Introduction
Industrial wastewater from diverse industrial
processes, including steel manufacturing, electroplating, leather
tanning and wood preservation, are responsible for the discharge of
chromium (Cr) into the environment (1). Cr exists in a number of oxidation
states in nature, of which the hexavalent [Cr(VI)] and trivalent
[Cr(III)] states are the most stable forms with biological
significance (2). Cr(III) is an
essential trace nutrient that is necessary for lipid and glucose
metabolism, whereas Cr(VI), a class IA human carcinogen that was
recognized in 1990, is the most toxic form of Cr due to its rapid
permeability through biological membranes and subsequent
interactions with intracellular proteins, lipids, DNA and other
biological macromolecules (3).
Cr(VI) and its compounds have long been considered carcinogens,
that primarily cause lung cancer via the inhalation route. In 2015,
Karagiannis et al (4)
conducted an epidemiological study in Greece which confirmed that
exposure to Cr(VI) by drinking water elevated the incidence of
primary liver cancer. Since then, general interest surrounding
Cr(VI)-induced hepatotoxicity has consistently increased, with a
shift in the prinicpal focus of investigation from lung cancer to
liver cancer.
The ability of the mitochondria to produce reactive
oxygen species (ROS) was first demonstrated in 1961 by Jensen
(5), and it is certain that
mitochondria are the primary source of cellular superoxide and
hydrogen peroxide in the majority of cell types. The production of
mitochondrial ROS, including oxygen free radicals, such as
superoxide anion radicals (O2˙−) and hydroxyl
radicals (˙OH), and non-radical oxidants, such as
singlet oxygen (1O2) and hydrogen peroxide
(H2O2), is involved in the pathogenesis of
various diseases and disorders, including Alzheimer's disease,
Parkinson's disease and amyotrophic lateral sclerosis; this is due
to its harmful effects on proteins, lipids and DNA that may cause
cell damage and even death (6). A
previous study demonstrated that mitochondrial respiratory chain
complex (MRCC) I (NADH-ubiquinone oxidoreductase) was the principal
source of ROS as a consequence of electron leakage during
respiration in mitochondria under pathological conditions; however,
not under resting and healthy conditions; therefore, it is not
unexpected that >40% of all mitochondrial-associated disorders
are associated with mutations in subunits of MRCC I (7). MRCC III [cytochrome c (Cyt C)
reductase] is additionally considered to be the primary site on the
electron transfer chain to generate ROS (8); however, there remains a lack of exact
mechanistic knowledge of the architecture of mitochondrial
ROS-generation systems, including MRCC I and III, and of detailed
insights into the molecular mechanisms controlling their expression
or activities. The mitochondrial permeability transition pore
(mPTP) is a voltage- and Ca2+-dependent channel, the
prolonged opening of which maintains the permeability of the inner
mitochondrial membrane to solutes with a molecular weight <1,500
Da (9). It is widely recognized
that brief mPTP openings have an essential physiological role in
maintaining healthy mitochondrial homeostasis and functions.
Mitochondrial membrane potential (MMP) disruption has been
confirmed to be involved in a variety of apoptotic phenomena,
including cytochrome c (Cyt C) release and caspase
activation. ROS are key inducers of mPTP opening, which ultimately
progress to MMP collapse, and initiate apoptotic pathways by
promoting the release of Cyt C and apoptosis inducing factor (AIF)
(10).
The endoplasmic reticulum (ER) is a complex,
specialized organelle with functions including the synthesis and
posttranslational modification of proteins, metabolism of lipids
and carbohydrates, and homeostatic control of intracellular
Ca2+ and the redox system. It has been confirmed that
the principal Ca2+ release channels from the ER are
ryanodine receptors in excitable cells and inositol
1,4,5-trisphosphate (IP3) receptors (IP3R) in non-excitable cells,
including hepatocytes; ER Ca2+ release via IP3R is
initiated by binding of the signaling molecule IP3.
Glucose-regulated protein 78 (GRP78), an ER chaperone that is
involved in protein processing and the provision of cellular
protection, is used as a monitor of ER stress (ERS) (11). ERS may be alleviated by the
unfolded protein response (UPR) in the early stage. A previous
study identified that the apoptotic response is mediated through
activation of the ERS-associated pro-apoptotic marker
CCAAT/enhancer-binding protein homologous protein (CHOP/GADD153)
primarily by three UPR signal pathways of the RNA-activated protein
kinase-like ER kinase (PERK), inositol-requiring
enzyme-1/X-box-binding protein (IRE1/XBP-1) and activating
transcription factor 6 (ATF6) (12). Therefore, CHOP is considered to be
the target gene of the UPR signal pathways and pro-apoptosis during
ERS.
Although it is well demonstrated that mitochondrial
damage and ERS are involved in heavy metal-induced cytotoxicity
(13), the role of mitochondrial
damage in Cr(VI)-induced ERS, and the correlation between the two,
have not been described and remain to be elucidated. In the present
study, the ability of Cr(VI) to induce ERS in L-02 hepatocytes was
first evaluated by describing mitochondrial damage and the
associated mechanism, following which the role of ROS-mediated
mitochondria damage in Cr(VI)-induced ERS was investigated. The
present data indicate the role of mitochondrial damage in ERS and
provide novel insight into the elucidation of Cr(VI)-induced
cytotoxicity.
Materials and methods
Reagents
RPMI-1640 medium, trypsin-EDTA (0.25%) and fetal
bovine serum (FBS) were obtained from Gibco (Thermo Fisher
Scientific, Inc.; Waltham, MA, USA). Potassium dichromate was
obtained from Changsha Chemical Reagents Company (Changsha, China).
Antibodies specific for CHOP (L63F7; cat. no. 2895), PERK (D11A8;
cat. no. 5683), IRE1α (14C10; cat. no. 3294), GRP78/BiP (C50B12;
cat. no. 3177) and GAPDH (D16H11; cat. no. 5174) were obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies
specific for IP3R2 (C-20; cat. no. sc-7278), AIF (B-9; cat. no.
sc-55519), Cyt C (6H2; cat. no. sc-13561), caveolin-1 (Cav-1;
4H312; cat. no. sc-70516), phosphoinositide 3-kinase (PI3K; 4F3;
cat. no. sc-293172), protein kinase B (AKT) 1 (cat. no. sc-135829),
phospho-AKT (p-AKT; Ser 473; cat. no. sc-7985-R), goat anti-rabbit
immunoglobulin G (IgG)-horseradish peroxidase (HRP; cat. no.
sc-2004), rabbit anti-goat IgG-HRP (cat. no. sc-2768) and goat
anti-mouse IgG-HRP (cat. no. sc-2005) were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Caspase-3 antibody
(cat. no. 19677-1-AP) was obtained from ProteinTech Group, Inc.
(Wuhan, China). The primary antibodies for the MRCCs, including
Complex I subunit NDUFS3 (cat. no. MS110), Complex II (succinate
dehydrogenase) subunit 70 kDa Fp (cat. no. MS204), Complex III
subunit Core 2 (cat. no. MS304), Complex IV (Cyt C oxidase) subunit
II (cat. no. MS405), and complex V (ATP synthase) subunit α (cat.
no. MS502) were purchased from MitoSciences, Inc. (Eugene, OR,
USA). All other chemicals and solvents were of analytical grade or
superior pharmaceutical grade.
Cell culture
The immortalized human L-02 hepatocyte cell line was
provided by The China Center for Type Culture Collection of Wuhan
University (Wuhan, China). The cells were maintained in RPMI-1640
with 10% (v/v) FBS, 2 mM L-glutamine, and antibiotics (50 U/ml
penicillin and 50 µg/ml streptomycin) in a 5% CO2
environment at 37°C, as previously described (14).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) for gene expression
analysis
RT-qPCR analysis was performed as previously
described (15). Total cellular
RNA of L-02 hepatocytes was prepared using an RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany) according to the manufacturer's
protocol. Total cDNA was synthesized using a PrimeScript RT
reagents kit (Takara Biotechnology, Co., Ltd., Dalian, China). qPCR
analysis was performed using a LightCycler® 96 Sequence
Detection System (Roche Diagnostics, Basel, Switzerland) in a 20 µl
reaction containing 10 µM of each primer, 1 µl template cDNA, 10 µl
SYBR Premix EX Taq (SYBR® Premix Ex Taq™ II (Tli RNaseH
Plus; Takara Bio, Inc., Otsu, Japan), and 0.25 µl ROX reference
dye. The PCR was run at 95°C for 30 sec followed by 45 cycles of
95°C for 5 sec and 60°C for 34 sec (15). ACTB was used as a control. Gene
expression was calculated using the comparative threshold cycle
2−∆∆Cq method (16).
The data were collected from three separate experiments. The
forward (F) and reverse (R) primer sequences were as follows: CHOP,
(F) 5′-TTCTCGGGCAGGGCGTACTGA-3′ and (R) 5′-TGGTGCCCTTCTTCCTTCCC-3′;
PERK, (F) 5′-TCAGCCTTCACCTTAGGCCGA-3′ and (R)
5′-AAGCCTCTGCTCCCTTTCCTAC-3′; IRE1, (F)
5′-TGCATTAGGACATATGCGCCCTAA-3′ and (R) 5′-CTAAGGCTGCTCCACGTGCA-3′
(R); GRP78, (F) 5′-CATACCCCGTATCCTGTCG-3′ and (R)
5′-CGAATCAGATGCCGTTCGCT-3′; ACTB, (F) 5′-CACGACGGCGTGTAGGT-3′ and
(R) 5′-CTCCAAAATATGCTGGGTCAT-3′.
Immunoblot for protein expression
The hepatocytes were lysed using a Mammalian Cell
Lysis kit obtained from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany). The protein concentrations of the mitochondrial
suspensions were determined using the Bradford Coomassie blue
protein-binding (17). Protein
samples containing 50 µg protein were separated by 12% SDS-PAGE and
subsequently transferred onto a polyvinylidene difluoride membrane.
The membranes were blocked for 1 h at room temperature with 4%
non-fat milk and immunostained with primary antibodies (1:1,000)
overnight at 4°C. The membranes were incubated with the appropriate
secondary antibodies (1:1,000) for 1 h at room temperature,
developed with the Super Enhanced Chemiluminescence Detection kit
(Applygen Technologies, Inc., Beijing, China) and subsequently
exposed onto films.
Flow cytometry analysis for apoptotic
cells
For the determination of apoptotic cell death, the
L-02 hepatocytes were stained with Annexin V-Fluorescein
Isothiocyanate (FITC; 0.5 µg/ml final concentration) and propidium
iodide (PI; 1 µg/ml final concentration), analyzed on a flow
cytometer equipped with a 488 nm argon laser light source, and
evaluated using CellQuest software version 5.1 (BD Biosciences,
Franklin Lakes, NJ, USA). Apoptotic cell death was determined by
quantifying the population of Annexin V-FITC-positive cells (early
apoptotic) and Annexin V-FITC/PI-positive cells (late
apoptotic).
ER Ca2+ concentration
determination
Ca2+ concentration in the ER was detected
using the Intracellular Ca2+ Concentration in Cell
Endoplasmic Reticulum Detection kit (Genmed Scientifics, Inc.,
Shanghai, China). All procedures were performed according to the
manufacturer's protocol. The result was quantified using a
fluorescence spectrophotometer with excitation at 490 nm and
emission at 525 nm.
MMP assay
Variations in MMP were assessed using the
fluorescent cationic dye Rhodamine 123 (Rh123), which is a cationic
membrane-permeant fluorescent probe that accumulates in
mitochondria and is released upon membrane depolarization. The
treated hepatocytes were harvested, washed twice with PBS and
stained with Rh123 (2 µg/ml) for 30 min in the dark. The
fluorescence intensity was analyzed with a fluorescence
spectrometer at an excitation wavelength of 495 nm and an emission
wave length of 535 nm.
Measurement of mPTP opening
The treated L-02 hepatocytes were collected and
processed for mitochondrial isolation. The pellets of the treated
hepatocytes were washed twice with ice-cold PBS and resuspended
with five volumes of buffer A (250 mM sucrose, 20 mM HEPES, 10 mM
KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM
dithiothreitol, and 0.1 mM phenylmethylsulfonyl fluoride; pH 7.5).
They were left on ice for 2 min and subsequently homogenized with a
syringe (20–30 times, confirmed ~90% cells breakage occurred). The
homogenates were centrifuged twice at 1,500 × g for 15 min at 4°C.
The supernatant obtained was centrifuged at 10,000 × g for 15 min
at 4°C. The resulting obtained mitochondrial pellets were
resuspended in buffer A. The protein content in the mitochondrial
suspensions was determined using Coomassie Brilliant Blue (G-250)
by the Bradford method (17). The
opening of the PTPs was determined using a mitochondrial
permeability transition pore detection kit (Genmed Scientifics,
Inc.) according to the manufacturer's protocol. The results were
evaluated using a fluorescence spectrophotometer, with an
excitation wavelength of 488 nm and an emission wave length of 505
nm.
Measurement of the activity of MRCC
I–V
The activities of MRCC I–V were determined with
Mitochondrial Respiratory Chain Complexes Activity Assay kits
(Genmed Scientifics, Inc.). All experiments were performed
according to the protocol provided by the manufacturer. All
measurements were performed in triplicate.
Measurement of ROS production
The hepatocytes were exposed to different
concentrations of Cr(VI) (0, 8 and 16 µM) for 24 h with or without
the pretreatment with 10 mM N-acetyl-cysteine (NAC) for 1 h at
37°C. The production of ROS was measured using hydroethidine (HE;
Molecular Probes; Thermo Fisher Scientific, Inc.) in the
hepatocytes. HE is a non-fluorescent compound that is able to
diffuse through cell membranes and may be rapidly oxidized to
ethidium under the action of O2˙−. The
hepatocytes were treated with 2 µM HE for 15 min at 37°C, and were
subsequently analyzed by flow cytometry. For each sample,
104 cells were analyzed.
Statistical analysis
For all quantitative data collected, all values are
expressed as the mean ± standard deviation of at least three
independent experiments. Statistical significance was determined by
one-way analysis of variance followed by Dunnett's post-hoc test.
All statistical analyses were performed using SPSS 19.0 (IBM Corp,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Cr(VI) exposure induces ERS in
hepatocytes
The L-02 hepatocytes were exposed to different
concentrations of Cr(VI) (0, 8 and 16 µM) for 24 h and processed
for mRNA expression determination of ERS-associated genes, CHOP,
PERK, IRE1 and GRP78. As presented in Fig. 1A, Cr(VI) upregulated the mRNA
expression levels of all the detected genes in a
concentration-dependent manner. The western blot results, presented
in Fig. 1B, confirmed that Cr(VI)
additionally upregulated the relative protein expression levels. A
previous study suggested that ER stress is pivotal in cellular
apoptosis (18), the present study
examined whether Cr(VI)-induced ERS was accompanied by apoptotic
cell death. Caspase-3 is the primary executioner caspase in
apoptosis, and it was identified that Cr(VI) increased the
expression of caspase-3 in a concentration-dependent manner
(Fig. 1C). The flow cytometry
results (Fig. 1D) demonstrated
that Cr(VI) increased the population of early [Annexin V-FITC (+)]
and late [(Annexin V-FITC (+)/PI (+)] apoptotic cells.
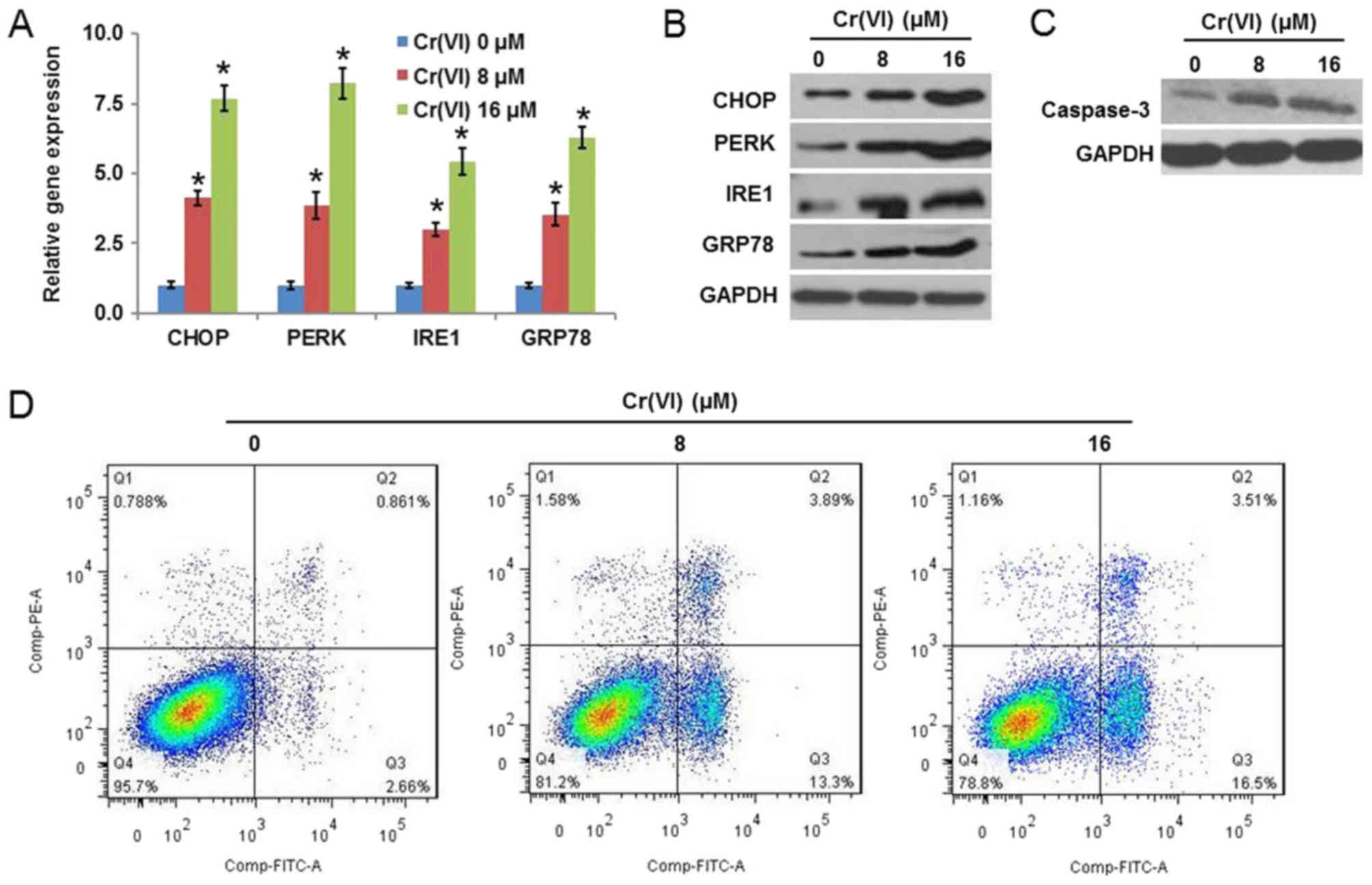 | Figure 1.Cr(VI) exposure induces ERS and
apoptosis in hepatocytes. The L-02 hepatocytes were exposed to
different concentrations of Cr(VI) (0, 8 and 16 µM) for 24 h. The
mRNA and protein expression levels of ERS-associated genes,
CHOP, PERK, IRE1 and GRP78 were determined using (A)
reverse transcription-quantitative polymerase chain reaction
analysis and (B) western blotting, respectively. (C) Expression of
caspase-3 was examined by western blotting. (D) Population of early
apoptotic cells [Annexin V-FITC (+)] and late apoptotic cells
[(Annexin V-FITC (+)/PI (+)] was determined by flow cytometry. All
values are expressed as the mean ± standard deviation and each
experiment was repeated at least three times. *P<0.05 vs. the
control. Cr(VI); hexavalent chromium; ERS, endoplasmic reticulum
stress; CHOP, CCAAT/enhancer-binding protein homologous protein;
PERK, RNA-activated protein kinase-like ER kinase; IRE1,
inositol-requiring enzyme-1; GRP78, glucose-regulated protein 78;
FITC, fluorescein isothiocyanate; PI, propidium iodide. |
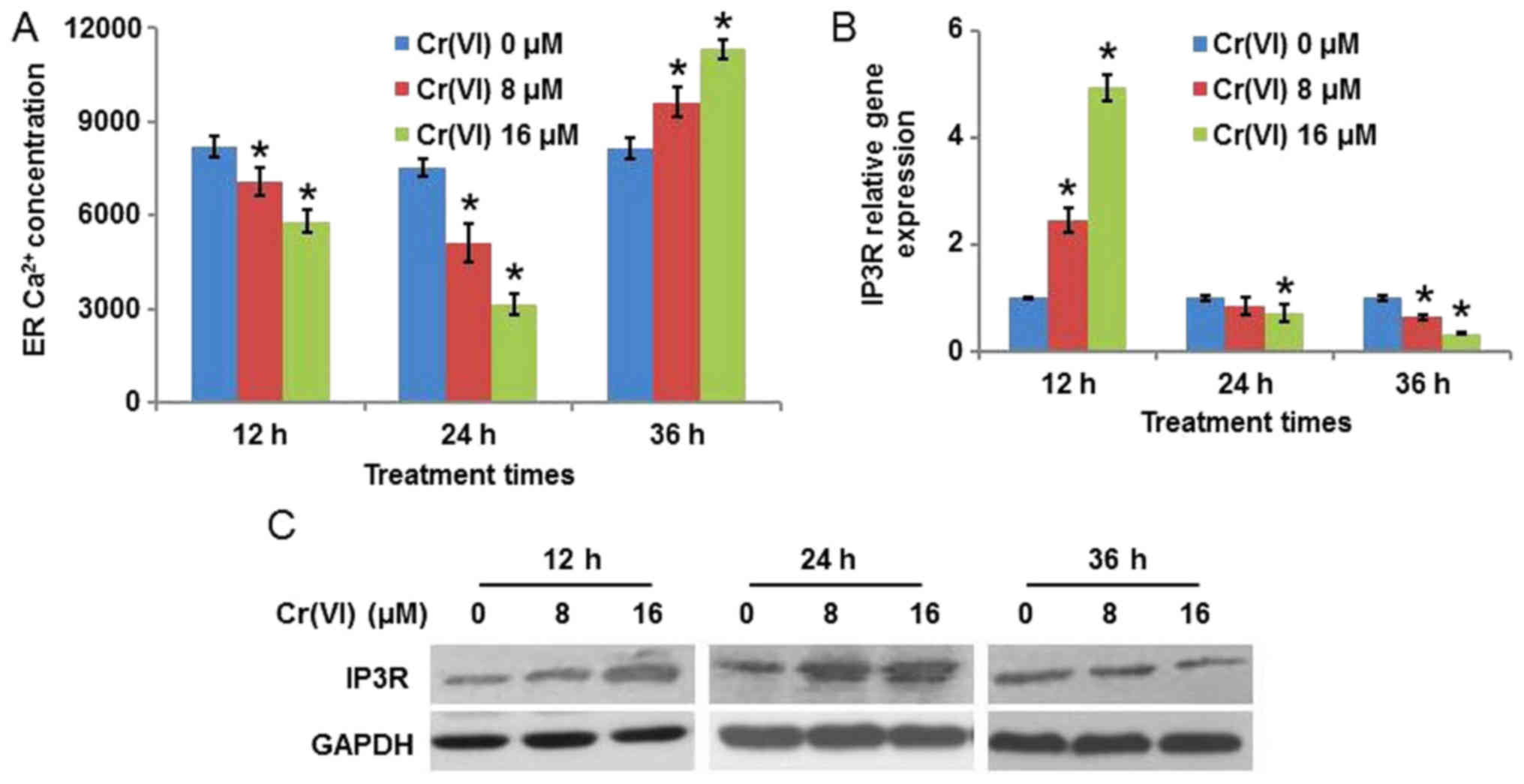
It is known that the disturbance of ER
Ca2+ homeostasis may lead to ER stress (19). As the concentration of
Ca2+ in the ER varies with time during ERS, the present
study determined ER Ca2+ concentration following
treatment with different concentrations (0, 8 and 16 µM) and
treatment durations (12, 24 and 36 h) of Cr(VI). As presented in
Fig. 2A, Cr(VI) decreased ER
Ca2+ concentration in a concentration-dependent manner
when the treatment times were 12 and 24 h, with the decrease more
marked at 24 h. Cr(VI) increased ER Ca2+ concentration
when the treatment time was 36 h. As the principal Ca2+
release channels from the ER are IP3R, the expression of IP3R in
Cr(VI)-treated hepatocytes was detected. It was identified that
following exposure to different concentrations of Cr(VI), the mRNA
expression level of IP3R was upregulated at 12 h, was downregulated
at 36 h, and demonstrated no obvious alteration at 24 h compared
with the control (Fig. 2B). The
protein expression levels, presented in Fig. 2C, demonstrated that the expression
of IP3R was increased at 12 and 24 h, with the increase being more
marked at 24 h, and decreased at 36 h following Cr(VI) exposure;
this suggested that Ca2+ release from the ER was
increased at 12 and 24 h, and decreased at 36 h, which explains the
result in Fig. 2A.
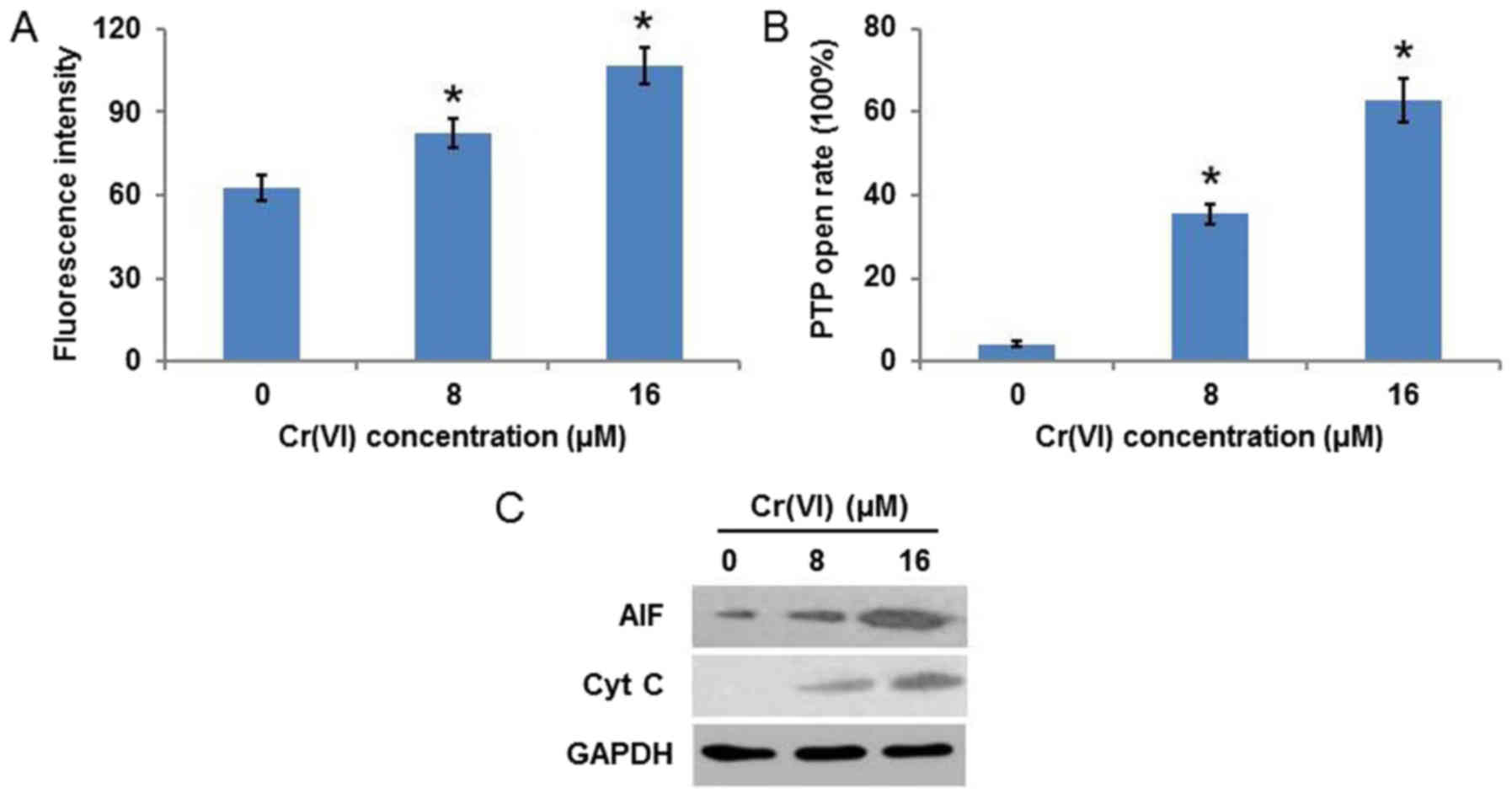
Cr(VI) induces mitochondrial
damage
Mitochondrial damage is frequently accompanied by a
decrease of MMP and an increase of mPTP opening. As demonstrated in
Fig. 3A, Cr(VI) increased the
fluorescence intensity in a concentration-dependent manner,
indicating the collapse of MMP. It was additionally identified that
Cr(VI) increased the mPTP opening rate (Fig. 3B). As the release of AIF and Cyt C
is associated with the loss of MMP and the increase in mPTP opening
(20), the expression of these
proteins was examined and it was observed that the protein
expression levels of AIF and Cyt C were increased in a
concentration-dependent manner (Fig.
3C). These results suggested that Cr(VI) induced a
mitochondrial-mediated apoptotic pathway in human L-02
hepatocytes.
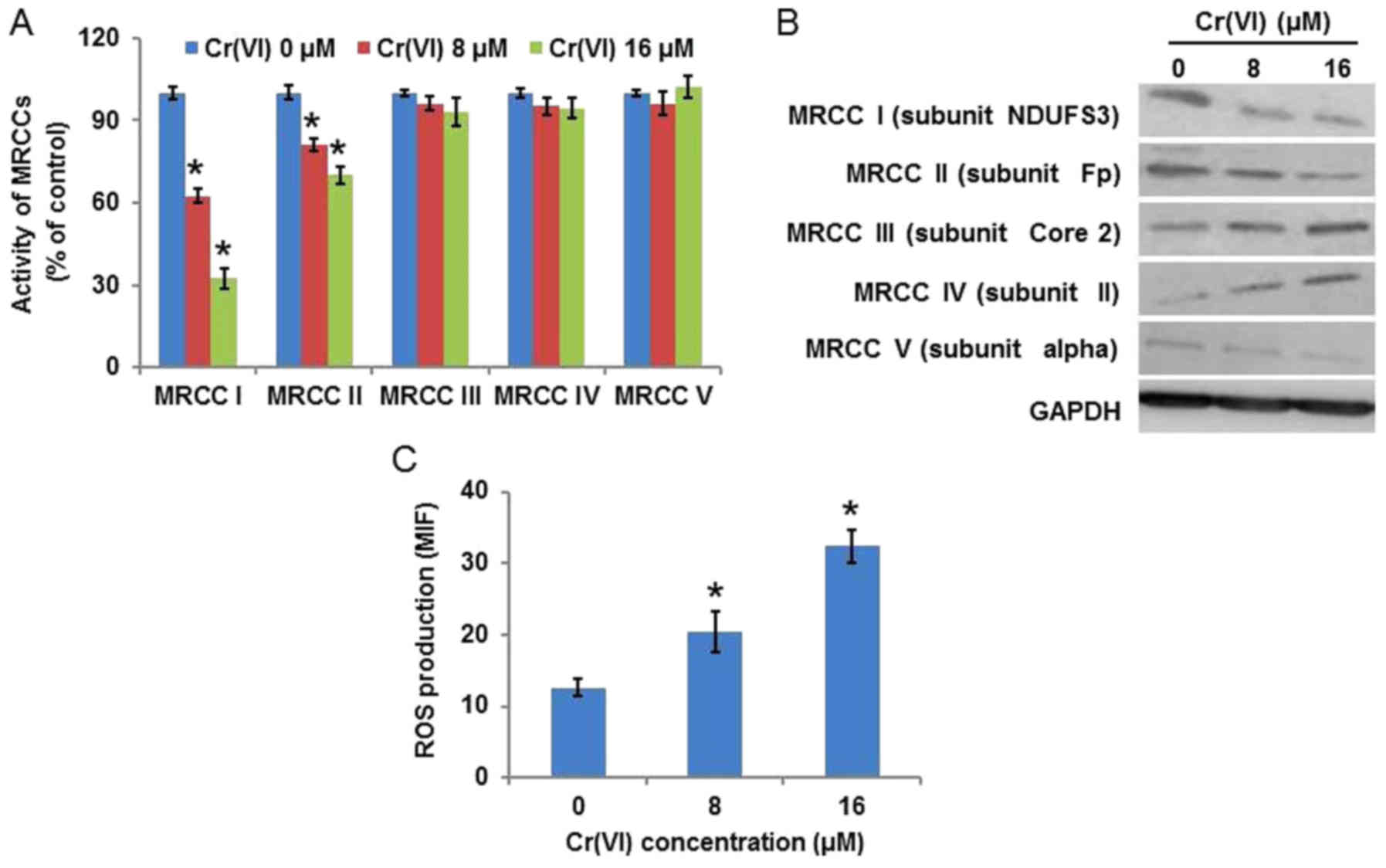
Cr(VI)-induced mitochondria damage is
associated with ROS
To elucidate the source of ROS, the activity of
MRCCs was examined (Fig. 4A), and
it was identified that Cr(VI) significantly inhibited the activity
of MRCC I, marginally inhibited the activity of MRCC II in a
concentration-dependent manner, and demonstrated no regulatory
effect on MRCC III, IV or V. The protein expression levels of MRCCs
(Fig. 4B) demonstrated similar
results. Increasing evidence suggests that ROS are essential in
various cytotoxic mechanisms induced by exogenous toxicant
exposure. ROS production was measured by HE staining and the result
was quantified by flow cytometry using the mean intensity of
fluorescence. As presented in Fig.
4C, Cr(VI) treatment increased ROS production in a
concentration-dependent manner, indicating the generation of a
large quantity of intracellular ROS.
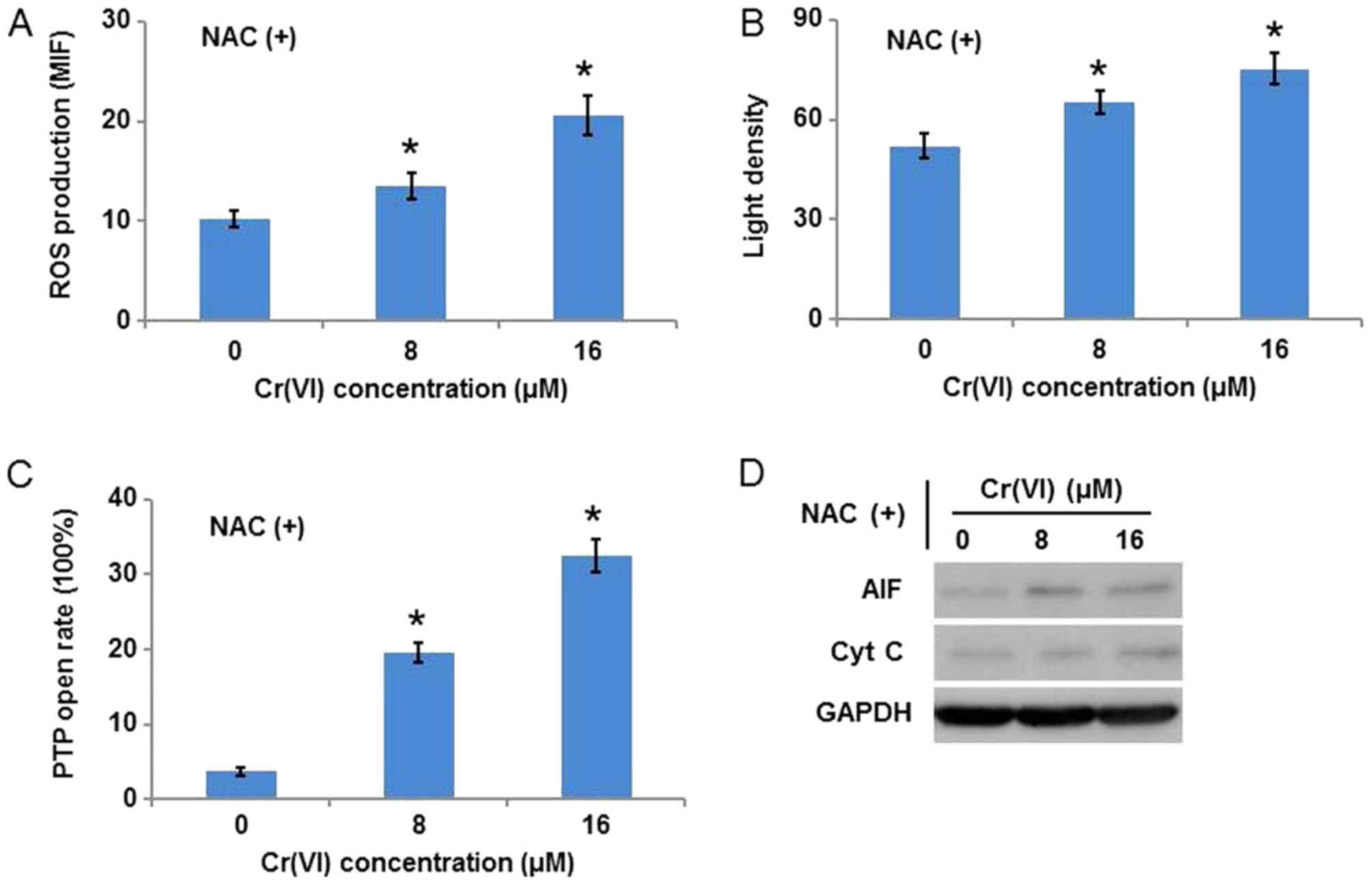
As it was inferred that ROS are key in
Cr(VI)-induced mitochondrial damage, the ROS scavenger, NAC, was
used to inhibit ROS. The hepatocytes were exposed to different
concentrations of Cr(VI) following pretreatment with 10 mM NAC for
1 h. As presented in Fig. 5A, ROS
production was inhibited following Cr(VI) exposure, which confirmed
the specificity of NAC. Notably, NAC application alleviated the
Cr(VI)-induced collapse of MMP (Fig.
5B), decreased the susceptibility of the hepatocytes to mPTP
opening (Fig. 5C), and inhibited
the release of AIF and Cyt C from mitochondria (Fig. 5D).
Role of ROS-mediated mitochondria
damage in Cr(VI)-induced ERS
The effect of NAC application on the mRNA expression
levels of ERS-associated genes following Cr(VI) exposure were
examined. The results (Fig. 6A)
demonstrated that NAC alleviated the Cr(VI)-induced increase of the
mRNA expression levels of CHOP, PERK, IRE1 and GRP78.
The protein expression levels (Fig.
6B) exhibited similar results. It was additionally identified
that NAC decreased the population of early and late apoptotic cells
following Cr(VI) exposure, indicating the inhibition of apoptotic
cell death (Fig. 6C). Cav-1 is the
most important component of caveolae that has been demonstrated to
regulate intracellular Ca2+ signals (21) and the PI3K/AKT pathway (22), and AKT is reported to regulate IP3R
(23). As presented in Fig. 6D, Cr(VI) treatment increased the
protein expression of Cav-1 and IP3R; however, decreased the
protein expression of PI3K, p-AKT (Ser 473) and AKT, indicating the
regulatory effect of the Cav-1/PI3K/AKT pathway on the expression
of IP3R. NAC application alleviated the Cr(VI)-induced alterations
of these proteins. These results suggested that ROS activated
Cav-1, which further abrogated the inhibitory effect of AKT on
IP3R. These findings suggest a cross-talk of apoptotic signaling
between the mitochondria and the ER, which is involved in
Cr(VI)-induced apoptosis.
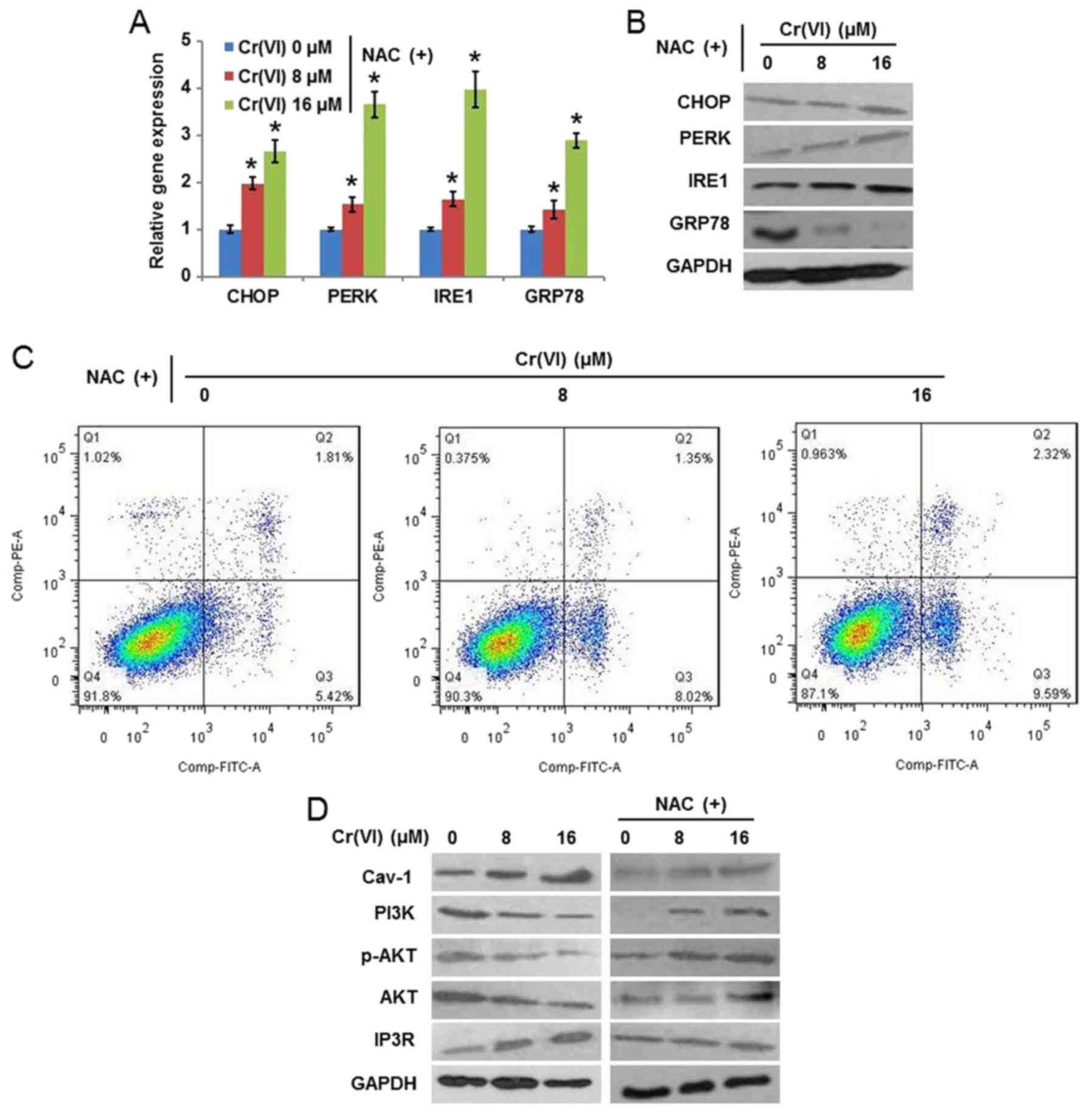 | Figure 6.Role of reactive oxygen
species-mediated mitochondrial damage in Cr(VI)-induced ERS. The
hepatocytes were pretreated with 10 mM NAC for 1 h and then exposed
to different concentrations of Cr(VI) (0, 8 and 16 µM) for 24 h.
The (A) mRNA and (B) protein expression of ERS-associated genes
were determined using reverse transcription-quantitative polymerase
chain reaction analysis and western blotting, respectively. (C)
Apoptotic cell death was examined by flow cytometry using Annexin
V-FITC/PI double staining. (D) Protein expression of Cav-1, PI3K,
p-AKT, AKT and IP3R was determined by western blotting. *P<0.05
vs. the control. Cr(VI); hexavalent chromium; NAC,
N-acetyl-cysteine; CHOP, CCAAT/enhancer-binding protein homologous
protein; PERK, RNA-activated protein kinase-like ER kinase; IRE1,
inositol-requiring enzyme-1; GRP78, glucose-regulated protein 78;
FITC, fluorescein isothiocyanate; PI, propidium iodide; Cav-1,
caveolin-1; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B;
p-AKT, phospho-AKT; IP3R, inositol 1,4,5-trisphosphate. |
Discussion
In the present study, it was demonstrated that
Cr(VI) upregulated ERS-associated genes, CHOP, PERK, IRE1
and GRP78, indicating that Cr(VI) was capable of inducing
ERS in L-02 hepatocytes. It was additionally identified that the
concentration of Ca2+ in the ER varied with time during
Cr(VI)-induced ERS. Cr(VI) decreased the ER Ca2+
concentration when the treatment durations were 12 and 24 h, and
increased the ER Ca2+ concentration when the treatment
lasted for 36 h. These were consistent with the alterations in the
protein expression of IP3R, which was increased at 12 and 24 h, and
decreased at 36 h. The mRNA and protein expression levels of IP3R
were decreased at 36 h post-Cr(VI) exposure, indicating the
existence of cellular self-protection mechanisms which alleviate
the marked Cr(VI)-induced release of Ca2+ from the ER.
Mitochondrial damage may be characterized by the collapse of MMP
and the increase of mPTP opening. To elucidate the source of ROS,
the activities and protein expression of MRCC I–V were detected. It
was identified that, contrary to a previous study that suggested
that MRCC I and III in mitochondria are important sites of ROS
generation in mammalian cells (24), MRCC I and II were inhibited,
whereas, other complexes were not affected by Cr(VI) exposure. It
was confirmed that Cr(VI) induced burst generation of ROS in the
hepatocytes. As redox homeostasis appears to be a critical factor
for the maintenance of normal functioning of mitochondria, a high
level of intracellular ROS (oxidative stress) is deleterious and
apparently had a causative effect on mitochondrial damage (25). Therefore, the balance between ROS
formation and removal allows for normal cellular function, whereas,
the imbalance causes oxidative stress and results in
pathobiological consequences (25). mPTP opening and MMP collapse are
additionally viewed as the mitochondrial response to various
oxidative stresses resulting in the amplified ROS signal (25). In the present study, the rapid loss
of MMP, the increase of mPTP opening, the release of Cyt C and AIF,
and the activation of caspase-3 were observed in hepatocytes
following Cr(VI) exposure.
It has been demonstrated that mitochondrial ROS
production increases ER Ca2+ release and mitochondrial
Ca2+ loading (26). In
our previous study, it was observed that Cr(VI) additionally
induced Ca2+ overload in the mitochondria, and the
increased levels of mitochondrial Ca2+ additionally
stimulated mitochondrial ROS production (27). The increased mitochondrial ROS
demonstrated damaging effects on mitochondrial membrane lipids,
which consequently resulted in the disruption of MMP and the
pathological opening of mPTPs; the increased mPTP opening
additionally led to further collapse of the MMP (28). This vicious circle may damage
mitochondria and result in rupture of the outer mitochondrial
membrane, with the consequent release of Cyt C and AIF from the
mitochondria into the cytosol, which ultimately activates the
caspase cascade and induces apoptosis (29). Additionally, it has been confirmed
that ROS-damaged mitochondria tend to produce more ROS in order to
activate mitochondrial-mediated apoptotic or necrotic pathways
(30).
Previous studies suggested that mitochondrial ROS
generation-induced altered redox homeostasis in the cell is
sufficient to initiate ERS, which may in turn induce the production
of ROS in the ER and mitochondria (31,32).
In the present study, it was demonstrated that mitochondrial ROS
generation initiates a destructive cycle involving mitochondrial
damage and ERS, which further increases ROS production and leads to
apoptotic cell death. Extensive evidence is accumulating that burst
generation of ROS can disturb ER protein folding and thus induce
ERS, which may activate the UPR to resolve this protein-folding
defect (33). ERS is known to
trigger three principal branches of the UPR, including the PERK,
IRE1 and ATF6 pathways, which serve as proximal sensors of the
protein folding status in the ER (34). Increasing literature suggests the
mitochondria and ER build a dynamic network where they cooperate in
the generation of Ca2+ signals (35). In addition, ROS accumulation
affects Ca2+ homeostasis in the ER, which is followed by
the activation of ER chaperone gene GRP78 to prevent intracellular
Ca2+ overload-induced cytotoxicity (36). Therefore, when ERS and ER
dysfunction occur beyond possibility of restoration, the activation
of pro-apoptotic signaling pathways can be viewed as the mechanism
to protect the organism by eliminating damaged cells. To further
confirm the functional role of ROS in Cr(VI)-mediated mitochondrial
damage and ERS in the hepatocytes, the ROS scavenger NAC was used
in the present study.
Cav-1, an oncoprotein and tumor suppressor, is known
to be the essential structural protein component of the plasma
membrane microdomains called caveolae and is associated with
various membranous structures, including the ER (37). Numerous ion channels and key
proteins involved in Ca2+ signals are located in
caveolae, indicating that caveolae may be important in the
regulation of Ca2+ signals (38). Cav-1 is a marker protein and the
most important structural component of caveolae. It has been
reported that ROS generation can promote the phosphorylation of
Cav-1 on tyrosine-14 and then result in the activation of Cav-1
(39). Previous evidence suggested
that Cav-1 positively regulates the PI3K/Akt pathway (40), whereas others demonstrated the
opposite result that Cav-1 negatively regulates the PI3K/AKT
pathway (41). It has additionally
been confirmed that AKT phosphorylates IP3R and subseuqently
suppresses the pro-apoptotic Ca2+-release function
(42), indicating that AKT may
negatively regulate IP3R. Therefore, it was suggested that Cr(VI)
induced IP3R-mediated Ca2+ release from the ER through
ROS/Cav-1/AKT signaling. It was confirmed in the present study that
Cr(VI) increased the protein expression levels of Cav-1 and IP3R;
however, decreased the protein expression levels of PI3K, p-AKT
(Ser 473) and AKT, indicating that Cav-1 was activated, whereas the
AKT pathway was inhibited following ROS accumulation induced by
Cr(VI). Therefore, it was demonstrated in the present study that
Cr(VI) induced Ca2+ release from the ER through
ROS/Cav-1/AKT/IP3R signaling. As it has additionally been reported
that Cav-1 may directly interact with IP3R (43), further investigations are required
to elucidate the possible molecular mechanism involved in
Cr(VI)-induced IP3R-mediated Ca2+ release from the
ER.
In conclusion, the present findings demonstrated
that Cr(VI) induced ERS in L-02 hepatocytes, which was
characterized by the upregulation of ERS-associated CHOP, PERK,
IRE1 and GRP78, and by the mass release of Ca2+ from the
ER. The Cr(VI)-induced mitochondrial production of ROS by
disturbing MRCC I and II was the key inducer of a destructive cycle
of mitochondrial damage involving mPTP opening and MMP collapse,
which further increased ROS production. These results additionally
provide detailed mechanistic information on how Cr(VI)-induced ROS
exerts its regulatory effects on principal Ca2+ release
channels from the ER, IP3R (activation of Cav-1 and inhbition of
the PI3K/AKT pathway), which confirmed that Cr(VI) induced the
release of Ca2+ from the ER through ROS/Cav-1/AKT/IP3R
signaling. These data demonstrated the role of mitochondrial damage
in Cr(VI)-induced ERS and provide novel insight into the
elucidation of Cr(VI)-induced cytotoxicity.
Acknowledgements
The authors would like to thank Professor Caigao
Zhong (Xiangya School of Public Health, Central South University,
Changsha, China) for his valuable ideas and suggestions.
Funding
The present study was financially supported by The
National Natural Science Foundation of China (grant no.
81773478).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FX designed the experiments, supervised the project
and wrote the manuscript. QL, YX and MH performed the experiments
and analyzed the data. YZ contributed to the conception of the
study and interpreted the data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
MRCC
|
mitochondrial respiratory chain
complex
|
|
mPTP
|
mitochondrial permeability transition
pore
|
|
MMP
|
mitochondrial membrane potential
|
|
AIF
|
apoptosis inducing factor
|
|
IP3
|
inositol 1,4,5-trisphosphate
|
|
CHOP/GADD153
|
CCAAT/enhancer-binding protein
homologous protein
|
|
PERK
|
RNA-activated protein kinase-like ER
kinase
|
|
IRE1/XBP-1
|
inositol-requiring
enzyme-1/X-box-binding protein
|
|
ATF6
|
activating transcription factor 6
|
References
|
1
|
Tekerlekopoulou AG, Tsiflikiotou M,
Akritidou L, Viennas A, Tsiamis G, Pavlou S, Bourtzis K and Vayenas
DV: Modelling of biological Cr(VI) removal in draw-fill reactors
using microorganisms in suspended and attached growth systems.
Water Res. 47:623–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thacker U, Shouche PY and Madamwar D:
Hexavalent chromium reduction by Providencia sp. Pro Biochemistry.
41:1332–1337. 2006. View Article : Google Scholar
|
|
3
|
Shrivastava R, Upreti RK, Seth PK and
Chaturvedi UC: Effects of chromium on the immune system. FEMS
Immunol Med Microbiol. 34:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karagiannis D, Deliveliotis C,
Papadimitriou E, Riza E, Lykou A, Petralias A, Papatsoris A and
Linos A: Oral exposure to hexavalent chromium through drinking
water and urologic morbidity in an industrial area of Greece. J
Public Health. 23:249–255. 2015. View Article : Google Scholar
|
|
5
|
Jensen PK: Antimycin-insensitive oxidation
of succinate and reduced nicotinamide-adenine dinucleotide in
electron-transport particles II. Steroid effects. Biochim Biophys
Acta. 122:167–174. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu H, Jia X, Luo Z, Guan H, Jiang H, Li X
and Yan M: Inhibition of store-operated Ca(2+) channels prevent
ethanol-induced intracellular Ca(2+) increase and cell injury in a
human hepatoma cell line. Toxicol Lett. 208:254–261. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bernardi P, Krauskopf A, Basso E,
Petronilli V, Blachly-Dyson E, Di Lisa F and Forte MA: The
mitochondrial permeability transition from in vitro artifact to
disease target. FEBS J. 273:2077–2099. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Oliveira F, Chauvin C, Ronot X,
Mousseau M, Leverve X and Fontaine E: Effects of permeability
transition inhibition and decrease in cytochrome c content on
doxorubicin toxicity in K562 cells. Oncogene. 25:2646–2655. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsutsumi S, Gotoh T, Tomisato W, Mima S,
Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M and Mizushima
T: Endoplasmic reticulum stress response is involved in
nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death
Differ. 11:1009–1016. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Xiao F, Liu X, Liu K, Zhou X and
Zhong C: Cr(VI) induces cytotoxicity in vitro through activation of
ROS-mediated endoplasmic reticulum stress and mitochondrial
dysfunction via the PI3K/Akt signaling pathway. Toxicol In Vitro.
41:232–244. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo L, Wang F, Zhang Y, Zeng M, Zhong C
and Xiao F: In vitro cytotoxicity assessment of roundup
(glyphosate) in L-02 hepatocytes. J Environ Sci Health B.
52:410–417. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhong X, Zeng M, Bian H, Zhong C and Xiao
F: An evaluation of the protective role of vitamin C in reactive
oxygen species-induced hepatotoxicity due to hexavalent chromium in
vitro and in vivo. J Occup Med Toxicol. 12:152017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jing G, Wang JJ and Zhang SX: ER stress
and apoptosis: A new mechanism for retinal cell death. Exp Diabetes
Res. 2012:5895892012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soboloff J and Berger SA: Sustained ER
Ca2+ depletion suppresses protein synthesis and induces
activation-enhanced cell death in mast cells. J Biol Chem.
277:13812–13820. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou L, Jiang L, Xu M, Liu Q, Gao N, Li P
and Liu EH: Miltirone exhibits antileukemic activity by
ROS-mediated endoplasmic reticulum stress and mitochondrial
dysfunction pathways. Sci Rep. 6:205852016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sathish V, Abcejo AJ, Thompson MA, Sieck
GC, Prakash YS and Pabelick CM: Caveolin-1 regulation of
store-operated Ca(2+) influx in human airway smooth muscle. Eur
Respir J. 40:470–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhan Y, Wang L, Liu J, Ma K, Liu C, Zhang
Y and Zou W: Choline plasmalogens isolated from swine liver inhibit
hepatoma cell proliferation associated with caveolin-1/Akt
Signaling. PLoS One. 8:e773872013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Khan MT, Wagner L, Yule DI, Bhanumathy C
and Joseph SK: Akt kinase phosphorylation of inositol
1,4,5-trisphosphate receptors. J Biol Chem. 281:3731–3737. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Daniel PL, Amadou K, David FS, Ryan L and
Mohammed A: Differential effects of buffer pH on Ca(2+)-induced ROS
emission with inhibited mitochondrial complexes I and III. Front
Physiol. 6:582015.PubMed/NCBI
|
|
25
|
Kocyigit A and Guler EM: Curcumin induce
DNA damage and apoptosis through generation of reactive oxygen
species and reducing mitochondrial membrane potential in melanoma
cancer cells. Cell Mol Biol (Noisy-le-grand). 63:97–105. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ
and Camara AK: Inhibited mitochondrial respiration by amobarbital
during cardiac ischaemia improves redox state and reduces matrix
Ca2+ overload and ROS release. Cardiovasc Res. 77:406–415.
2008.PubMed/NCBI
|
|
27
|
Yi X, Zhang Y, Zhong C, Zhong X and Xiao
F: Role of STIM1 in Cr(VI)-induced [Ca2+]i increase and cell injury
in L-02 hepatocytes. Metallomics. 8:1273–1282. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mo ZT, Fang YQ, He YP and Zhang S:
β-asarone protects PC12 cells against OGD/R-induced injury via
attenuating Beclin-1-dependent autophagy. Acta Pharmacol Sin.
33:737–742. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agrawal M, Kumar V, Singh AK, Kashyap MP,
Khanna VK, Siddiqui MA and Pant AB: trans-Resveratrol protects
ischemic PC12 cells by inhibiting the hypoxia associated
transcription factors and increasing the levels of antioxidant
defense enzymes. Acs Chem Neurosci. 4:285–294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Diwakar L, Kenchappa RS, Annepu J and
Ravindranath V: Downregulation of glutaredoxin but not glutathione
loss leads to mitochondrial dysfunction in female mice CNS:
Implications in excitotoxicity. Neurochem Int. 51:37–46. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ozgur R, Turkan I, Uzilday B and Sekmen
AH: Endoplasmic reticulum stress triggers ROS signalling, changes
the redox state, and regulates the antioxidant defence of
Arabidopsis thaliana. J Exp Bot. 65:1377–1390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A
and Agostinis P: PERK is required at the ER-mitochondrial contact
sites to convey apoptosis after ROS-based ER stress. Cell Death
Differ. 19:1880–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:892012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hajnóczky G, Csordás G, Das S,
Garcia-Perez C, Saotome M, Sinha Roy S and Yi M: Mitochondrial
calcium signalling and cell death: Approaches for assessing the
role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium.
40:553–560. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshioka Y, Ishii Y, Ishida T, Yamada H,
Oguri K and Motojima K: Suppression of stress proteins, GRP78,
GRP94, calreticulin and calnexin in liver endoplasmic reticulum of
rat treated with a highly toxic coplanar PCB. Fukuoka Igaku Zasshi.
92:201–216. 2001.(In Japanese). PubMed/NCBI
|
|
37
|
Luanpitpong S, Talbott SJ, Rojanasakul Y,
Nimmannit U, Pongrakhananon V, Wang L and Chanvorachote P:
Regulation of lung cancer cell migration and invasion by reactive
oxygen species and caveolin-1. J Biol Chem. 285:38832–38840. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel HH, Murray F and Insel PA:
G-protein-coupled receptor-signaling components in membrane raft
and caveolae microdomains. Handb Exp Pharmacol. 186:167–184. 2008.
View Article : Google Scholar
|
|
39
|
Wehinger S, Ortiz R, Díaz MI, Aguirre A,
Valenzuela M, Llanos P, Mc Master C, Leyton L and Quest AF:
Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS
enhances palmitate-induced death of beta-pancreatic cells. Biochim
Biophys Acta. 1852:693–708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shack S, Wang XT, Kokkonen GC, Gorospe M,
Longo DL and Holbrook NJ: Caveolin-induced activation of the
phosphatidylinositol 3-kinase/Akt pathway increases arsenite
cytotoxicity. Mol Cell Biol. 23:2407–2414. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guan J, Yuan Z, He J, Wu Z, Liu B, Lin X,
Mo L and Mo H: Overexpression of caveolin-1 reduces Taxol
resistance in human osteosarcoma cells by attenuating PI3K-Akt-JNK
dependent autophagy. Exp Ther Med. 12:2815–2822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szado T, Vanderheyden V, Parys JB, De
Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U,
Söderberg O, et al: Phosphorylation of inositol 1,4,5-trisphosphate
receptors by protein kinase B/Akt inhibits Ca2+ release and
apoptosis. Proc Natl Acad Sci USA. 105:2427–2432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sundivakkam PC, Kwiatek AM, Sharma TT,
Minshall RD, Malik AB and Tiruppathi C: Caveolin-1 scaffold domain
interacts with TRPC1 and IP3R3 to regulate Ca2+ store
release-induced Ca2+ entry in endothelial cells. Am J Physiol Cell
Physiol. 296:403–413. 2009. View Article : Google Scholar
|