Introduction
During cerebral ischemia, mitogen-activated protein
kinases and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)
signaling cascades are key in the regulation of cell proliferation,
survival and apoptosis, and in the translation of anti-apoptotic
factors B-cell lymphoma-2 (Bcl-2) and Bcl-extra-large (Bcl-xL),
leading to neuroprotection against ischemic injury (1–4).
Three closely related Akt isoforms, namely Akt1, Akt2 and Akt3,
directly phosphorylate and activate mammalian target of rapamycin
(mTOR) signaling and its downstream effectors in response to
activation of the PI3K/Akt signaling pathway (5). Activated Akt (mainly Akt1 and Akt3)
protects against ischemia-induced neuronal injury by upregulating
the activity of mTOR, which in turn triggers the phosphorylation of
Akt in the ischemia region, thus creating a positive feedback loop
for the activation of Akt/mTOR signaling following cerebral
ischemic injury (6). mTOR is a
289-kDa serine/threonine protein kinase that regulates various
cellular activities, including cell proliferation, growth and
survival (7,8). Akt/mTOR signaling protects against
apoptosis by promoting the expression of anti-apoptotic proteins
Bcl-2 and Bcl-xL, and by inhibiting the expression of pro-apoptotic
proteins Bcl-2-associated X protein (Bax) and Bcl-2 antagonist of
cell death (Bad) in the ischemic region following focal cerebral
ischemia (8). There is compelling
evidence that a significant increase in the Bcl-2 (Bcl-xL)/Bax
ratio promotes neuronal cell survival following cerebral
ischemic-reperfusion (I/R) injury (9). mTOR forms two protein complexes, mTOR
Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2), and is involved in
the regulation of cell growth and metabolism (7,10).
mTORC1 function has been investigated extensively, whereas the
function of mTORC2 remains to be fully elucidated. The major mTORC1
components include mTOR, proline-rich Akt substrate of 40 kDa and
Raptor (11). mTOR activates its
downstream target, p70 S6 kinase (p70S6K), which subsequently
phosphorylates the 40S ribosomal protein S6 (S6) to control the
initiation of translation (10,11).
The phosphorylation of S6 is also crucial in the regulation of mRNA
translation and protein synthesis (12). The mTOR pathway is activated
following ischemic brain injury. Activation of the mTOR/p70S6K/S6
signaling cascade regulates ribosome biogenesis and protein
translation, and subsequently exerts neuroprotective effects
against infarction following focal cerebral ischemia (5,11,13).
By contrast, a decrease in the activity of mTOR has been linked to
the induction of apoptosis in in vitro and in vivo
models of cerebral ischemia (14).
The family of eukaryotic initiation factor 4E
(eIF4E)-binding proteins (4E-BPs) comprises three members, 4E-BP1,
4E-BP2 and 4E-BP3. As another downstream target of mTOR, 4E-BP1 is
a translational repressor that binds to eIF4E and thereby inhibits
its interaction with eIF4G (15,16).
The availability of eIF4E is a rate-limiting step in the initiation
of translation and is an important factor in the regulation of gene
expression. The interaction between 4E-BP1 and eIF4E inhibits
protein synthesis; however, protein synthesis is essential for
neuronal cell survival following cerebral ischemic insults
(16). The phosphorylation of
4E-BP1 by mTOR causes its dissociation from eIF4E, leading to the
subsequent formation of an eIF4E-eIF4G complex and thereby
promoting anti-apoptotic protein translation (7). Activation of the Akt/mTOR/4E-BP1
signaling pathway significantly ameliorates cerebral infarction and
improves neurological outcomes following ischemic postconditioning
in rats, and the beneficial effects can be attributed to the
inhibition of mitochondria-mediated apoptosis (11,17).
Ferulic acid (4-hydroxy-3-methoxycinnamic acid,
FerA) is a major active ingredient derived from Angelica
sinensis (Oliv.) Diels (1). In
traditional Chinese medicine, A. sinensis has been used for
centuries to improve blood circulation and as a treatment for
stroke (18–20). Our previous studies demonstrated
that FerA exerts neuroprotective effects against cerebral I/R
injury by downregulating the inflammatory response and oxidative
stress in the ischemic region during the acute phase (21,22),
and by inhibiting apoptosis in the penumbral cortex during the
subacute phase (1) following
transient middle cerebral artery occlusion (MCAo). Other studies
have shown that FerA prevents ischemia-induced apoptosis by
phosphorylating Akt/Bad or Akt/phosphoprotein enriched in
astrocytes-15 signaling in the acute phase of permanent MCAo
(23,24). However, the precise mechanisms
underlying the neuroprotective effects of FerA on cerebral ischemic
injury in the subacute phase of permanent MCAo remain to be
elucidated.
Therefore, the present study evaluated the effects
of various doses of FerA administered 7 days following permanent
MCAo and examined the involvement of Akt signaling cascades in the
penumbral cortex.
Materials and methods
Experimental animals
A total of 106 male Sprague-Dawley (SD) rats
(BioLASCO Taiwan Co., Ltd., Taipei, Taiwan), 8–9 weeks of age and
weighing 300–350 g, were used in the present study. The rats were
housed in standard plastic cages, fed a constant provision of solid
food, provided with water ad libitum, and maintained in 12-h
light-dark cycles under a relative humidity of 55±5% at 22±2°C. All
study procedures were approved by the Institutional Animal Care and
Use Committee of China Medical University (Taichung, Taiwan; permit
no. 2016-321).
Permanent MCAo
The permanent MCAo model was performed on adult SD
rats as described previously with modifications (25). The rats were initially anesthetized
with 5% isoflurane in a plastic chamber. Subsequently, general
anesthesia was maintained using a 2% isoflurane-oxygen mixture
delivered via facemask inhalation. The anesthetized rat was removed
from the plastic chamber, and its head was fixed on the
stereotactic frame. Following dissecting the scalp, a burr hole was
drilled into the skull 2.5 mm lateral and 2.0 mm posterior to the
bregma in order to expose the right distal end of the MCA. The rat
was placed in the supine position, and a neck incision was
performed to expose the right common, external carotid artery (ECA)
and internal carotid artery (ICA). A 3-0 nylon suture was inserted
from the ECA into the ICA for a distance of 21–22 mm. During MCAo,
the blood flow in the MCA was continually evaluated using laser
Doppler flowmetry (DRT4, Moor Instruments, Inc., Devon, UK) through
the burr hole in the skull. Blood flow reduction to 20–25% of the
pre-ischemic value was used to validate the success of the
permanent MCAo model.
Evaluation of neurological
functions
The neurological status of each rat was examined 1,
3, and 7 days following cerebral ischemia using a modified
neurological deficit score (NDS; score of 0–18) as described
previously (26). A laboratory
assistant blinded to the group assignment evaluated the items of
neurological examination, which included motor, sensory, balance
and reflex tests.
Experiment A
Grouping
The rats were randomly divided into five groups:
Sham, Vehicle, FerA-60 mg, FerA-80 mg, and FerA-100 mg groups
(n=4-5). In the FerA-60 mg, FerA-80 mg, and FerA-100 mg groups, the
rats underwent MCAo surgery and were simultaneously administered
FerA intravenously (iv) at doses of 60, 80 and 100 mg/kg,
respectively. Following 7 days of cerebral ischemia, the rats were
examined for neurological functions and then sacrificed. In the
Vehicle group, the rats underwent the same procedures as that of
the FerA-100 mg group, but the rats were administered saline
instead of FerA. In the Sham group, the rats underwent the same
procedures as that of the Vehicle group; however, the rats did not
undergo MCAo.
Evaluation of cerebral infarct
area
Following 7 days of cerebral ischemia, the rats were
examined for their neurological status and subsequently sacrificed.
Their brains were carefully removed from the skull and divided into
six serial 2-mm thick coronal slices using a brain slicer matrix.
The brain slices were placed in a constant temperature box and
incubated with 2% solution of 2,3,5-triphenyltetrazolium chloride
(TTC; Merck KGaA, Darmstadt, Germany) for 5 min at 37°C. Following
TTC staining, the cerebral infarct in the MCA territory turned pale
white, whereas the healthy brain tissue turned dark red. The TTC
stained brain sections were visualized and photographed using a
digital camera (Nikon Coolpix 775; Nikon Corporation, Tokyo,
Japan). The areas of infarction were evaluated using image analysis
software (ImageJ version 1.46; National Institutes of Health,
Bethesda, MD, USA), and the percentage of cerebral infarction was
obtained by determining the ratio of the cerebral infarct area to
the total brain area.
Experiment B
Grouping
The rats were randomly divided into the following
five groups: Sham, Vehicle, FerA-60 mg, FerA-80 mg, and FerA-100 mg
groups (n=4-5). They underwent the same procedures as described in
Experiment A.
Western blot analysis
Following 7 days of cerebral ischemia, the rats were
sacrificed and their brains were carefully removed. The coronal
sections of the brains obtained from 1.7 mm anterior to 4.3 mm
posterior to the bregma were used for western blot analysis. The
penumbral cortex was homogenized in cold Cytosol Extraction Buffer
mix (BioVision, Inc., Milpitas, CA, USA) using a tissue homogenizer
(Kinematia Polytron RT-3000). The samples were further divided into
cytosolic and mitochondrial fractions using the
Mitochondria/Cytosol Fractionation kit (cat. no. K256-100
BioVision, Inc.). The concentrations of the cytosolic and
mitochondrial proteins were evaluated using a Bio-Rad protein
assay. Equal quantities (15 µg/well) of protein were loaded into
the gel wells and separated according to their molecular weight by
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, as
described previously (27).
Subsequently, the gel-separated proteins were transferred onto
nitrocellulose (NC) membranes. Based on the molecular weight
marker, the NC membranes were carefully split into pieces and
incubated with the following primary antibodies: Akt (rabbit, cat.
no. 4685; Cell Signaling Technology, Inc., Danvers, MA, USA,
1:1,000), p-Akt (rabbit, cat. no. 9271; Cell Signaling Technology,
Inc., 1:1,000), mTOR (rabbit, cat. no. 2972; Cell Signaling
Technology, Inc., 1:1,000), p-mTOR (rabbit, cat. no. 2971; Cell
Signaling Technology, Inc., 1:1,000), 4E-BP1 (rabbit, cat. no.
9644; Cell Signaling Technology, Inc., 1:1,000), p-4E-BP1 (rabbit,
cat. no. 2855; Cell Signaling Technology, Inc., 1:1,000), p70S6K
(rabbit, cat. no. 9202; Cell Signaling Technology, Inc., 1:1,000),
p-p70S6K (rabbit, cat. no. 9208; Cell Signaling Technology, Inc.,
1:1,000), Bcl-2 (rabbit, cat. no. 2876; Cell Signaling Technology,
Inc., 1:1,000), Bax (rabbit, cat. no. 2772; Cell Signaling
Technology, Inc., 1:1,000), Bcl-xL (rabbit, cat. no. 2762; Cell
Signaling Technology, Inc., 1:1,000), actin (mouse, cat. no.
NB600-501; Novus Biologicals, LLC, Littleton, CO, USA, 1:5,000) and
heat shock protein 60 (HSP60; rabbit, cat. no. 4870, Cell Signaling
Technology, Inc., 1:1,000) overnight at 4°C. The membranes were
subsequently incubated with an anti-rabbit horseradish peroxidase
(HRP)-conjugated Immunoglobulin G (IgG; cat. no. 123450; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or
anti-mouse HRP-conjugated IgG (cat. no. 127442; Jackson
ImmunoResearch Laboratories, Inc.) secondary antibody for 1 h at
room temperature. Densitometric analyses were performed using
analysis software (ImageJ). The immunoblotting results were
determined as the optical density ratios of target proteins to
actin or phosphorylated proteins to non-phosphorylated
proteins.
Immunohistochemical (IHC)
analysis
Following 7 days of ischemia, the rats were
sacrificed (n=3-4). Their brains were carefully removed, postfixed
in 4% paraformalaldehyde, and cut into 15-µm-thick sections, as
described previously (28). The
brain sections were then incubated with primary antibodies
overnight at 4°C to detect the expression of cytochrome c
(rabbit; cat. no. 10993-1-AP; ProteinTech Group, Inc., Chicago, IL,
USA, 1:50) and cleaved caspase-3 (rabbit; cat. no., 9664; Cell
Signaling Technology, Inc., 1:100). Following incubation with the
primary antibodies, the sections were subsequently incubated with
the appropriate secondary antibody-avidin-biotin peroxidase
complexes (Leica Biosystems, Newcastle Ltd., Newcastle, UK),
3,3′-diaminobenzidine (Leica Biosystems Newcastle, Ltd.) and
hematoxylin (Merck KGaA). The immunoreactive cells in the penumbral
cortex were detected under a light microscope (Axioskop 40; Carl
Zeiss AG, Oberkochen, Germany). The brain sections obtained from
the rats of the Vehicle group that were stained without primary
antibodies were used as negative controls.
Terminal deoxynucleotidyl
transferase-mediated dUTP-biotin nick-end labeling (TUNEL)
assay
TUNEL staining was performed in accordance with the
manufacturer's protocol (QIA33; Calbiochem; Merck KGaA). The brain
slides of the adjacent sections used in IHC analysis were first
incubated with 20 µg/ml proteinase K for 20 min at room
temperature, as described previously (29). The TUNEL-reactive cells in the
penumbral cortex were examined under a light microscope.
Experiment C
Grouping
The rats were randomly divided into four groups: DS
+ Sham, DS + Vehicle, DS + FerA-100 mg, and LY + FerA-100 mg groups
(n=4-5). In the LY + FerA-100 mg group, the rats underwent the same
surgical procedures as that of the FerA-100 mg group (Experiment
A); however, the rats also received an intracerebroventricular
(ICV) infusion of LY294002 (LY), a PI3K/Akt inhibitor, 30 min prior
to MCAo. In the DS + FerA-100 mg group, the rats underwent the same
surgical procedures as that of the LY + FerA-100 mg group, but the
rats received an ICV infusion of 1% dimethyl sulfoxide (DMSO). In
the DS + Vehicle group, the rats underwent the same surgical
procedures as that of the Vehicle group (Experiment A), but the
rats also received an ICV infusion of 1% DMSO 30 min prior to MCAo.
In the DS + Sham group, the rats underwent the same surgical
procedures as that of the DS + Vehicle group, but the rats did not
undergo MCAo.
ICV infusion of LY or 1% DMSO
The rats were anesthetized with 5% isoflurane, and
anesthesia was maintained via facemask inhalation using 2%
isoflurane. The rats received an ICV infusion of 10 µl of a
solution containing LY (10 mM in DMSO, cat. no. 1747-5, BioVision,
Inc.) or 1% DMSO into their right hemisphere. The infusions were
performed over a 6-min period using a 10 µl Hamilton microliter
syringe (Hamilton Company, Reno, NV, USA). The infusion was
administered into the right cerebral ventricle (0.8 mm posterior to
the bregma, 1.5 mm lateral on the right side, and 3.5 mm below the
skull surface).
Western blot analysis
Following 7 days of ischemia, the rats were
sacrificed, and their brains were immediately removed for
incubation with antibodies for western blot analysis of the
expression of Akt, p-Akt, mTOR, p-mTOR, 4E-BP1, p-4E-BP1, eIF4E
(rabbit, cat. no. 9742; Cell Signaling Technology, Inc., 1:1,000),
Bcl-2 and Bax. The protocol used for analysis of the protein
samples using western blotting was the same as that described for
Experiment B.
Experiment D
Grouping
The rats were randomly divided into the following
four groups: DS + Sham, DS + Vehicle, DS + FerA-100 mg, and LY +
FerA-100 mg groups (n=3-4). They underwent the same experimental
protocols as described in Experiment C.
Evaluation of cerebral infarct
area
At 7 days post-ischemia, the rats were sacrificed.
They underwent the same experimental protocols for cerebral infarct
detection as described in Experiment A.
Statistical analysis
All data in the present study are expressed as the
mean ± standard deviation. Comparisons were performed using one-way
analysis of variance with a Scheffe post hoc test. P<0.05 was
considered to indicate a statistically significant difference. All
data were analyzed using SPSS 13.0 software (SPSS, Inc., Chicago,
IL, USA).
Results
Effects of FerA treatment on cerebral
infarct area
At 7 days post-ischemia, the percentage of the
cerebral infarct area was significantly increased in the Vehicle
group compared with that in the Sham group (P<0.05) and was
significantly reduced in the FerA-80 mg and FerA-100 mg groups
compared with that in the Vehicle group (both P<0.05; Figs. 1 and 2A). However, no significant difference
was found in the percentage of cerebral infarct area between the
Vehicle and FerA-60 mg groups (P>0.05).
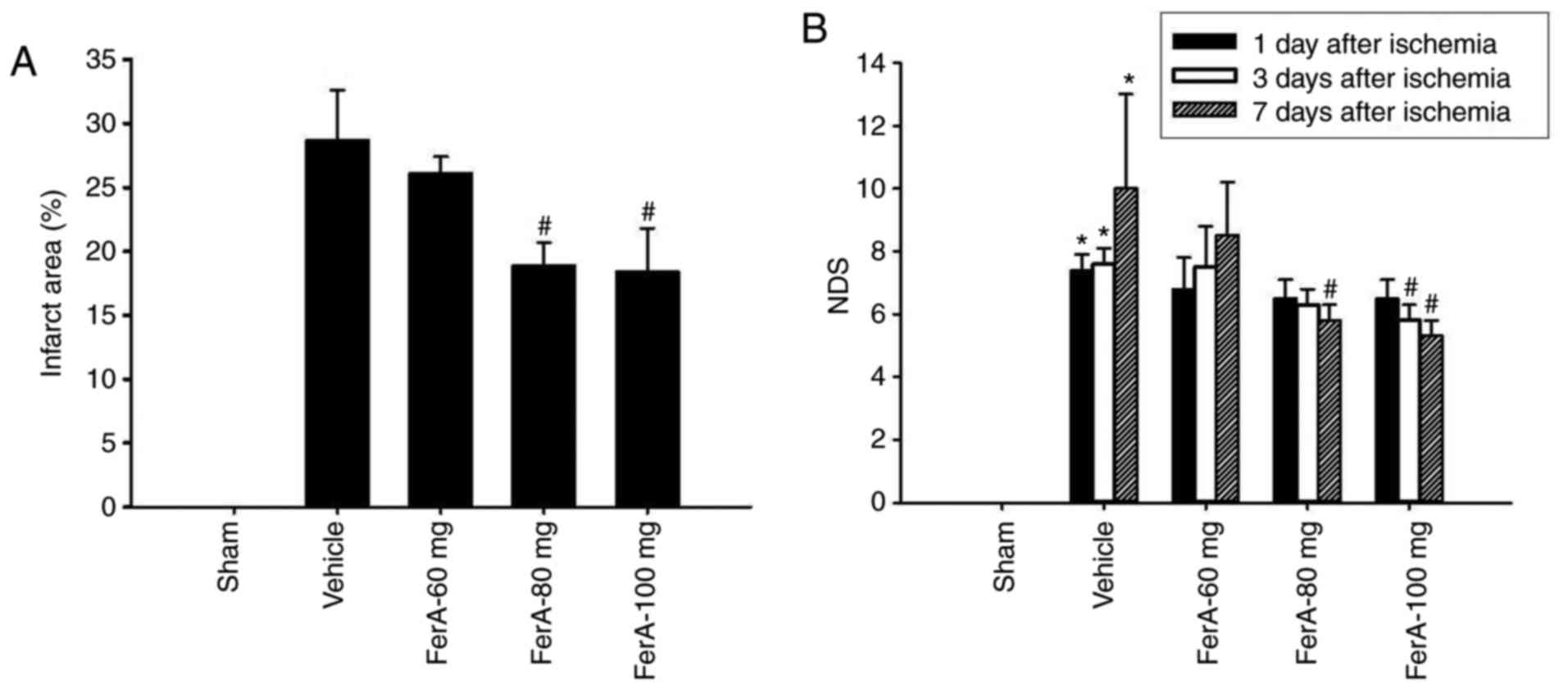 | Figure 2.Effects of FerA treatment on cerebral
infarct area and neurological deficits 7 days following cerebral
ischemia. (A) Percentages of cerebral infarct areas in the Sham,
Vehicle, FerA-60 mg, FerA-80 mg, and FerA-100 mg groups were
evaluated at 7 days post-ischemia (n=4-5). (B) NDS of the Sham,
Vehicle, FerA-60 mg, FerA-80 mg, and FerA-100 mg groups were
evaluated at 1, 3, and 7 days post-ischemia. *P<0.05 vs. the
Sham group; #P<0.05 vs. the Vehicle group. FerA,
ferulic acid; NDS, neurological deficit score. |
Effects of FerA treatment on
neurological function
At 1 day post-ischemia, the NDS of the Vehicle,
FerA-60 mg, FerA-80 mg, and FerA-100 mg groups did not differ
significantly (P>0.05; Fig.
2B). At 3 days post-ischemia, the NDS of the FerA-100 mg group
was significantly reduced compared with that of the Vehicle group
(P<0.05; Fig. 2B). No
significant differences were observed in the NDS among the Vehicle,
FerA-60 mg and FerA-80 mg groups (P>0.05). At 7 days
post-ischemia, the NDS of the FerA-80 mg and FerA-100 mg groups
were significantly reduced compared with that of the Vehicle group
(both P<0.05; Fig. 2B), whereas
the NDS of the Vehicle and the FerA-60 mg groups did not differ
significantly (P>0.05).
Effects of FerA treatment on the
cytosolic expression of p-Akt, Akt, p-mTOR, mTOR, p-4E-BP1, 4E-BP1,
p-p70S6K, and p70S6K
The results of the western blot analysis showed that
the ratios of cytosolic p-Akt/Akt and p-mTOR/mTOR expression in the
penumbral cortex were significantly reduced in the Vehicle group
(both 0.3-fold) compared with those in the Sham group (both
P<0.05) and were significantly increased in the FerA-80 mg (3.4-
and 5.1-fold, respectively) and FerA-100 mg (5.0- and 5.1-fold,
respectively) groups compared with the Vehicle group (all
P<0.05; Fig. 3A-C) at 7 days
post-ischemia. The ratios of cytosolic p-Akt/Akt and p-mTOR/mTOR
expression in the Vehicle and FerA-60 mg groups did not differ
significantly (P>0.05). The ratio of cytosolic 4E-BP1/actin
expression was significantly reduced in the Vehicle group
(0.4-fold) compared with that in the Sham group (P<0.05) and was
significantly increased in the FerA-80 mg (2.6-fold) and FerA-100
mg (2.9-fold) groups compared with that in the Vehicle group (both
P<0.05; Fig. 3A and D). The
ratios of cytosolic 4E-BP1/actin expression in the Vehicle and
FerA-60 mg groups did not differ significantly (P>0.05). The
ratios of cytosolic p-4E-BP1/4E-BP1 expression were significantly
increased in the FerA-80 mg (13.3- and 2.4-fold, respectively) and
FerA-100 mg (11.7- and 2.1-fold, respectively) groups compared with
those in the Sham and Vehicle groups (all P<0.05; Fig. 3A and E). The ratios of cytosolic
p-4E-BP1/4E-BP1 expression in the Sham, Vehicle, and FerA-60 mg
groups did not differ significantly (P>0.05). The ratios of
cytosolic p70S6K/actin and p-p70S6K/p70S6K expression in the
penumbral cortex among the Sham, Vehicle, and FerA-treated groups
did not differ significantly (P>0.05; Fig. 3A, F and G).
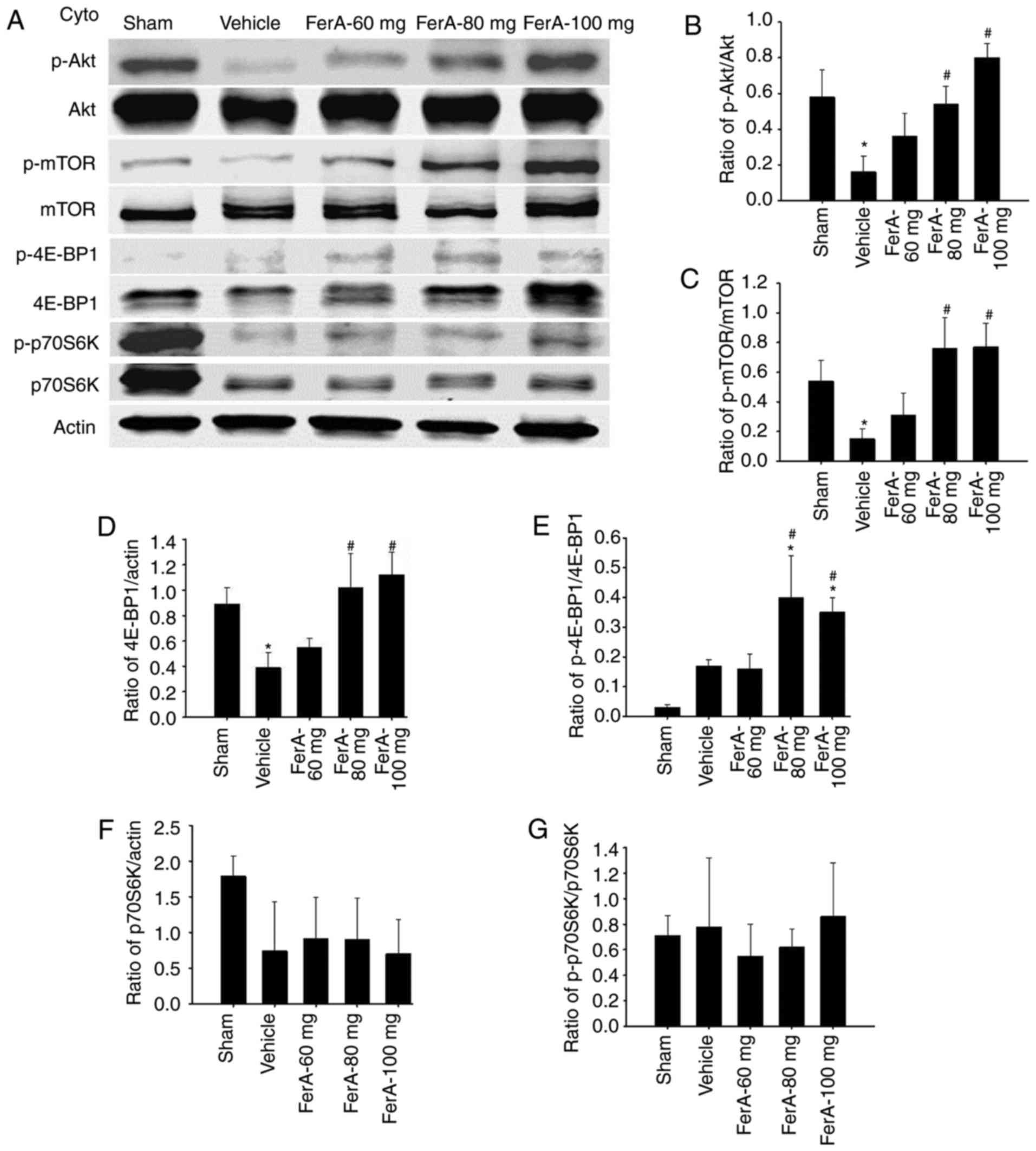 | Figure 3.Effects of FerA treatment on the
cytosolic expression of p-Akt, Akt, p-mTOR, mTOR, p-4E-BP1, 4E-BP1,
p-p70S6K, and p70S6K in the penumbral cortex. (A) Representative
western blot images revealed the cytosolic expression of p-Akt,
Akt, p-mTOR, mTOR, p-4E-BP1, 4E-BP1, p-p70S6K, and p70S6K in the
penumbral cortex of the Sham, Vehicle, FerA-60 mg, FerA-80 mg, and
FerA-100 mg groups at 7 days post-ischemia. Actin was used as an
internal control for cytosolic extracts. The ratios of (B)
p-Akt/Akt, (C) p-mTOR/mTOR, (D) 4E-BP1/actin, (E) p-4E-BP1/4E-BP1,
(F) p70S6K/actin, and (G) p-p70S6K/p70S6K expression were measured
in the penumbral cortex in the Sham, Vehicle, FerA-60 mg, FerA-80
mg, and FerA-100 mg groups (n=4-5). *P<0.05 vs. the Sham group;
#P<0.05 vs. the Vehicle group. FerA, ferulic acid;
cyto, cytosolic fraction; mTOR, mammalian target of rapamycin;
4E-BP1, eukaryotic initiation factor 4E-binding protein 1; p-,
phosphorylated. |
Effects of FerA treatment on the ratio
of mitochondrial expression of Bcl-2, Bcl-xL and Bax
The ratio of mitochondrial Bcl-2/Bax expression in
the penumbral cortex was markedly reduced in the Vehicle group
(0.2-fold) compared with that in the Sham group (P<0.05) and was
markedly increased in the FerA-80 mg (10.5-fold) and FerA-100 mg
(11.6-fold) groups compared with that in the Vehicle group (both
P<0.05; Fig. 4A and B) at 7
days post-ischemia. The ratios of mitochondrial Bcl-2/Bax
expression in the Vehicle and FerA-60 mg groups did not differ
significantly (P>0.05). No significant differences were observed
in the ratio of mitochondrial Bcl-xL/Bax expression among the Sham,
Vehicle and FerA-treated groups (P>0.05; Fig. 4A and C).
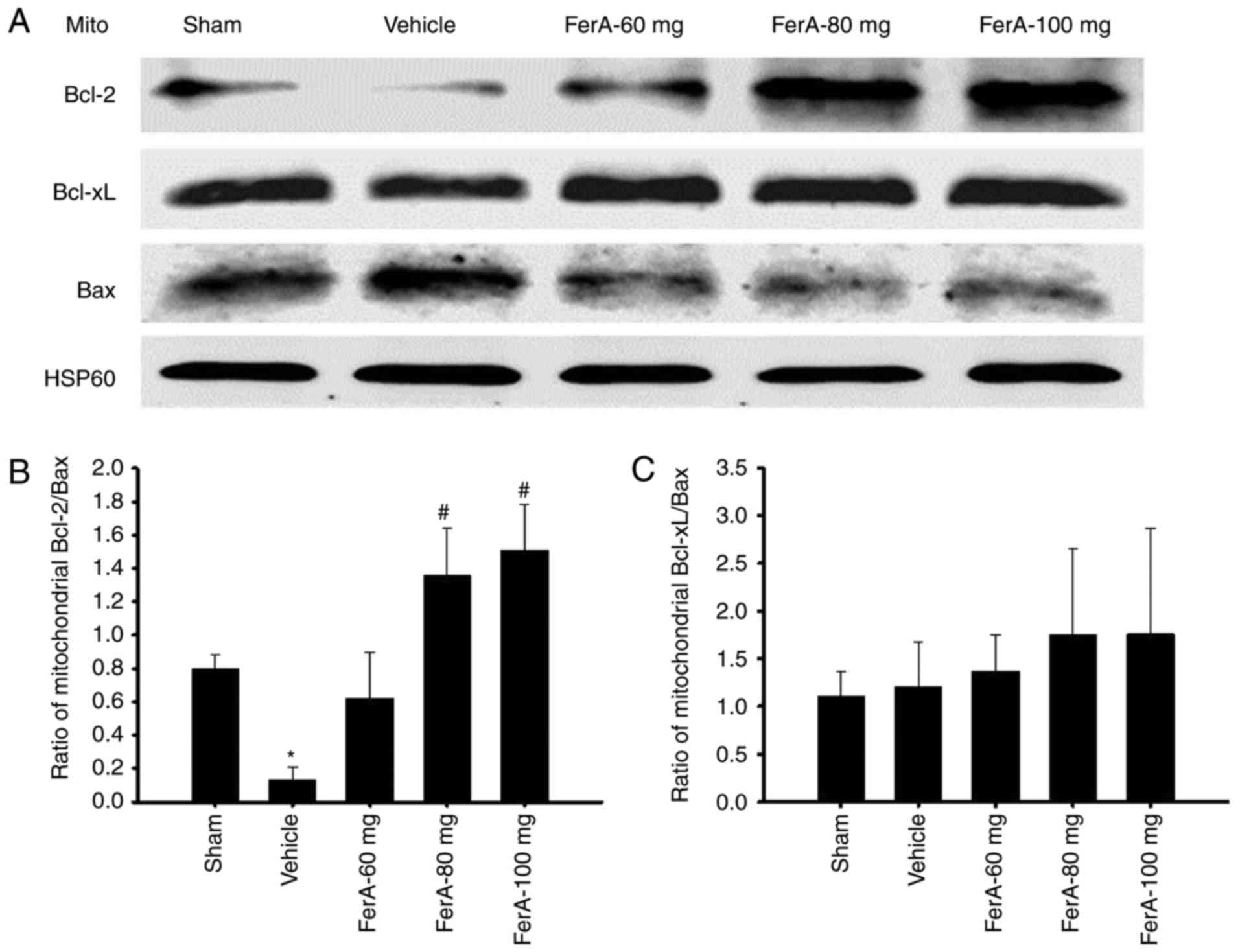 | Figure 4.Effects of FerA treatment on the
mitochondrial expression of Bcl-2, Bcl-xL, and Bax in the penumbral
cortex. (A) Representative western blot images revealed the
mitochondrial expression of Bcl-2, Bcl-xL and Bax in the penumbral
cortex in the Sham, Vehicle, FerA-60 mg, FerA-80 mg, and FerA-100
mg groups at 7 days post-ischemia. HSP60 was used as an internal
control for mitochondrial extracts. The ratios of (B) mitochondrial
Bcl-2/Bax and (C) mitochondrial Bcl-xL/Bax expression were measured
in the penumbral cortex in the Sham, Vehicle, FerA-60 mg, FerA-80
mg, and FerA-100 mg groups (n=4-5). *P<0.05 vs. the Sham group;
#P<0.05 vs. the Vehicle group. FerA, ferulic acid;
mito, mitochondrial fraction; Bcl-2, B-cell lymphoma-2; Bax,
Bcl-2-associated X protein; Bcl-xL, Bcl-extra-large. |
Effects of FerA treatment on the
expression of cytochrome c-, cleaved caspase-3-, and
TUNEL-immunoreactive cells
The numbers of cytochrome c-, cleaved
caspase-3-, and TUNEL-immunoreactive cells in the penumbral cortex
were calculated (counts/1 mm2) at 7 days post-ischemia.
The numbers of cytochrome c-, cleaved caspase-3-, and
TUNEL-immunoreactive cells were significantly increased in the
Vehicle group compared with those in the Sham group (all P<0.05)
and were significantly reduced in the FerA-80 mg and FerA-100 mg
groups compared with those in the Vehicle group (all P<0.05;
Fig. 5A-C; Table I). No significant differences were
observed in the numbers of cytochrome c-, cleaved caspase-3-, and
TUNEL-immunoreactive cells between the Vehicle and FerA-60 mg
groups (P>0.05). The penumbral cortex region in which cells were
counted is indicated in Fig.
5D.
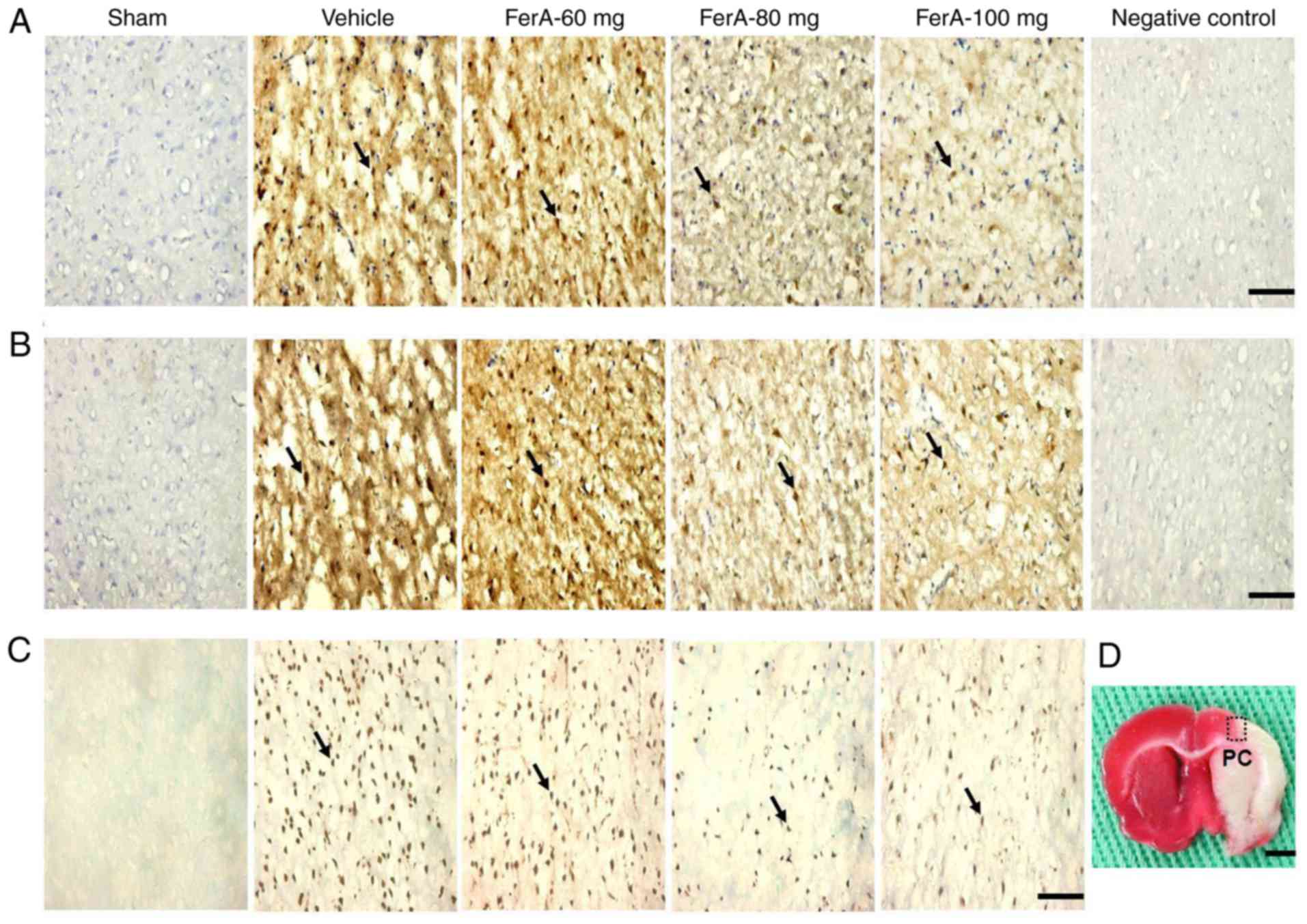 | Figure 5.Effects of FerA treatment on the
expression of cytochrome c-, cleaved caspase-3-, and
TUNEL-immunoreactive cells in the PC. Representative images show
(A) cytochrome c-, (B) cleaved caspase-3- and (C)
TUNEL-immunoreactive cells in the PC in the Sham, Vehicle, FerA-60
mg, FerA-80 mg and FerA-100 mg groups at 7 days post-ischemia.
Arrows indicate these immunoreactive cells (scale bars=10 µm). (D)
Representative image shows the 2,3,5-triphenyltetrazolium
chloride-stained brain coronal section. The dotted square indicates
the area of measurement of immunoreactive cells. Dotted square=1
mm2; scale bar=200 µm). FerA, ferulic acid; TUNEL,
terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end
labeling; PC, penumbral cortex. |
 | Table I.Expression of cytochrome c-,
cleaved caspase-3-, and TUNEL-immunoreactive cells (count/1
mm2). |
Table I.
Expression of cytochrome c-,
cleaved caspase-3-, and TUNEL-immunoreactive cells (count/1
mm2).
| Factor | Sham | Vehicle | FerA-60 mg | FerA-80 mg | FerA-100 mg |
|---|
| Cytochrome
c | 0±0 | 322±26a | 272±28 | 129±21b | 95±15b |
| Cleaved
caspase-3 | 0±0 | 258±55a | 316±50 | 112±5b | 83±24b |
| TUNEL | 0±0 |
818±110a | 699±107 | 366±65b | 291±42b |
Effects of DS + FerA-100 mg and LY +
FerA-100 mg on the ratios of cytosolic expression of p-Akt/Akt,
p-mTOR/mTOR, p-4E-BP1/actin, 4E-BP1/actin, and eIF4E/actin
The ratios of cytosolic p-Akt/Akt, p-mTOR/mTOR,
4E-BP1/actin, and eIF4E/actin expression in the penumbral cortex
were significantly reduced in the DS + Vehicle group (0.4-, 0.3-,
0.4-, and 0.2-fold, respectively) compared with those in the DS +
Sham group (all P<0.05) and were significantly increased in the
DS + FerA-100 mg group (3.2-, 3.7-, 3.5-, and 3.3-fold,
respectively) compared with those in the DS + Vehicle group (all
P<0.05 Fig. 6A-E) at 7 days
post-ischemia. No significant differences were observed in the
ratios of cytosolic p-Akt/Akt, p-mTOR/mTOR, 4E-BP1/actin and
eIF4E/actin expression between the DS + Vehicle and LY + FerA-100
mg groups (P>0.05). The ratio of cytosolic p-4E-BP1/actin
expression was significantly increased in the DS + FerA-100 mg
group (11.8- and 4.7-fold, respectively) compared with the ratios
in the DS + Sham and DS + Vehicle groups (both P<0.05; Fig. 6A and F). No significant differences
were observed in the ratios of cytosolic p-4E-BP1/actin expression
among the DS + Sham, DS + Vehicle, and LY + FerA-100 mg groups
(P>0.05).
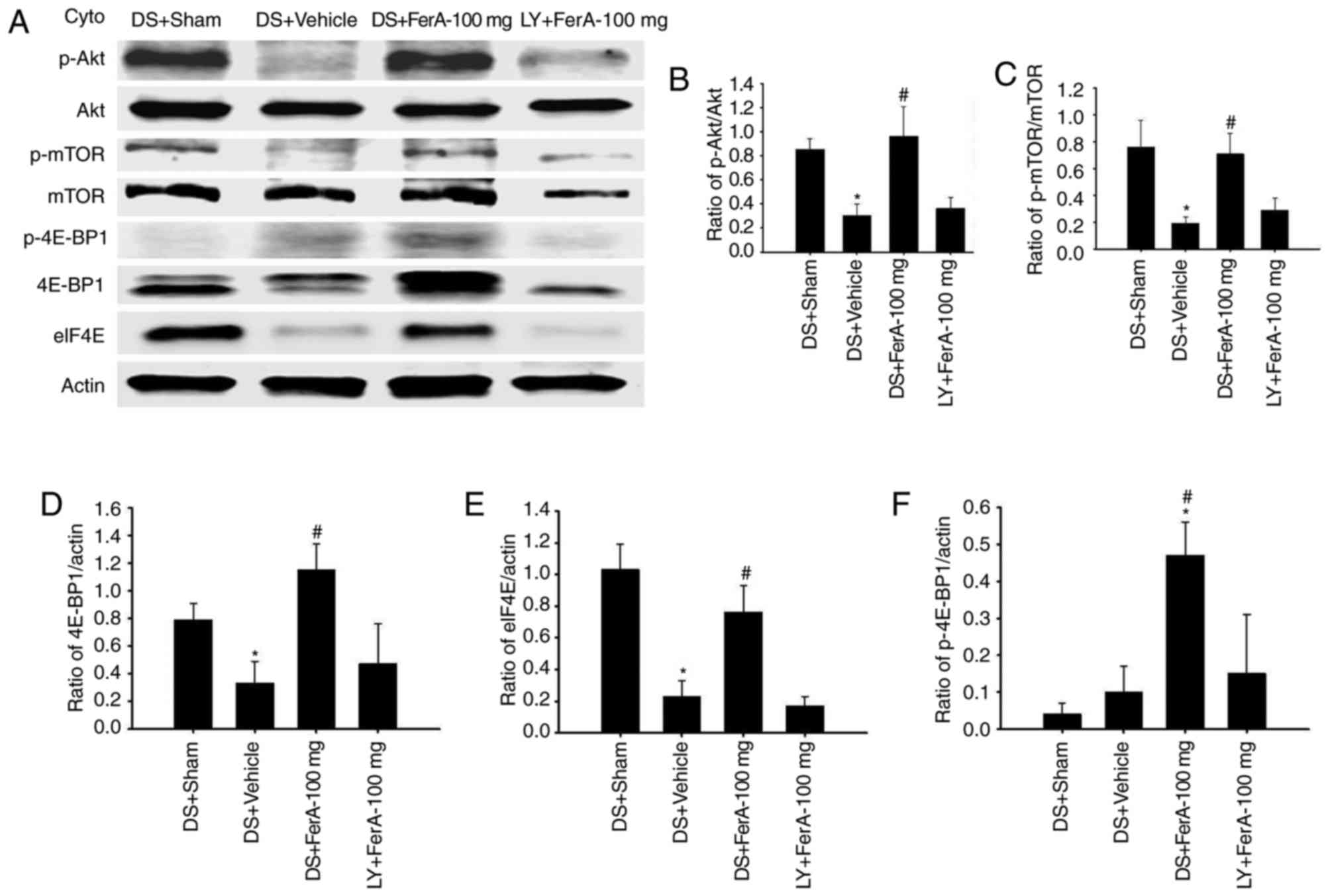 | Figure 6.Effects of DS + FerA-100 mg and LY +
FerA-100 mg on the cytosolic expression of p-Akt, Akt, p-mTOR,
mTOR, p-4E-BP1, 4E-BP1 and eIF4E in the penumbral cortex. (A)
Representative images show cytosolic expression of p-Akt, Akt,
p-mTOR, mTOR, p-4E-BP1, 4E-BP1, and eIF4E in the penumbral cortex
in the DS + Sham, DS + Vehicle, DS + FerA-100 mg, and LY + FerA-100
mg groups at 7 days post-ischemia. The ratios of (B) p-Akt/Akt (C)
p-mTOR/mTOR (D) 4E-BP1/actin (E) eIF4E/actin, and (F)
p-4E-BP1/actin expression were measured in the penumbral cortex in
the DS + Sham, DS + Vehicle, DS + FerA-100 mg, and LY+FerA-100 mg
groups (n=4-5). *P<0.05 vs. the DS + Sham group;
#P<0.05 vs. the DS + Vehicle group. FerA, ferulic
acid; DS, dimethyl sulfoxide; LY, LY294002; cyto, cytosolic
fraction; mTOR, mammalian target of rapamycin; eIF4E, eukaryotic
initiation factor 4E; 4E-BP1, eIF4E-binding protein 1; p-,
phosphorylated. |
Effects of DS + FerA-100 mg and LY +
FerA-100 mg on the ratio of mitochondrial expression of
Bcl-2/Bax
The ratio of mitochondrial Bcl-2/Bax expression in
the penumbral cortex was markedly reduced in the DS + Vehicle group
(0.2-fold) compared with that in the DS + Sham group (P<0.05)
and was markedly increased in the DS + FerA-100 mg group (3.5-fold)
compared with that in the DS + Vehicle group (P<0.05; Fig. 7A and B) at 7 days post-ischemia. No
significant differences were observed in the ratios of
mitochondrial Bcl-2/Bax expression between the DS + Vehicle and LY
+ FerA-100 mg groups (P>0.05).
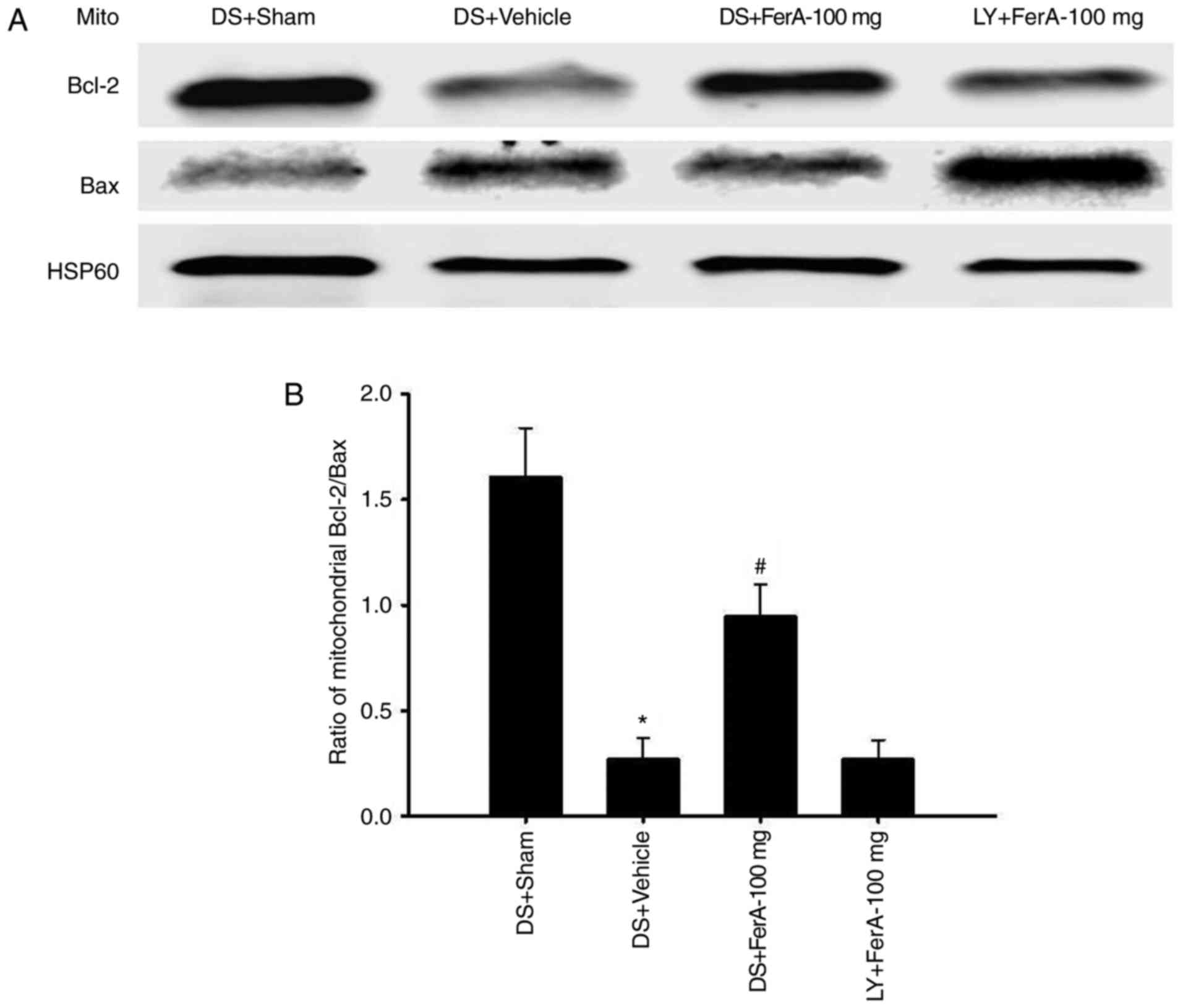 | Figure 7.Effects of DS + FerA-100 mg and LY +
FerA-100 mg on the mitochondrial expression of Bcl-2 and Bax in the
penumbral cortex. (A) Representative images show the mitochondrial
expression of Bcl-2 and Bax in the penumbral cortex in the DS +
Sham, DS + Vehicle, DS + FerA-100 mg, and LY + FerA-100 mg groups
at 7 days post-ischemia. The ratio of (B) mitochondrial Bcl-2/Bax
expression was measured in the penumbral cortex in the DS + Sham,
DS + Vehicle, DS + FerA-100 mg, and LY + FerA-100 mg groups
(n=4-5). *P<0.05 vs. the DS + Sham group; #P<0.05
vs. the DS + Vehicle group. FerA, ferulic acid; DS, dimethyl
sulfoxide; LY, LY294002; Mito, mitochondrial fraction; Bcl-2,
B-cell lymphoma-2; Bax, Bcl-2-associated X protein. |
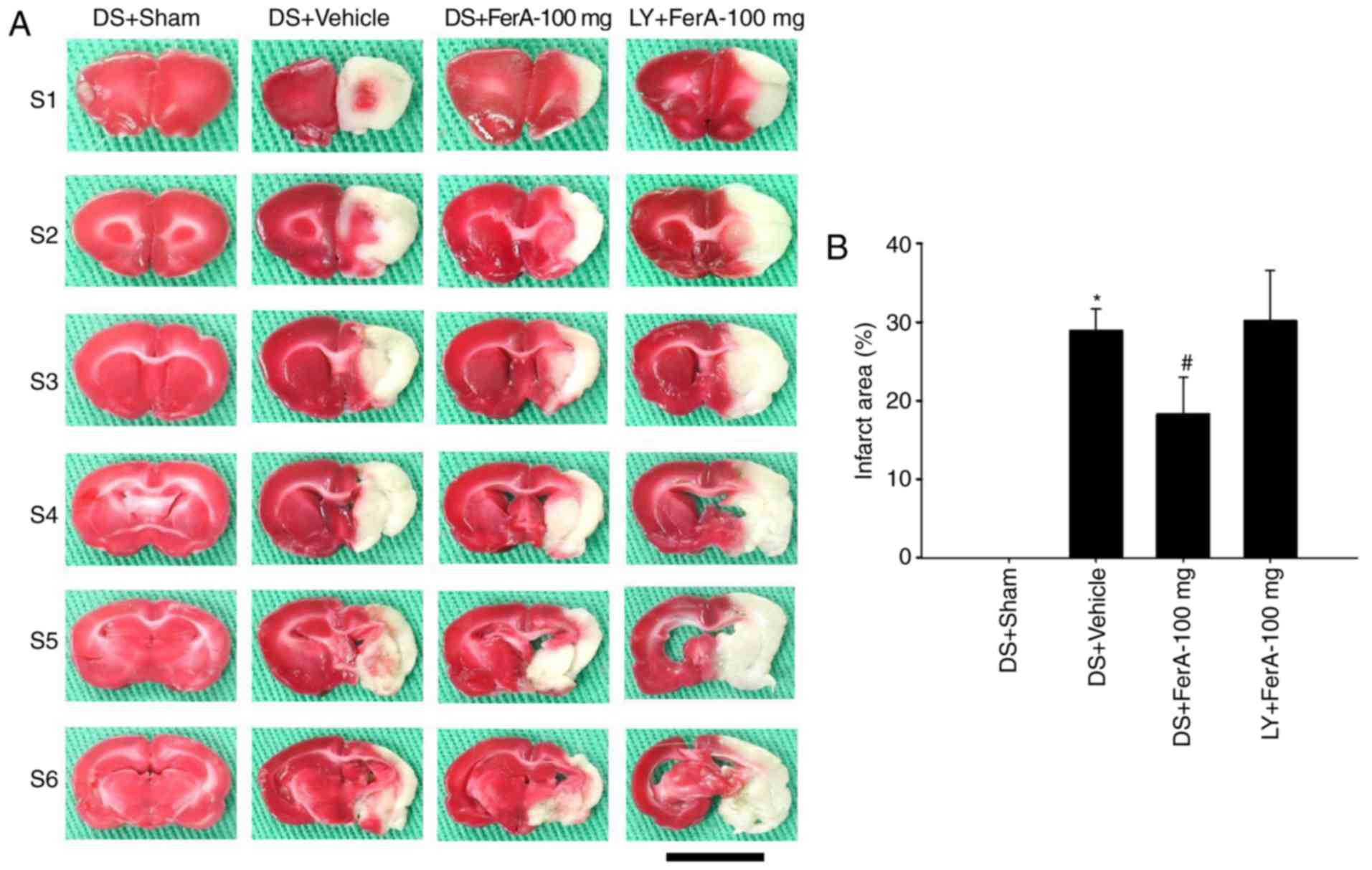
Effects of DS + FerA-100 mg and LY +
FerA-100 mg on the cerebral infarct area
The percentage cerebral infarct area was
significantly increased in the DS + Vehicle group compared with
that in the DS + Sham group (P<0.05) and was significantly
reduced in the DS + FerA-100 mg group compared with that in the DS
+ Vehicle group (P<0.05; Fig. 8A
and B) at 7 days post-ischemia. No significant differences were
observed in the percentage of cerebral infarct areas between the DS
+ Vehicle and LY + FerA-100 mg groups (P>0.05).
Discussion
In permanent focal cerebral ischemia, the infarct
area rapidly increases within 1 day following the onset of ischemia
and then undergoes delayed infarct expansion between 1 and 7 days
post-ischemia (30,31). In the present study, the cerebral
infarction areas were calculated in a rat model of permanent MCAo
at 7 days post-ischemia. The results, consistent with previous
findings on cerebral infarction, revealed that the total infarct
area involving the cortex and striatum predominantly developed 7
days following cerebral ischemia and that the neurological status
declined between the acute phase (day 1) and the subacute phase
(day 7) following permanent MCAo. Previous studies have shown that
FerA (100 mg/kg) administered iv immediately following the onset of
ischemia exerted neuroprotective effects against cerebral
infarction 1 day following permanent cerebral ischemia (23,32).
In the present study, the effects of FerA during the subacute phase
of ischemic injury were further investigated, and it was found that
FerA administered iv at a dose of 80 (FerA-80 mg) or 100 mg/kg
(FerA-100 mg), but not 60 mg/kg (FerA-60 mg), immediately following
ischemia effectively ameliorated cerebral infarction and improved
neurological outcomes 7 days following permanent MCAo.
Increasing evidence has revealed that apoptotic
cells present in the ischemic penumbra contribute to infarct
expansion during permanent (30,31,33)
and transient (1,28) focal cerebral ischemia. Therefore,
inhibition of apoptosis in the peri-infarct region leads to the
mitigation of delayed infarct expansion in the subacute stage of
cerebral ischemia (1,33). Akt signaling is a key regulator of
cell death and survival through the activation of various
anti-apoptotic pathways during cerebral ischemic injury (34,35).
Previous studies have demonstrated that the pharmacological
activation of Akt signaling provides neuroprotective effects
against apoptosis in the ischemic penumbra and subsequent cerebral
infarction in permanent MCAo (36,37).
In the present study, the results of the western blot analysis
showed that p-Akt was significantly downregulated in the penumbral
cortex following permanent MCAo. However, this decreased expression
of p-Akt was effectively restored in the FerA-80 mg and FerA-100 mg
groups 7 days following cerebral ischemia. These findings suggest
that FerA treatment protects against cerebral ischemic injury in
the subacute phase of ischemia, and the neuroprotective effects on
cerebral infarction are at least partially attributed to the
activation of Akt signaling in the penumbral cortex 7 days
following permanent MCAo.
Activated Akt can rapidly phosphorylate certain
downstream proteins, including mTOR, and the activity of mTOR is
regulated through the phosphorylation of its serine 2448 (14). Akt-dependent activation of the
multi-protein complex, including mTOR (mTORC1) causes the
phosphorylation of p70S6K and 4E-BP1, its two major downstream
targets, which regulate the initiation of translation and protein
synthesis (38). During cerebral
ischemia, the activation of Akt/mTOR/p70S6K/S6 signaling results in
a marked reduction in the ischemic area, leading to the suppression
of protein synthesis and exacerbation of neuronal cell death
(12). By contrast, the
pharmacological upregulation of mTOR/p70S6K signaling protects
against apoptosis and cerebral infarction in the acute phase of
permanent (5,13) and transient (39) MCAo. Unphosphorylated 4E-BP1 binds
to eIF4E with high affinity and prevents it from interacting with
eIF4G for the initiation of mRNA translation. By contrast, p-4E-BP1
dissociates from eIF4E, which subsequently binds to eIF4G,
resulting in the assembly of translation initiation factors
(40,41). Activation of mTOR/4E-BP1 signaling
in the peri-infarct region exerts neuroprotective effects against
cerebral ischemic injuries in the early phase of ischemic
postconditioning (17). The
immunoblotting results in the present study indicated that the
ratio of p-mTOR/mTOR, the expression of 4E-BP1, and the ratio of
p-4E-BP1/4E-BP1 were markedly decreased in the cytosolic fraction
in the penumbral cortex, whereas these expression levels were
effectively upregulated in the FerA-80 mg and FerA-100 mg groups 7
days following permanent cerebral ischemia. These results are
consistent with those of a previous study, in which the upregulated
expression of p-mTOR, 4E-BP1 and p-4E-BP1 in the peri-infarct
cortex markedly reduced ischemic lesions and improved neurological
outcomes in the late phase of permanent MCAo (17). Koh (42) reported that FerA exerted
neuroprotective effects against cerebral infarction by activating
mTOR/p70S6K/S6 signaling in the ischemic cortex 1 day following
permanent MCAo (42). However, in
the present study, FerA treatment did not affect the expression of
p70S6K or p-p70S6K in the cytosolic fraction in the penumbral
cortex 7 days following permanent focal cerebral ischemia. On the
basis of these results, it was hypothesized that FerA treatment
effectively activates Akt/mTOR survival signaling, and that the
beneficial effects of FerA treatment on ischemia-induced cerebral
infarction are most likely attributed to the activation of
Akt/mTOR/4E-BP1-, but not Akt/mTOR/p70S6K-, induced survival
signaling in the penumbral cortex at 7 days following cerebral
ischemia.
Accumulating evidence indicates that eIF4E is
critical for translational control and is inactivated by stress,
including ischemic stress, and activated by survival factors. In
addition, the overexpression of eIF4E exerts beneficial effects on
cytochrome c-mediated apoptosis through the upregulation of
anti-apoptotic proteins, including Bcl-2 and Bcl-xL, in various
cell lines in vitro (43–45).
Fan et al (45) reported
that pharmacological inhibition of the eIF4E-eIF4G interaction
induced the apoptosis of human lung cancer cell lines in
vitro. Under cerebral ischemia conditions, the translocation of
pro-apoptotic Bax from the cytosol to the mitochondria disrupts the
integrity of the mitochondrial outer membrane, resulting in the
release of cytochrome c (46). Cytosolic cytochrome c
combines with apoptotic protease activating factor-1 and dATP to
form the apoptosome. This complex recruits and activates caspase-9,
which subsequently cleaves the downstream effector caspase-3,
thereby triggering cytochrome c-mediated apoptosis (47). It has been suggested that the
anti-apoptotic Bcl-2 protein (Bcl-xL) binds to the mitochondrial
membrane and preserves mitochondrial outer membrane integrity by
preventing the translocation of Bax; thus, Bcl-2 directly inhibits
the release of cytochrome c from the mitochondria into the
cytosol (48). An increased ratio
of mitochondrial Bcl-2 (Bcl-xL)/Bax prevents the translocation of
Bax to the mitochondria, promoting cell survival, whereas a
decreased ratio of mitochondrial Bcl-2 (Bcl-xL)/Bax induces
mitochondrial Bax homo-oligomerization and subsequently causes
mitochondrial damage, leading to cytochrome c-mediated
apoptosis in the ischemic region following cerebral ischemia
(1,46). The results of the present study
revealed that the ratio of mitochondrial Bcl-2/Bax expression was
significantly decreased in the penumbral cortex following MCAo,
whereas the reduced ratio of Bcl-2/Bax expression was effectively
upregulated in the FerA-80 mg and FerA-100 mg groups at 7 days
post-ischemia. However, FerA treatment did not affect the ratios of
mitochondrial Bcl-xL/Bax expression in the penumbral cortex. In the
IHC and TUNEL assays, the expression of cytochrome c,
cleaved caspase-3, and TUNEL immunoreactivity was significantly
upregulated in the penumbral cortex following permanent MCAo,
however, these increased immunoreactivity levels were effectively
downregulated in the FerA-80 mg and FerA-100 mg groups at 7 days
post-ischemia. These results appear to coincide with those of a
previous study, in which pharmacological treatment exerted a
neuroprotective effect against cerebral infarction through the
inhibition of cytochrome c-mediated apoptosis in the
ischemic region, and the anti-apoptotic effect was possibly due to
upregulation of the mitochondrial Bcl-2/Bax ratio in the acute
phase of permanent cerebral ischemia (49). These findings suggest that the
upregulation of Akt/mTOR/4E-BP1-mediated Bcl-2, but not Bcl-xL,
anti-apoptotic cascade may be involved in the neuroprotective
effects of FerA-80 mg and FerA-100 mg treatment, and that the
effects of FerA against mitochondrial Bax-related apoptosis can be
further attributed to inhibition of the cytochrome
c-mediated caspase-3 activation pathway in the penumbral
cortex 7 days following permanent MCAo.
To determine the precise mechanism underlying the
involvement of Akt-mediated anti-apoptotic signaling in FerA
treatment, another experiment was performed in the FerA-100 mg
group as the representative group of FerA treatment to evaluate the
action of LY, a selective inhibitor of PI3K/Akt signaling, 7 days
following cerebral ischemia. It was found that 1% DMSO (vehicle
control) pretreatment (DS + Sham, DS + Vehicle, and DS + FerA-100
mg) did not alter the expression of p-Akt, p-mTOR or p-4E-BP1.
Furthermore, the expression of eIF4E was markedly decreased in the
DS + Vehicle group and the reduced expression of eIF4E was
effectively restored in the DS + FerA-100 mg group. However, LY
pretreatment (LY + FerA-100 mg) effectively abrogated the
upregulating effects of FerA-100 mg treatment on the expression of
p-Akt. LY + FerA-100 mg treatment consequently suppressed
mTOR/4E-BP1/eIF4E signaling and activated the mitochondrial
Bax-related apoptotic signaling cascade in the penumbral cortex,
resulting in the exacerbation of the cerebral infarct size 7 days
following permanent cerebral ischemia. On the basis of these
results, it was hypothesized that FerA treatment exerts beneficial
effects on cerebral infarction by activating Akt signaling, and
that the downregulating effects of FerA treatment on mitochondrial
Bax-induced apoptosis are attributed to the upregulation of the
Akt/mTOR/4E-BP1/eIF4E/Bcl-2 anti-apoptotic signaling in the
penumbral cortex 7 days following cerebral ischemia. To the best of
our knowledge, the present study is the first to show that FerA
treatment exerts neuroprotective effects against Bax-induced
apoptosis by upregulating Akt/mTOR/4E-BP1-mediated, but not
Akt/mTOR/p70S6K-mediated Bcl-2 anti-apoptotic signaling in the
subacute phase of permanent MCAo.
Compelling evidence shows that Akt-mediated
signaling downregulates the expression of inducible nitric oxide
synthase, which elicits nitric oxide (NO)-induced apoptosis in the
ischemic region following transient MCAo (50,51).
Previous studies have shown that FerA protects against cerebral
ischemic injury by inhibiting NO-induced apoptotic signaling in the
acute phase of cerebral ischemia (18,52).
However, the role of FerA-induced Akt-mediated signaling in the
regulation of nitric oxide synthase during the subacute phase of
permanent MCAo requires elucidation in the future.
In conclusion, the findings of the present study
suggest that FerA administered at a dose of 80 or 100 mg/kg
immediately following the onset of cerebral ischemia effectively
reduces cerebral infarction and improves neurological functions 7
days following cerebral ischemia, and that the anti-infarction
effects of FerA treatment are associated with the activation of
Akt-mediated anti-apoptotic signaling in the penumbral cortex. The
effects of FerA treatment on mitochondrial Bax-induced apoptosis
can be further attributed to the upregulation of
Akt/mTOR/4E-BP1/Bcl-2 anti-apoptotic signaling, which inhibits the
cytochrome c/caspase-3-dependent apoptotic pathway in the
penumbral cortex 7 days following permanent focal cerebral
ischemia. Evidence indicates that FerA exerts neuroprotective
effects against cerebral infarct in the acute phase of permanent
cerebral ischemia (23,32). The results of the present study
further indicate that FerA treatment exerts beneficial effects on
cerebral infarction in the subacute phase of permanent MCAo.
Therefore, FerA treatment offers a potential strategy in the
subacute phase of permanent focal cerebral ischemia. Previous
studies have demonstrated that upregulation of the upstream
components of Akt signaling, including tropomyosin receptor kinase
B (53,54) and receptor tyrosine kinase
(55), effectively attenuates
cerebral ischemic injury in rat models of cerebral ischemia.
Therefore, further investigations are warranted to characterize the
effects of FerA on the regulation of Akt upstream signaling and in
the chronic phase of cerebral ischemia for determining its future
clinical application.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from China
Medical University (grant no. CMU105-S-40) and China Medical
University Hospital (grant nos. DMR-105-007 and DMR-107-165),
Taichung, Taiwan.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CYC and YCL designed experiments. CYC performed the
experiments, analyzed the data and wrote the manuscript. STK
participated in the conception and design of the study, and helped
to draft the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All study procedures were approved by the
Institutional Animal Care and Use Committee of China Medical
University (Taichung, Taiwan; permit no. 2016-321).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cheng CY, Tang NY, Kao ST and Hsieh CL:
Ferulic acid administered at various time points protects against
cerebral infarction by activating p38 MAPK/p90RSK/CREB/Bcl-2
anti-apoptotic signaling in the subacute phase of cerebral
ischemia-reperfusion injury in rats. PLoS One. 11:e01557482016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang H, Zhong R, Xia Z, Song J and Feng
L: Neuroprotective effects of rhynchophylline against ischemic
brain injury via regulation of the Akt/mTOR and TLRs signaling
pathways. Molecules. 19:11196–11210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang M, Li YJ, Ding Y, Zhang HN, Sun T,
Zhang K, Yang L, Guo YY, Liu SB, Zhao MG and Wu YM: Silibinin
prevents autophagic cell death upon oxidative stress in cortical
neurons and cerebral ischemia-reperfusion injury. Mol Neurobiol.
53:932–943. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y,
Ruan Y and Xu A: The neuroprotection of liraglutide against
ischaemia-induced apoptosis through the activation of the PI3K/AKT
and MAPK pathways. Sci Rep. 6:268592016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koh PO: Melatonin prevents ischemic brain
injury through activation of the mTOR/p70S6 kinase signaling
pathway. Neurosci Lett. 444:74–78. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie R, Cheng M, Li M, Xiong X, Daadi M,
Sapolsky RM and Zhao H: Akt isoforms differentially protect against
stroke-induced neuronal injury by regulating mTOR activities. J
Cereb Blood Flow Metab. 33:1875–1885. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maiese K, Chong ZZ, Wang S and Shang YC:
Oxidant stress and signal transduction in the nervous system with
the PI 3-K, Akt, and mTOR cascade. Int J Mol Sci. 13:13830–13866.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang C, Wang Z, Zhang X, Dong L, Xing Y,
Li Y, Liu Z, Chen L, Qiao H, Wang L and Zhu C: Protection by
silibinin against experimental ischemic stroke: Up-regulated pAkt,
pmTOR, HIF-1alpha and Bcl-2, down-regulated Bax, NF-kappaB
expression. Neurosci Lett. 529:45–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu C, Fujihara H, Yao J, Qi S, Li H,
Shimoji K and Baba H: Different expression patterns of Bcl-2,
Bcl-xl, and Bax proteins after sublethal forebrain ischemia in
C57Black/Crj6 mouse striatum. Stroke. 34:1803–1808. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chi OZ, Barsoum S, Vega-Cotto NM, Barsoum
S, Vega-Cotto NM, Jacinto E, Liu X, Mellender SJ and Weiss HR:
Effects of rapamycin on cerebral oxygen supply and consumption
during reperfusion after cerebral ischemia. Neuroscience.
316:321–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xie R, Wang P, Cheng M, Sapolsky R, Ji X
and Zhao H: Mammalian target of rapamycin cell signaling pathway
contributes to the protective effects of ischemic postconditioning
against stroke. Stroke. 45:2769–2776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koh PO: Gingko biloba extract (EGb 761)
prevents cerebral ischemia-induced p70S6 kinase and S6
phosphorylation. Am J Chin Med. 38:727–734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koh PO, Cho JH, Won CK, Lee HJ, Sung JH
and Kim MO: Estradiol attenuates the focal cerebral ischemic injury
through mTOR/p70S6 kinase signaling pathway. Neurosci Lett.
436:62–66. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chong ZZ, Yao Q and Li HH: The rationale
of targeting mammalian target of rapamycin for ischemic stroke.
Cell Signal. 25:1598–1607. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ayuso MI, Hernández-Jiménez M, Martin ME,
Salinas M and Alcázar A: New hierarchical phosphorylation pathway
of the translational repressor eIF4E-binding protein 1 (4E-BP1) in
ischemia-reperfusion stress. J Biol Chem. 285:34355–34363. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ayuso MI, Martinez-Alonso E, Cid C, Alonso
de Leciñana M and Alcázar A: The translational repressor
eIF4E-binding protein 2 (4E-BP2) correlates with selective delayed
neuronal death after ischemia. J Cereb Blood Flow Metab.
33:1173–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie R, Wang P, Ji X and Zhao H: Ischemic
post-conditioning facilitates brain recovery after stroke by
promoting Akt/mTOR activity in nude rats. J Neurochem. 127:723–732.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koh PO: Ferulic acid modulates nitric
oxide synthase expression in focal cerebral ischemia. Lab Anim Res.
28:273–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Wang H, Wang T, Jiang N, Yu P,
Chong Y and Fu F: Ferulic acid ameliorates nerve injury induced by
cerebral ischemia in rats. Exp Ther Med. 9:972–976. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu YC and Hsieh CL: Pharmacological
effects of radix angelica sinensis (Danggui) on cerebral
infarction. Chin Med. 6:322011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng CY, Ho TY, Lee EJ, Su SY, Tang NY
and Hsieh CL: Ferulic acid reduces cerebral infarct through its
antioxidative and anti-inflammatory effects following transient
focal cerebral ischemia in rats. Am J Chin Med. 36:1105–1119. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng CY, Su SY, Tang NY, Ho TY, Chiang SY
and Hsieh CL: Ferulic acid provides neuroprotection against
oxidative stress-related apoptosis after cerebral
ischemia/reperfusion injury by inhibiting ICAM-1 mRNA expression in
rats. Brain Res. 1209:136–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koh PO: Ferulic acid prevents the cerebral
ischemic injury-induced decrease of Akt and Bad phosphorylation.
Neurosci Lett. 507:156–160. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koh PO: Ferulic acid prevents the cerebral
ischemic injury-induced decreases of astrocytic phosphoprotein
PEA-15 and its two phosphorylated forms. Neurosci Lett.
511:101–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Sanberg PR, Li Y, Wang L, Lu M,
Willing AE, Sanchez-Ramos J and Chopp M: Intravenous administration
of human umbilical cord blood reduces behavioral deficits after
stroke in rats. Stroke. 32:2682–2688. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsiang CY, Wu SL and Ho TY: Morin inhibits
12-O-tetradecanoylphorbol-13-acetate-induced hepatocellular
transformation via activator protein 1 signaling pathway and cell
cycle progression. Biochem Pharmacol. 69:1603–1611. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheng CY, Lin JG, Tang NY, Kao ST and
Hsieh CL: Electroacupuncture-like stimulation at the Baihui (GV20)
and Dazhui (GV14) acupoints protects rats against subacute-phase
cerebral ischemia-reperfusion injuries by reducing S100B-mediated
neurotoxicity. PLoS One. 9:e914262014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng CY, Lin JG, Su SY, Tang NY, Kao ST
and Hsieh CL: Electroacupuncture-like stimulation at Baihui and
Dazhui acupoints exerts neuroprotective effects through activation
of the brain-derived neurotrophic factor-mediated
MEK1/2/ERK1/2/p90RSK/bad signaling pathway in mild transient focal
cerebral ischemia in rats. BMC Complement Altern Med. 14:922014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsui T, Mori T, Tateishi N, Kagamiishi
Y, Satoh S, Katsub N, Morikawa E, Morimoto T, Ikuta F and Asano T:
Astrocytic activation and delayed infarct expansion after permanent
focal ischemia in rats. Part I: Enhanced astrocytic synthesis of
s-100beta in the periinfarct area precedes delayed infarct
expansion. J Cereb Blood Flow Metab. 22:711–722. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mori T, Town T, Kobayashi M, Tan J, Fujita
SC and Asano T: Augmented delayed infarct expansion and reactive
astrocytosis after permanent focal ischemia in apolipoprotein E4
knock-in mice. J Cereb Blood Flow Metab. 24:646–656. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koh PO: Ferulic acid attenuates the
injury-induced decrease of protein phosphatase 2A subunit B in
ischemic brain injury. PLoS One. 8:e542172013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tateishi N, Mori T, Kagamiishi Y, Satoh S,
Katsube N, Morikawa E, Morimoto T, Matsui T and Asano T: Astrocytic
activation and delayed infarct expansion after permanent focal
ischemia in rats. Part II: Suppression of astrocytic activation by
a novel agent (R)-(−)-2-propyloctanoic acid (ONO-2506) leads to
mitigation of delayed infarct expansion and early improvement of
neurologic deficits. J Cereb Blood Flow Metab. 22:723–734. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tang Q, Han R, Xiao H, Shen J, Luo Q and
Li J: Neuroprotective effects of tanshinone IIA and/or
tetramethylpyrazine in cerebral ischemic injury in vivo and in
vitro. Brain Res. 1488:81–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi J, Gu JH, Dai CL, Gu J, Jin X, Sun J,
Iqbal K, Liu F and Gong CX: O-GlcNAcylation regulates
ischemia-induced neuronal apoptosis through AKT signaling. Sci Rep.
5:145002015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu J, Li J, Hu H, Liu P, Fang Y and Wu D:
Rho-kinase inhibitor, fasudil, prevents neuronal apoptosis via the
Akt activation and PTEN inactivation in the ischemic penumbra of
rat brain. Cell Mol Neurobiol. 32:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ishrat T, Sayeed I, Atif F, Hua F and
Stein DG: Progesterone is neuroprotective against ischemic brain
injury through its effects on the phosphoinositide 3-kinase/protein
kinase B signaling pathway. Neuroscience. 210:442–450. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bracho-Valdés I, Moreno-Alvarez P,
Valencia-Martinez I, Robles-Molina E, Chavez-Vargas L and
Vázquez-Prado J: mTORC1- and mTORC2-interacting proteins keep their
multifunctional partners focused. IUBMB Life. 63:896–914. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li W, Yang Y, Hu Z, Ling S and Fang M:
Neuroprotective effects of DAHP and Triptolide in focal cerebral
ischemia via apoptosis inhibition and PI3K/Akt/mTOR pathway
activation. Front Neuroanat. 9:482015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Josse L, Xie J, Proud CG and Smales CM:
mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor
stoichiometry influence recombinant protein productivity from
GS-CHOK1 cells. Biochem J. 473:4651–4664. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koh PO: Ferulic acid attenuates focal
cerebral ischemia-induced decreases in p70S6 kinase and S6
phosphorylation. Neurosci Lett. 555:7–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Attar-Schneider O, Pasmanik-Chor M,
Tartakover-Matalon S, Drucker L and Lishner M: eIF4E and eIF4GI
have distinct and differential imprints on multiple myeloma's
proteome and signaling. Oncotarget. 6:4315–4329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li S, Perlman DM, Peterson MS, Burrichter
D, Avdulov S, Polunovsky VA and Bitterman PB: Translation
initiation factor 4E blocks endoplasmic reticulum-mediated
apoptosis. J Biol Chem. 279:21312–21317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan S, Li Y, Yue P, Khuri FR and Sun SY:
The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments
TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5
induction independent of inhibition of cap-dependent protein
translation. Neoplasia. 12:346–356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao G, Minami M, Pei W, Yan C, Chen D,
O'Horo C, Graham SH and Chen J: Intracellular Bax translocation
after transient cerebral ischemia: Implications for a role of the
mitochondrial apoptotic signaling pathway in ischemic neuronal
death. J Cereb Blood Flow Metab. 21:321–333. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Noshita N, Sugawara T, Fujimura M,
Morita-Fujimura Y and Chan PH: Manganese superoxide dismutase
affects cytochrome c release and caspase-9 activation after
transient focal cerebral ischemia in mice. J Cereb Blood Flow
Metab. 21:557–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li L, Peng L and Zuo Z: Isoflurane
preconditioning increases B-cell lymphoma-2 expression and reduces
cytochrome c release from the mitochondria in the ischemic
penumbra of rat brain. Eur J Pharmacol. 586:106–113. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu D, Lu C, Wan R, Auyeung WW and Mattson
MP: Activation of mitochondrial ATP-dependent potassium channels
protects neurons against ischemia-induced death by a mechanism
involving suppression of Bax translocation and cytochrome c
release. J Cereb Blood Flow Metab. 22:431–443. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee HK, Jang JY, Yoo HS and Seong YH:
Neuroprotective effect of phytoceramide against transient focal
ischemia-induced brain damage in rats. Arch Pharm Res.
38:2241–2350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zheng L, Ding J, Wang J, Zhou C and Zhang
W: Effects and mechanism of action of inducible nitric oxide
synthase on apoptosis in a rat model of cerebral
ischemia-reperfusion injury. Anat Rec. 299:246–255. 2016.
View Article : Google Scholar
|
|
52
|
Cheng CY, Su SY, Tang NY, Ho TY, Lo WY and
Hsieh CL: Ferulic acid inhibits nitric oxide-induced apoptosis by
enhancing GABA(B1) receptor expression in transient focal cerebral
ischemia in rats. Acta Pharmacol Sin. 31:889–899. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yao RQ, Qi DS, Yu HL, Liu J, Yang LH and
Wu XX: Quercetin attenuates cell apoptosis in focal cerebral
ischemia rat brain via activation of BDNF-TrkB-PI3K/Akt signaling
pathway. Neurochem Res. 37:2777–2786. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qi D, Ouyang C, Wang Y, Zhang S, Ma X,
Song Y, Yu H, Tang J, Fu W, Sheng L, et al: HO-1 attenuates
hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt
signaling pathway in stroke. Brain Res. 1577:69–76. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao H, Shimohata T, Wang JQ, Sun G,
Schaal DW, Sapolsky RM and Steinberg GK: Akt contributes to
neuroprotection by hypothermia against cerebral ischemia in rats. J
Neurosci. 25:9794–9806. 2005. View Article : Google Scholar : PubMed/NCBI
|