
were excluded. The series GSE6631 and GSE83519 included 22 paired
samples each, and GSE58911 contained 15 paired samples. These data
series were downloaded (retrieval date, January 7, 2018), and the
DEGs between the HNSCC and matched control samples included in the
three data series were filtered using GEO2R (www.ncbi.nlm.nih.gov/geo/info/geo2r.html), an
NCBI-GEO integration tool. The default parameters used for DEG
identification was a false discovery rate (Benjamini-Hochberg
procedure) and P<0.05 (17,22).
Then, MultiExperiment Viewer (23)
and R package ‘ggplot2’ were applied for cluster analysis and
visualization of DEGs. With these software, the overall
distribution of DEGs in cancer tissues and paired normal tissues
may be presented using a hierarchical clustering graph. The
distribution of each DEG in different samples and the distribution
of all DEGs in a given sample may also be distinguished using a
clustering graph. The distribution tendency of DEGs may be
presented directly using a volcano diagram.
Screening of hub genes
Three sets of DEGs derived from the three data
series were further filtered according to rigorous screening
criteria, including a fold change (FC) of ≥2 and P≤0.05. The newly
acquired sets of DEGs were used in subsequent analyses. The
intersections of the three sets of DEGs originating from the data
series (GSE6631, GSE58911 and GSE83519) were presented using a Venn
diagram, which was implemented with the online software VENN
DIAGRAMS (http://bioinformatics.psb.ugent.be/beg/tools/venn-diagrams).
Subsequently, the common genes of the three DEG sets were extracted
and defined as hub genes if they were highly correlated with
HNSCC.
Validation of relative expression
levels of hub genes
In order to confirm and increase the reliability of
the data analysis, the relative expression levels of hub genes in
HNSCC were validated using GEPIA (http://gepia.cancer-pku.cn/index.html; retrieval date,
January 9, 2018), an online analysis software based on the TCGA and
Genotype-Tissue Expression (GTEx) databases, using |logFC|≥1 and
P≤0.05 as the cut-off criteria (19). The results are presented as box
plots.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway analyses
The GO project describes genes and gene products on
the basis of three different aspects, namely biological processes,
molecular functions and cellular components, in a
species-independent manner. GO has been widely used for genomics
and proteomics analyses in biomedical research (24,25).
KEGG, which primarily consists of various functions, including KEGG
PATHWAY and KEGG GENES, is used for understanding high-level
functions from molecular-level data (26). The KEGG PATHWAY, a manually
reference database for pathway mapping, represents the current
knowledge on molecular interactions, reactions and association
networks (26). To gain an
improved understanding of the hub genes, GO enrichment and KEGG
pathway analyses were performed using DAVID, an online integration
tool (retrieval date, January 9, 2018) (18,27).
Construction and analysis of
protein-protein interaction (PPI) network
For a more in-depth understanding of the
associations among hub genes, the PPI network of hub genes was
structured via online database STRING, according to the provided
manual (retrieval date, January 9, 2018) (28). PPIs may be evaluated and integrated
via the PPI networks (28).
Furthermore, the expression correlation of key nodes was verified
using GEPIA (retrieval date, January 9, 2018).
Extraction and analysis of potential
tumor markers
In order to identify tumor markers associated with
prognosis, the hub genes were further filtered according to the
score of nodes and the strength of interaction in the PPI network.
Next, a log-rank test of overall survival (200 months was the
longest follow-up period) associated with these selected hub genes
was performed using GEPIA (retrieval date, January 9, 2018).
Results
DEGs derived from three data series
associated with HNSCC
Initially, eight data series (including GSE83519,
GSE58911, GSE6631, GSE23036, GSE13397, GSE40185, GSE10774 and
GSE7073), which were able to be analyzed with GEO2R, were obtained
by the retrieval of mRNA expression profiles associated with HNSCC
in NCBI-GEO datasets. Among them, five data series were excluded
out according to the screening criteria, including the cell
line-based series GSE40185, GSE10774 and GSE7073, as well as the
unpaired tissue sample-based series GSE23036 and GSE13397. The
remaining three data series (GSE83519, GSE58911 and GSE6631) that
met the screening criteria were used in the subsequent
investigation.
GEO2R is a NCBI integration analysis tool for
expression profile data and may be used to screen DEGs of two or
more groups of sample sources (17,22).
Three sets of DEGs derived from the three included data series were
acquired by GEO2R analysis, and these are listed in Table I.
 | Table I.Differentially expressed genes in the
three data series. |
Table I.
Differentially expressed genes in the
three data series.
|
|
| Differentially
expressed genes |
|---|
|
|
|
|
|---|
| Series | Samples | Upregulated | Downregulated | Total |
|---|
| GSE6631 | 22 paired | 66 | 119 | 185 |
| GSE58911 | 15 paired | 204 | 450 | 654 |
| GSE83519 | 22 paired | 1,166 | 1,100 | 2,266 |
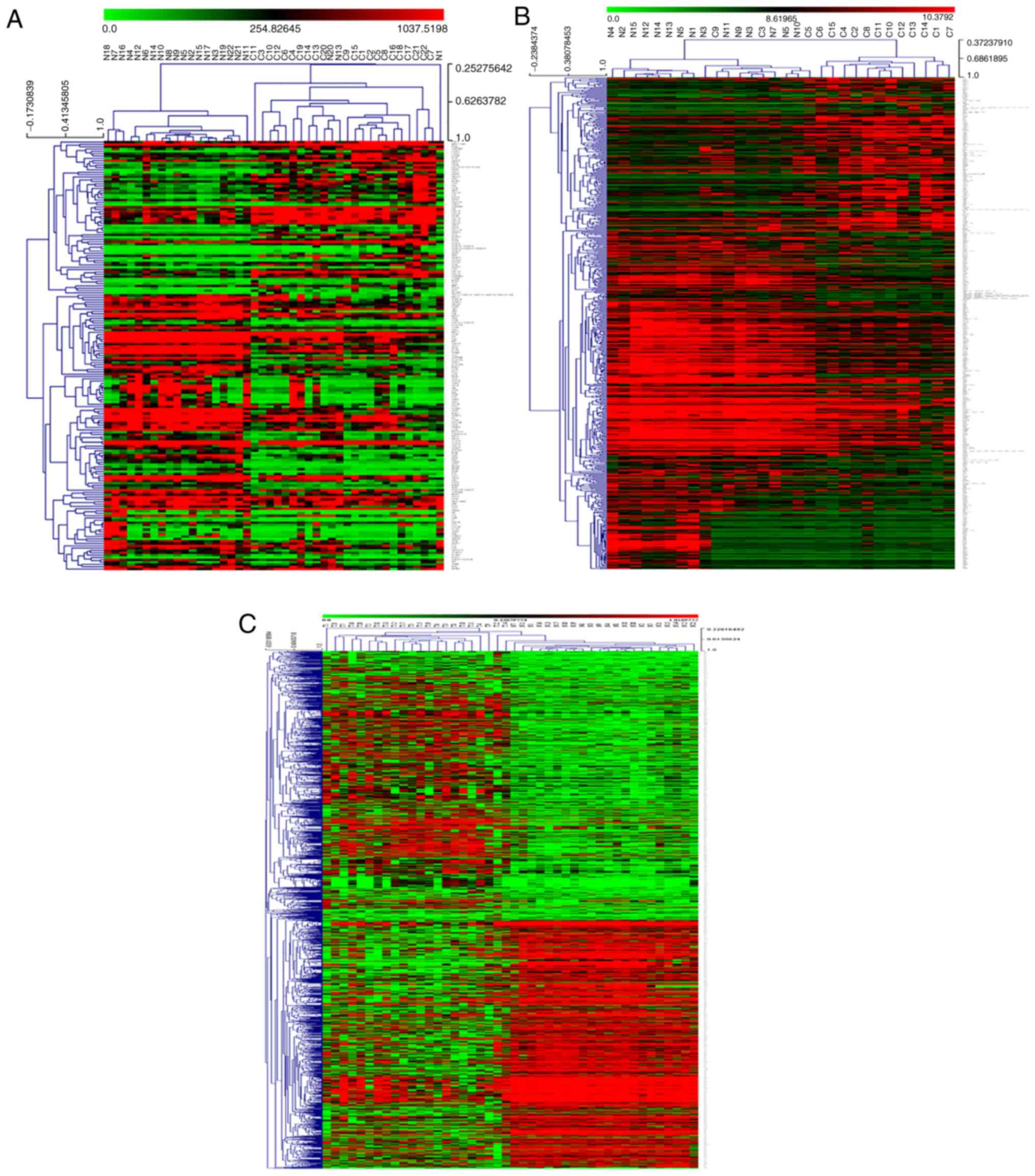
In order to present the distribution of DEGs in each
sample, the microarray data associated with the three NCBI-GEO
datasets were downloaded, and the relative gene expression values
of DEGs were extracted from the microarray data. Based on this,
clustering analysis was performed using MultiExperiment Viewer, an
open-source genomic analysis software (23). Subsequently, a volcano diagram was
developed using the R package ‘ggplot2’. The hierarchical
clustering graphs and volcano diagrams are presented in Fig. 1. Hierarchical clustering graphs and
volcano diagrams of DEGs of the three data series presented that
the DEGs are both upregulated and downregulated in cancerous
tissues, but mainly downregulated in cancerous tissues, which
suggested that the inactivation of tumor suppressor genes and the
activation of oncogenes played important roles in the development
of HNSCC, especially the inactivation of tumor suppressors.
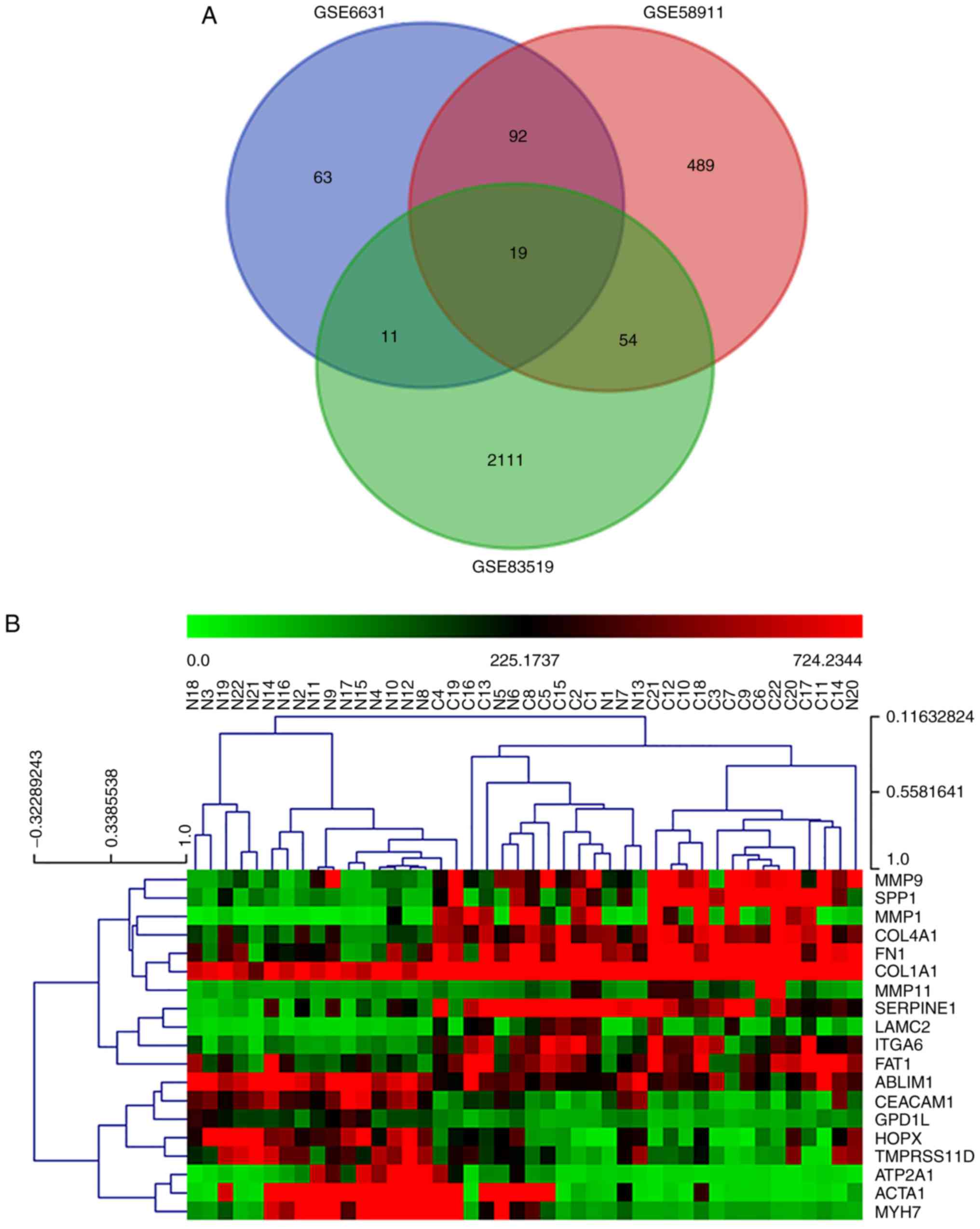
Nineteen hub genes are screened from
the DEGs of the three data series
The common DEGs from the three data series may be
associated with HNSCC and were defined as the hub genes. A total of
19 hub genes were extracted using the VENN DIAGRAMS drawing tool
(Fig. 2A), and were then
visualized as hierarchical clustering graphs for each data series
(Fig. 2B-D). Among the 19 hub
genes, 11 hub genes (including SPP1, COL4A1, COL1A1, FN1, ITGA6,
MMP1, MMP11, LAMC2, FAT1, SERPINE1 and MMP9) were overexpressed in
the HNSCC samples compared with the paired corresponding control
samples. Conversely, 8 hub genes (including GPD1 L, MYH7, ATP2A1,
HOPX, CEACAM1, TMPRSS11D, ABLIM1 and ACTA1) were downregulated in
the cancerous samples compared with the controls.
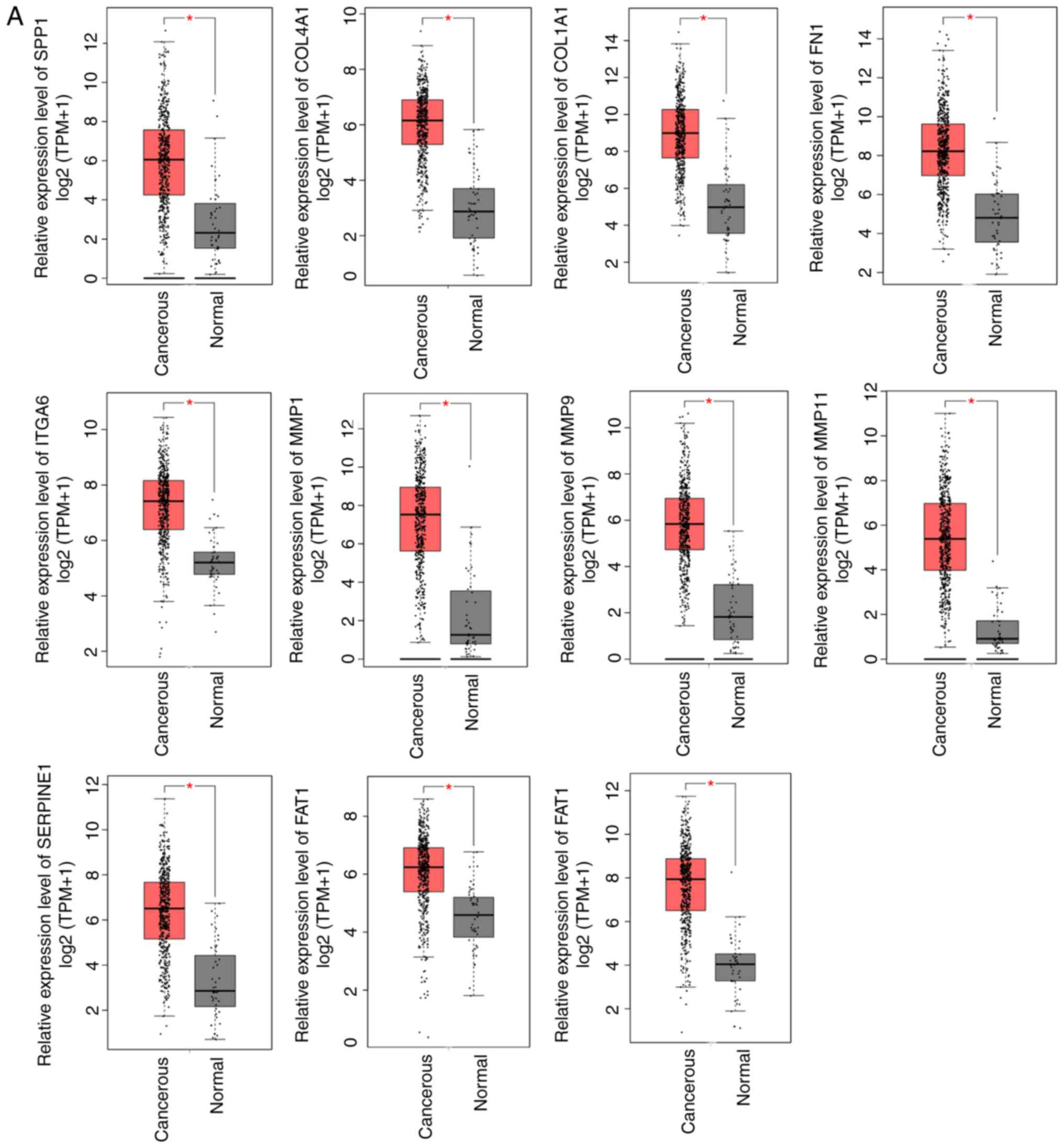
Relative expression levels of hub
genes in HNSCC samples against paired corresponding normal samples
are validated using GEPIA
Although the screening of hub genes performed in the
current study was vigorous and highly reliable, the relative
expression levels of 19 hub genes in tissues were further validated
through the online analysis tool GEPIA, which is based on TCGA and
GTEx databases (tumor, 519 cases; normal, 44 cases in GEPIA). The
results demonstrated that the relative expression trend of the 19
hub genes in GEPIA were consistent with the expression profiles
(Fig. 3), supporting the
reliability of the data analysis.
Nineteen hub genes are involved in
multiple GO terms and KEGG pathways
GO and KEGG pathway analyses of 19 hub genes were
further performed using the online analytical tool DAVID
(threshold, count ≥3; EASE score, ≤0.05), in order to achieve a
preliminary understanding of the biological functions in which
these hub genes may participate (Table II). The results of GO analysis
indicated that the main biological processes of the selected hub
genes consisted of extracellular matrix disassembly or
organization, leukocyte migration, collagen catabolic process, cell
adhesion, proteolysis, positive regulation of cell migration and
angiogenesis. The primary cellular component terms included the
extracellular region, extracellular matrix, stress fiber,
extracellular space, proteinaceous extracellular matrix,
filopodium, extracellular exosome, lamellipodium and perinuclear
region of the cytoplasm. Finally, the primary molecular function
terms included serine-type endopeptidase activity,
metalloendopeptidase activity, actin binding and calcium ion
binding. According to the GO terms and the distribution of hub
genes, the role of a protein in HNSCC may be preliminarily
hypothesized. For instance, MMP9 may participate in extracellular
matrix disassembly (GO:0022617) via exertion of
metalloendopeptidase activity (GO:0004222) in the extracellular
space (GO:0005615). Furthermore, KEGG pathway analysis indicated
that the screened hub genes were primarily involved in
extracellular matrix-receptor interaction (hsa04512), focal
adhesion (hsa04510), the PI3K-Akt signaling pathway (hsa04151),
pathways in cancer (hsa05200), small cell lung cancer (hsa05222)
and amoebiasis (hsa05146).
| Table II.GO and KEGG pathway analysis of hub
genes. |
Table II.
GO and KEGG pathway analysis of hub
genes.
| Term | Count | P-value | Genes | Fold
enrichment | FDR |
|---|
|
GOTERM_BP_DIRECT |
|
GO:0022617-extracellular
matrix disassembly | 6 |
1.36×10−8 | MMP9, MMP11, FN1,
MMP1, LAMC2, SPP1 | 69.77285319 |
1.78×10−5 |
|
GO:0030198-extracellular
matrix organization | 7 |
3.87×10−8 | FN1, SERPINE1,
LAMC2, SPP1, COL4A1, ITGA6, COL1A1 | 31.56390977 |
5.06×10−5 |
|
GO:0050900-leukocyte
migration | 6 |
1.48×10−7 | MMP9, FN1, MMP1,
CEACAM1, ITGA6, COL1A1 | 43.46505608 |
1.94×10−4 |
|
GO:0030574-collagen catabolic
process | 5 |
5.64×10−7 | MMP9, MMP11, MMP1,
COL4A1, COL1A1 | 69.04605263 |
7.37×10−4 |
|
GO:0007155-cell adhesion | 7 |
5.67×10−6 | FN1, LAMC2,
CEACAM1, SPP1, FAT1, ITGA6, COL1A1 | 13.47827084 | 0.007408 |
|
GO:0006508-proteolysis | 4 | 0.015348 | MMP9, MMP11,
TMPRSS11D, MMP1 | 7.070315789 | 18.30441 |
|
GO:0030335-positive regulation
of cell migration | 3 | 0.016282 | LAMC2, ITGA6,
COL1A1 | 14.40961098 | 19.31187 |
|
GO:0001525-angiogenesis | 3 | 0.023357 | FN1, SERPINE1,
CEACAM1 | 11.88954449 | 26.57648 |
|
GOTERM_CC_DIRECT |
|
GO:0005576-extracellular
region | 10 |
7.46×10−6 | MMP9, MMP11, FN1,
TMPRSS11D, SERPINE1, MMP1, LAMC2, SPP1, COL4A1, COL1A1 | 5.957502452 | 0.007625 |
|
GO:0031012-extracellular
matrix | 6 |
7.87×10−6 | MMP11, FN1,
SERPINE1, MMP1, COL4A1, COL1A1 | 19.44238976 | 0.008049 |
|
GO:0001725-stress fiber | 3 | 0.001279 | MYH7, ABLIM1,
ACTA1 | 53.28654971 | 1.300056 |
|
GO:0005615-extracellular
space | 7 | 0.001379 | MMP9, FN1,
SERPINE1, LAMC2, SPP1, COL1A1, ACTA1 | 4.984487946 | 1.401185 |
|
GO:0005578-proteinaceous
extracellular matrix | 4 | 0.002179 | MMP9, MMP11, FN1,
MMP1 | 14.31578947 | 2.205664 |
|
GO:0030175-filopodium | 3 | 0.002199 | FAT1, ITGA6,
ACTA1 | 40.52779837 | 2.225839 |
|
GO:0070062-extracellular
exosome | 9 | 0.003242 | MMP9, FN1,
TMPRSS11D, SERPINE1, CEACAM1, SPP1, FAT1, GPD1L, ACTA1 | 3.070943099 | 3.265211 |
|
GO:0030027-lamellipodium | 3 | 0.010687 | ABLIM1, FAT1,
ACTA1 | 17.98421053 | 10.40403 |
|
GO:0048471-perinuclear region
of cytoplasm | 4 | 0.021939 | ATP2A1, LAMC2,
SPP1, FAT1 | 6.178150691 | 20.29296 |
|
GOTERM_MF_DIRECT |
|
GO:0004252-serine-type
endopeptidase activity | 4 | 0.00235 | MMP9, MMP11,
TMPRSS11D, MMP1 | 13.93684211 | 2.240613 |
|
GO:0004222-metalloendopeptidase
activity | 3 | 0.006336 | MMP9, MMP11,
MMP1 | 23.58779693 | 5.938097 |
|
GO:0003779-actin binding | 3 | 0.034758 | MYH7, ABLIM1,
CEACAM1 | 9.587845513 | 28.8746 |
|
GO:0005509-calcium ion
binding | 4 | 0.038688 | MMP11, ATP2A1,
MMP1, FAT1 | 4.956617485 | 31.61489 |
| KEGG_PATHWAY |
|
hsa04512: ECM-receptor
interaction | 6 |
2.08×10−7 | FN1, LAMC2, SPP1,
COL4A1, ITGA6, COL1A1 | 36.65782493 |
1.93×10−4 |
|
hsa04510: Focal adhesion | 6 |
1.50×10−5 | FN1, LAMC2, SPP1,
COL4A1, ITGA6, COL1A1 | 15.48170276 | 0.013864 |
|
hsa04151: PI3K-Akt signaling
pathway | 6 |
1.78×10−4 | FN1, LAMC2, SPP1,
COL4A1, ITGA6, COL1A1 | 9.244147157 | 0.165019 |
|
hsa05200: Pathways in
cancer | 6 |
3.29×10−4 | MMP9, FN1, MMP1,
LAMC2, COL4A1, ITGA6 | 8.115091016 | 0.304375 |
|
hsa05222: Small cell lung
cancer | 4 |
3.65×10−4 | FN1, LAMC2, COL4A1,
ITGA6 | 25.01357466 | 0.33712 |
|
hsa05146: Amoebiasis | 4 |
6.98×10−4 | FN1, LAMC2, COL4A1,
COL1A1 | 20.05805515 | 0.644143 |
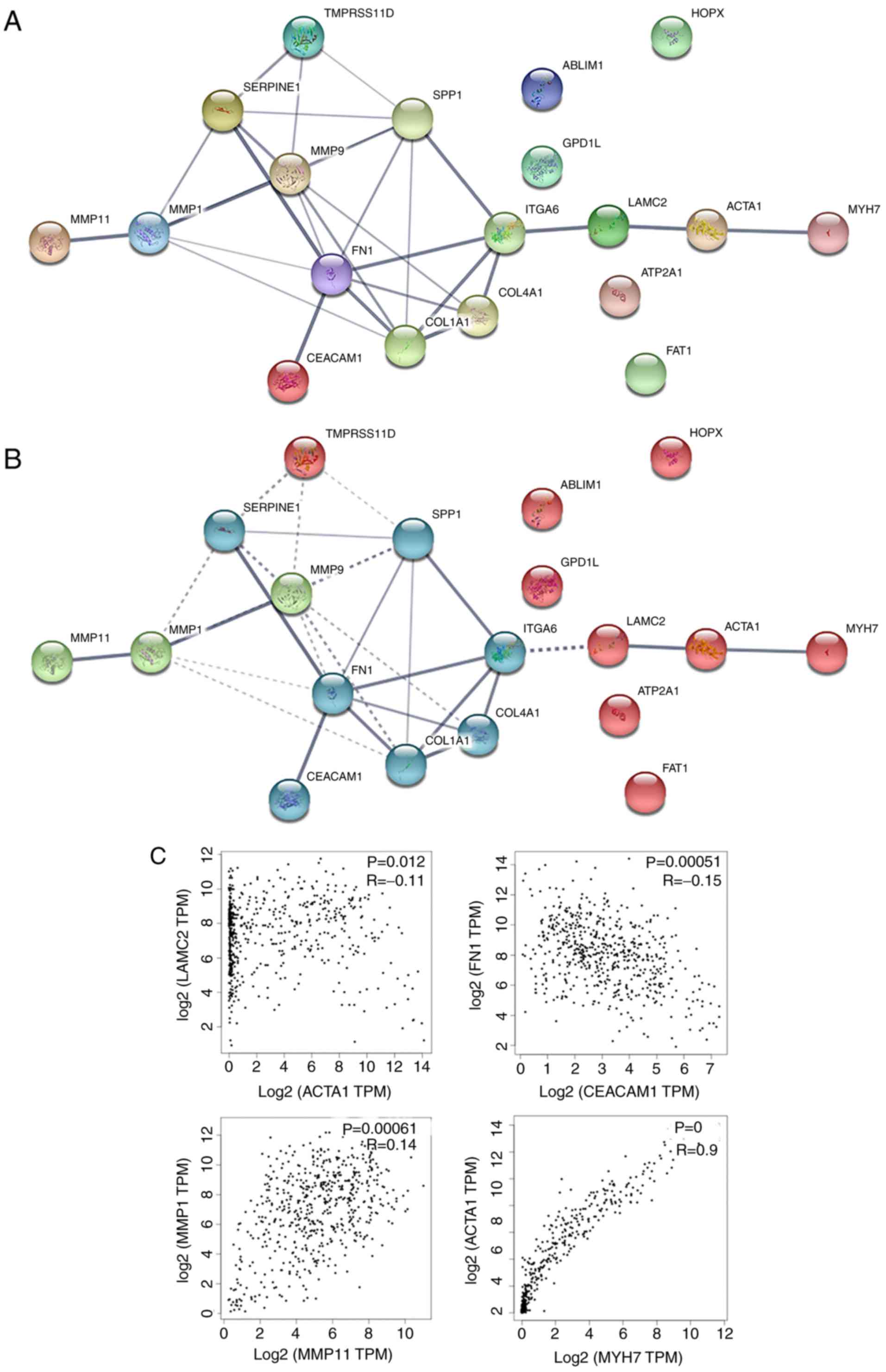
Construction and analysis of the PPI
network are implemented via STRING based on hub genes
In order to analyze the interaction of hub genes, a
PPI network was constructed using the online tool STRING. In total,
19 nodes and 29 edges were obtained (average node degree, 3.05; PPI
enrichment P-value, 7.55×10−15; meaning of network
edges, medium confidence score of 0.4), indicating a possibly high
correlation among these hub genes (Fig. 4A).
To confirm the possibility of interactions in the
PPI network, certain nodes of the PPI network were selected
according to the following aspects: Edge thickness (line thickness
indicates the strength of data support) (Fig. 4A), the results of the PPI
clustering (k-means clustering; number of clustering=3; Fig. 4B) and the principle that the
expression correlation means possible function interdependency.
Correlation analysis of the gene expression levels were further
verified using GEPIA, which is based on a large sample size from
TCGA and GTEx databases. The selected correlations consisted of
CEACAM1-FN1, MYH7-ACTA1, MMP11-MMP1 and ACTA1-LAMC2, and all the
correlations were verified using GEPIA (Fig. 4C). The aforementioned evidence
presumes that correlations among expression levels suggest that
associations exist on the functional level, and it is further
suggested that these genes may be associated with the development
or progression of HNSCC.
Nine hub genes are positively or
negatively associated with overall survival, and may be used as
potential tumor markers and prognostic indicators
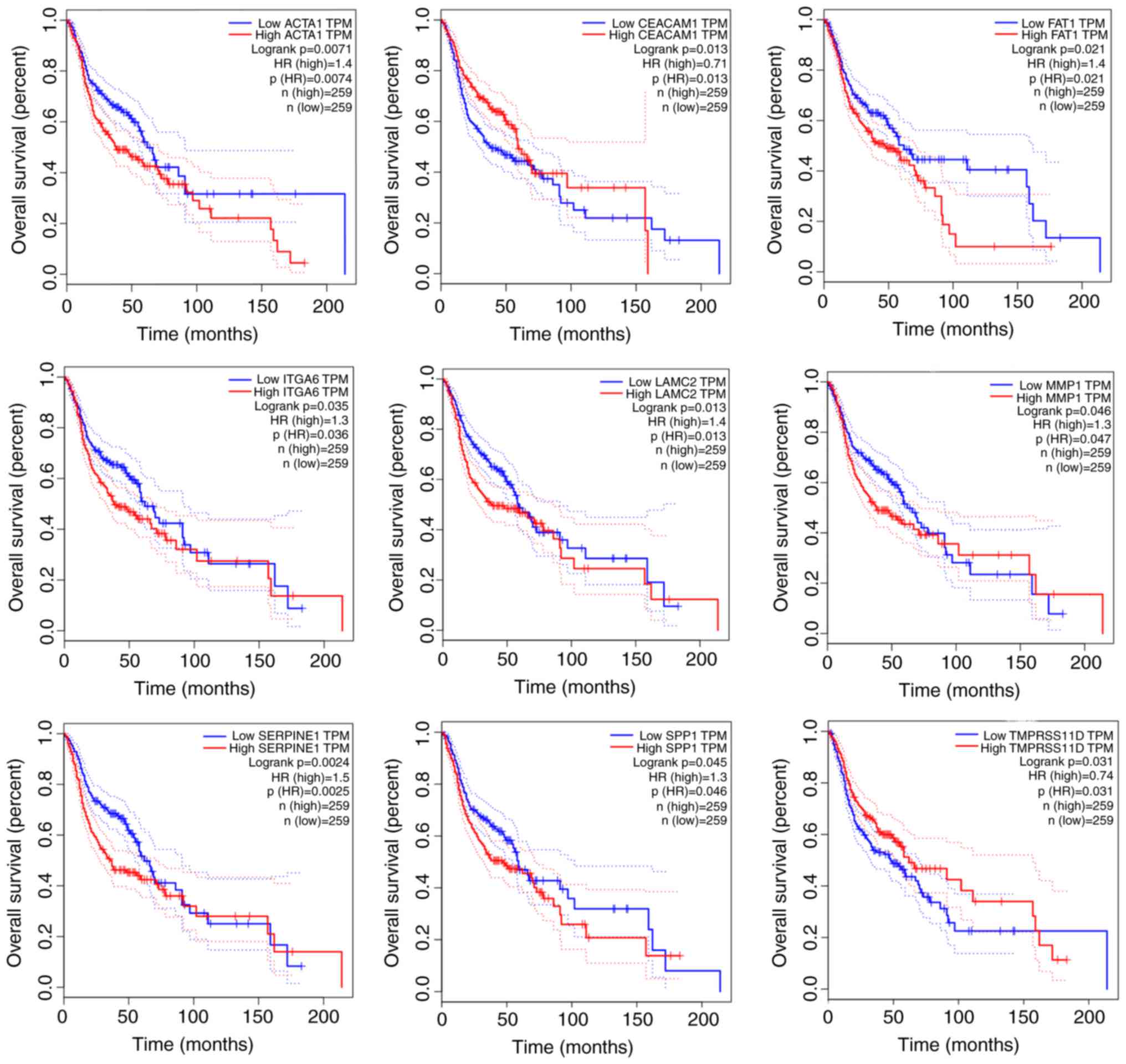
Based on the screening of tumor markers, overall
survival analysis of hub genes was performed using GEPIA. The
results suggested that nine hub genes (including SPP1, ITGA6,
TMPRSS11D, MMP1, LAMC2, FAT1, ACTA1, SERPINE1 and CEACAM1)
exhibited positive or negative associations with the overall
survival of patients with HNSCC, and these hub genes may be used as
potential tumor markers and prognostic indicators (Fig. 5). Nevertheless, the remaining hub
genes had no effect on the prognosis of patients and may be
involved in other processes of HNSCC development, which may be
attributed to the interaction of a variety of factors in the
occurrence of cancer. The nine hub genes validated by the online
database GEPIA, correlated with the prognosis of patients.
Furthermore, studies regarding the nine hub genes in HNSCC are
limited according to literature retrieval in NCBI/PubMed
(https://www.ncbi.nlm.nih.gov/pubmed;
key words, hub gene and HNSCC, ignore language, publication data
and article types). To the best of our knowledge, no previous
studies regarding the effects of TMPRSS11D and ACTA1 in HNSCC have
been performed to date.
Discussion
Microarray and high-throughput sequencing technology
provides valuable research information through profile screening of
small sample sizes. Gene expression profile microarrays provide
data from different samples, allowing for the identification of
DEGs using bioinformatics analysis that can be applied in further
studies (10,11,29).
Microarray data are stored in public databases for sharing and
re-mining, including GEO-datasets, TCGA and Oncomine (16,22,30).
Through data mining and bioinformatics analysis, researchers are
able to use public databases as valuable references for biological
research (11,17,31).
In the present study, three data series associated
with HNSCC (GSE83519, GSE58911 and GSE6631) were identified and
then deep mining of genomics data was performed with the aid of
bioinformatics software. The three included data series were based
on paired samples and the target genes were identified as the
common intersection of the three data series. The three data series
examined in the present study were all derived from paired tumor
and control samples of HNSCC patients, thus increasing the
reliability of the study. Furthermore, the common DEGs identified
demonstrated the same distribution tendency among the three data
series. These common DEGs may be regarded as results from three
experiment repetitions and an enlargement of the sample size, which
increases the reliability of the gene expression trend.
Comprehensive analysis of the above-mentioned databases confirmed
the reliability of data with regard to experimental reproducibility
and sample size. Furthermore, through screening and database
validation, the intersection of the selected DEGs was found to
include 19 mRNAs, which were defined as the hub genes and
considered to be highly correlated with HNSCC. The 19 hub genes
were differentially expressed in HNSCC simultaneously, suggesting
that these genes may independently or dependently be involved in
the occurrence or development of HNSCC. Thus, GO and KEGG pathway
analyses were adopted to perform functional enrichment of hub
genes, and these analyses categorized target genes in order to
identify genes of interest. Functional enrichment analysis
suggested that the highly correlated hub genes may participate in a
variety of biological processes. Additionally, a PPI network was
constructed based on the principle that proteins with associated
expression levels may interact with one another. The PPI network
and the network clustering results based on hub genes identified in
the present study indicated that MMP11, CEACAM1, MYH7 and ACTA1
were located in the first layer of the network. These genes were
suggested to be involved in the PPI network through higher binding,
as indicated by the edge thickness, which included
CEACAM1-FN1-SERPINE1, MYH7-ACTA1-LAMC2 and MMP11-MMP1-MMP9. In
order to further confirm these associations, the correlations among
connective nodes were verified based on large sample sizes using
GEPIA, following the principle that proteins with associated
expression levels may exhibit interactions, including CEACAM1-FN1,
MYH7-ACTA1, MMP11-MMP1 and ACTA1-LAMC2. The results suggested that
each protein in the primary line of the network exhibited a
positive or negative correlation. The results further demonstrated
that the hub genes participated in HNSCC through protein-protein
interactions.
The bioinformatics analysis conducted in the present
study provided important references for subsequent research;
however, it also has certain limitations. For instance, genes that
were differentially expressed among the three data sets were
screened as hub genes, which suggested that these genes may have
potential research value. However, useful information may be lost
in the screening process. For example, TP53, a routine tumor
suppressor, was screened out as it was only included in GSE83519,
but not in other two databases. However, as a conventional tumor
suppressor, TP53 has been demonstrated to be highly mutated and
hold important biological significance in HNSCC (32).
The principle aim of present study was to screen a
number of genes or proteins and provide a reference for clinical
diagnosis, treatment and prognosis of HNSCC. In line with the
purpose of screening tumor markers and prognostic indicators,
analysis of the association of survival with the expression of the
19 hub genes was performed using GEPIA. The results indicated that
nine of the hub genes (SPP1, ITGA6, TMPRSS11D, MMP1, LAMC2, FAT1,
ACTA1, SERPINE1 and CEACAM1) were identified as significant
prognostic indicators of HNSCC. To the best of our knowledge, few
studies have investigated these nine hub genes in HNSCC according
to literature retrieval using NCBI-PubMed. Previous studies have
revealed that the expression level of SPP1 in the plasma is
negatively associated with the survival time of the patients with
HNSCC (33), and this was
consistent with the results of the present study analysis; however,
these results remain controversial; another study concerning HNSCC
found no correlation between SPP1 expression level and prognosis of
HNSCC (34). In addition, it has
been reported that ITGA6 and LAMC2 promote the migration and
invasion of HNSCC cells (35), and
that MMP1 serves a role in the invasiveness of HNSCC (36). Furthermore, the mutation status of
FAT1 is associated with the prognosis of HPV-negative HNSCC
(37). A previous study also
reported that TMPRSS11D expression was reduced in squamous cell
carcinogenesis (38), whereas a
different study revealed that the expression of this gene was
significantly higher in non-small cell lung carcinoma tissues
compared with that in adjacent normal tissues (39). Nevertheless, previous studies
focusing on TMPRSS11D in HNSCC have not been identified. Notably,
the analysis of the present study suggested that the expression
level of TMPRSS11D was positively associated with the survival
time. Thus, it is necessary to perform further research regarding
the role of TMPRSS11D in HNSCC. A recent study reported that
CEACAM1 promotes the growth of HNSCC tumors (40). However, to the best of our
knowledge, no further studies on the underlying mechanisms have
been performed, and there appears to be only one other study
reporting that CEACAM1 is overexpressed in oral tumors and
correlated with carcinogenesis (41). In addition, previous data analysis
of colon cancer demonstrated that ACTA1 may be associated with the
methylation status of the oncogenome (42), but no further research has been
performed, particularly in HNSCC. Furthermore, a study reported
that SERPINE1 promotes cell migration, and overexpression of
SERPINE1 was revealed to be associated with poor survival in HNSCC
(43). All of the aforementioned
studies demonstrate that the mRNAs or proteins identified through
bioinformatics analysis in the present study are involved in the
biological process of HNSCC, which was in accordance with our
hypothesis. Notably, no studies investigating TMPRSS11D and ACTA1
in HNSCC have been identified. Although the aforementioned
literature analysis is only based on PubMed search, which is an
authoritative and comprehensive database, it also reflects the
research status of these genes to a certain extent. Therefore, the
present study also provided a practical and feasible direction for
further research.
Through rigorous statistical analysis,
bioinformatics analysis is able to provide theoretical biological
mechanisms and promote the development of biological research
(13,15). However, further verification is
required to confirm the results presented in the current study. In
the initial analysis, the sample size of the three enrolled
databases was small. If data from the TCGA or Oncomine databases,
which contain more samples, are reasonably integrated in the
initial analysis, the validity of the results of the present study
will be increased.
In conclusion, in the current study, a novel set of
core genes associated with HNSCC that may serve as potential
biomarkers was identified based on a series of bioinformatics
analyses and a large sample database. However, further validation
is required to verify these results.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Key Program of
Hebei Natural Science Foundation (grant no. H2017206391) and the
Project of Clinical Medical Talent Training and Basic Project
Research Funded by Government [grant Hebei finance society (2017)
no. 46].
Availability of data and materials
All data generated and analyzed during current study
were extracted from public databases (NCBI-GEO, GEPIA, DAVID and
STRING).
Authors' contributions
BSW conceived and designed the study; LZ contributed
to the acquisition, analysis and interpretation of data, and was
involved in drafting the manuscript; WWC and HC partially designed
and revised the manuscript for important intellectual content; WNC
and WXM participated in the data analysis and drafting of the
manuscript; WG was involved in the data analysis and the production
of figures.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
HPV
|
human papilloma virus
|
|
KDM5B
|
lysine (K)-specific demethylase 5B
|
|
GNG7
|
guanine nucleotide-binding protein
γ-7
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
GEO
|
Gene Expression Omnibus
|
|
DEGs
|
differentially expressed genes
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GEPIA
|
Gene Expression Profiling Interactive
Analysis
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DAVID
|
The Database for Annotation
Visualization and Integrated Discovery
|
|
PPI
|
protein-protein interaction
|
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN. Int J Cancer. 136:E359–E386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Magnes T, Egle A, Greil R and Melchardt T:
Update on squamous cell carcinoma of the head and neck: ASCO annual
meeting. Memo. 10:220–223. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tan H, Zhu G, She L, Wei M, Wang Y, Pi L,
Chen C, Zhang D, Tan P, Chen J, et al: MiR-98 inhibits malignant
progression via targeting MTDH in squamous cell carcinoma of the
head and neck. Am J Cancer Res. 7:2554–2565. 2017.PubMed/NCBI
|
|
4
|
Ardalan Khales S, Ebrahimi E, Jahanzad E,
Ardalan Khales S and Forghanifard MM: MAML1 and TWIST1
co-overexpression promote invasion of head and neck squamous cell
carcinoma. Asia Pac J Clin Oncol. Jan 15–2018.(Epub ahead of
print). doi: 10.1111/ajco.12843. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang D, Qiu Y, Li G, Liu C, She L, Zhang
D, Chen X, Zhu G, Zhang X, Tian Y, et al: KDM5B overexpression
predicts a poor prognosis in patients with squamous cell carcinoma
of the head and neck. J Cancer. 9:198–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trivedi S, Mattos J, Gooding W, Godfrey TE
and Ferris RL: Correlation of tumor marker expression with nodal
disease burden in metastatic head and neck cancer. Otolaryngol Head
Neck Surg. 149:261–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang H, Kiess A and Chung CH: Emerging
biomarkers in head and neck cancer in the era of genomics. Nat Rev
Clin Oncol. 12:11–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng L, Wang R, Lian M, Ma H, He N, Liu H,
Wang H and Fang J: Integrated analysis of long noncoding RNA and
mRNA expression profile in advanced laryngeal squamous cell
carcinoma. PLoS One. 11:e01692322016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li CQ, Huang GW, Wu ZY, Xu YJ, Li XC, Xue
YJ, Zhu Y, Zhao JM, Li M, Zhang J, et al: Integrative analyses of
transcriptome sequencing identify novel functional lncRNAs in
esophageal squamous cell carcinoma. Oncogenesis. 6:e2972017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seitz AK, Christensen LL, Christensen E,
Faarkrog K, Ostenfeld MS, Hedegaard J, Nordentoft I, Nielsen MM,
Palmfeldt J, Thomson M, et al: Profiling of long non-coding RNAs
identifies LINC00958 and LINC01296 as candidate oncogenes in
bladder cancer. Sci Rep. 7:3952017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zou AE, Ku J, Honda TK, Yu V, Kuo SZ,
Zheng H, Xuan Y, Saad MA, Hinton A, Brumund KT, et al:
Transcriptome sequencing uncovers novel long noncoding and small
nucleolar RNAs dysregulated in head and neck squamous cell
carcinoma. RNA. 21:1122–1134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Lena PG, Paz-Gallardo A, Paramio JM and
Garcia-Escudero R: Clusterization in head and neck squamous
carcinomas based on lncRNA expression: molecular and clinical
correlates. Clin Epigenetics. 9:362017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu S, Kong D, Chen Q, Ping Y and Pang D:
Oncogenic long noncoding RNA landscape in breast cancer. Mol
Cancer. 16:1292017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong N, Khwaja SS, Baker CM, Gay HA,
Thorstad WL, Daly MD, Lewis JS Jr and Wang X: Prognostic microRNA
signatures derived from The Cancer Genome Atlas for head and neck
squamous cell carcinomas. Cancer Med. 5:1619–1628. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laukens K, Naulaerts S and Berghe WV:
Bioinformatics approaches for the functional interpretation of
protein lists: From ontology term enrichment to network analysis.
Proteomics. 15:981–996. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar
R, et al: NCBI GEO: Mining tens of millions of expression
profiles-database and tools update. Nucleic Acids Res.
35:D760–D765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Demokan S, Chuang AY, Chang X, Khan T,
Smith IM, Pattani KM, Dasgupta S, Begum S, Khan Z, Liegeois NJ, et
al: Identification of guanine nucleotide-binding protein γ-7 as an
epigenetically silenced gene in head and neck cancer by gene
expression profiling. Int J Oncol. 42:1427–1436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang GJ, Luo MS, Chen GP and Fu MY:
MiRNA-mRNA crosstalk in laryngeal squamous cell carcinoma based on
the TCGA database. Eur Arch Otorhinolaryngol. 275:751–759. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saeed AI, Sharov V, White J, Li J, Liang
W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et
al: TM4: A free, open-source system for microarray data management
and analysis. Biotechniques. 34:374–378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene Ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
The Gene Ontology Consortium: Expansion of
the gene ontology knowledgebase and resources. Nucleic Acids Res.
45:D331–D338. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M, Araki M, Goto S, Hattori M,
Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T,
et al: KEGG for linking genomes to life and the environment.
Nucleic Acids Res. 36:D480–D484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yao J, Huang JX, Lin M, Wu ZD, Yu H, Wang
PC, Ye J, Chen P, Wu J and Zhao GJ: Microarray expression profile
analysis of aberrant long non-coding RNAs in esophageal squamous
cell carcinoma. Int J Oncol. 48:2543–2557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White NM, Cabanski CR, Silva-Fisher JM,
Dang HX, Govindan R and Maher CA: Transcriptome sequencing reveals
altered long intergenic non-coding RNAs in lung cancer. Genome
Biol. 15:4292014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stransky N, Egloff AM, Tward AD, Kostic
AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C,
McKenna A, et al: The mutational landscape of head and neck
squamous cell carcinoma. Science. 333:1157–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Le QT, Sutphin PD, Raychaudhuri S, Yu SC,
Terris DJ, Lin HS, Lum B, Pinto HA, Koong AC and Giaccia AJ:
Identification of osteopontin as a prognostic plasma marker for
head and neck squamous cell carcinomas. Clin Cancer Res. 9:59–67.
2003.PubMed/NCBI
|
|
34
|
Lim AM, Rischin D, Fisher R, Cao H, Kwok
K, Truong D, McArthur GA, Young RJ, Giaccia A, Peters L, et al:
Prognostic significance of plasma osteopontin in patients with
locoregionally advanced head and neck squamous cell carcinoma
treated on TROG 02.02 phase III trial. Clin Cancer Res. 18:301–307.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kinoshita T, Nohata N, Hanazawa T, Kikkawa
N, Yamamoto N, Yoshino H, Itesako T, Enokida H, Nakagawa M, Okamoto
Y and Seki N: Tumour-suppressive microRNA-29 s inhibit cancer cell
migration and invasion by targeting laminin-integrin signalling in
head and neck squamous cell carcinoma. Br J Cancer. 109:2636–2645.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kidacki M, Lehman HL, Green MV, Warrick JI
and Stairs DB: p120-catenin downregulation and PIK3CA mutations
cooperate to induce invasion through MMP1 in HNSCC. Mol Cancer Res.
15:1398–1409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim KT, Kim BS and Kim JH: Association
between FAT1 mutation and overall survival in patients with human
papillomavirus-negative head and neck squamous cell carcinoma. Head
Neck. 38 Suppl 1:E2021–E2029. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Duhaime MJ, Page KO, Varela FA, Murray AS,
Silverman ME, Zoratti GL and List K: Cell surface human airway
trypsin-like protease is lost during squamous cell carcinogenesis.
J Cell Physiol. 231:1476–1483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cao X, Tang Z, Huang F, Jin Q, Zhou X and
Shi J: High TMPRSS11D protein expression predicts poor overall
survival in non-small cell lung cancer. Oncotarget. 8:12812–12819.
2017.PubMed/NCBI
|
|
40
|
Tam K, Schoppy DW, Shin JH, Tay JK,
Moreno-Nieves U, Mundy DC and Sunwoo JB: Assessing the impact of
targeting CEACAM1 in head and neck squamous cell carcinoma.
Otolaryngol Head Neck Surg. 159:76–84. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang FF, Guan BX, Yang JY, Wang HT and
Zhou CJ: CEACAM1 is overexpressed in oral tumors and related to
tumorigenesis. Med Mol Morphol. 50:42–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu J, Li H, Sun L, Wang Z, Xing C and
Yuan Y: Aberrantly methylated-differentially expressed genes and
pathways in colorectal cancer. Cancer Cell Int. 17:752017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pavón MA, Arroyo-Solera I, Téllez-Gabriel
M, León X, Virós D, López M, Gallardo A, Céspedes MV, Casanova I,
López-Pousa A, et al: Enhanced cell migration and apoptosis
resistance may underlie the association between high SERPINE1
expression and poor outcome in head and neck carcinoma patients.
Oncotarget. 6:29016–29033. 2015. View Article : Google Scholar : PubMed/NCBI
|