Introduction
Myocardial infarction (MI) due to coronary occlusion
is the most common event in cardiovascular disease (1,2).
Following acute MI, cardiomyocyte apoptosis occurs; apoptosis may
be an indispensable pathway in cardiomyocyte death during acute
ischemia (3,4). The inhibition of cardiomyocyte
apoptosis following MI may be an effective method to ameliorate
left ventricular remodeling and promote cardiac function.
Carvedilol, a nonselective β-adrenergic receptor
(β-AR) antagonist, exhibits multiple pleiotropic properties,
including α-adrenergic receptor (α-AR) blocking and antioxidative
activities (5,6). Several studies using a variety of
in vitro and in vivo models have provided evidence
for the cardioprotective role of carvedilol; Nakamura et al
(7) demonstrated that the
administration of carvedilol improved cardiac function by
decreasing oxidative stress levels. In addition, carvedilol
decreased cardiomyocytic apoptosis by suppressing the expression of
inflammation-associated genes and apoptosis-associated proteins
through the phosphoinositide 3-kinase- and mitogen activated
protein kinase kinase-associated signaling pathways (8). However, the exact underlying
mechanisms of carvedilol are yet to be fully determined.
MicroRNAs (miRNAs) are a class of endogenous small
non-coding RNAs measuring ~22 nucleotides that negatively regulate
gene expression at the post-transcriptional level via the
3′-untranslated region (3′-UTR) of target mRNAs. Previously, an
emerging role of miRNAs in the development of cardiovascular
diseases has been explored (9,10).
Among the known miRNAs, cardiac-enriched and muscle-specific miR-1
has been demonstrated to be a key regulator of cardiac development
and disease (11–14). With the exception of regulating
cardiac development, miR-1 has been proposed to be involved in
regulating cardiomyocyte apoptosis. Notably, miR-1 levels were
significantly increased in response to oxidative stress-induced
apoptosis (15).
The aim of the present study was to investigate
whether carvedilol protects cardiomyocytes from apoptosis in an MI
rat model. Whether H2O2-induced cardiomyocyte
apoptosis may be associated with the effects of miR-1 expression
under the pathological conditions of cardiac disease was also
determined.
Materials and methods
Animal models of myocardial infarction
and drug administration
Prior to the initiation of the experimental
procedures, 30 healthy male Wistar rats (250–300 g) were obtained
from The Animal Center of the 2nd Affiliated Hospital of Harbin
Medical University (Harbin, China). Rats were housed under
temperature- (23±1°C) and humidity-(55±5%) controlled conditions,
with food and water ad libitum for 1 week. The rats used in
the present study were randomly divided into the sham, myocardial
infarction and carvedilol (Car) groups. As described previously,
left anterior descending coronary artery ligation was performed to
induce myocardial infarction (16). The rats in the Car group were
pretreated with an oral dose of 10 mg/kg carvedilol daily for 14
days prior to surgery. The rats in the sham group underwent open
chest procedures without coronary artery occlusion. The rats were
anesthetized with pentobarbital sodium (40 mg/kg) by
intraperitoneal injection prior to surgery and transthoracic
echocardiography. The physical methods of cervical dislocation or
decapitation were performed for euthanasia of the adult or neonatal
rats, respectively. Consistent with the American Veterinary Medical
Association Guidelines for the Euthanasia of Animals (2013 Edition)
(17), death was confirmed prior
to disposal of the rats. A combination of criteria, including lack
of pulse, breathing, corneal reflex or response to firm toe pinch,
inability to hear respiratory sounds and heartbeat by use of a
stethoscope, graying of the mucous membranes and rigor mortis were
used to verify animal death.
Echocardiography and infarct area
assessment
Transthoracic echocardiography was performed 24 h
after coronary artery ligation to measure the left ventricular
internal dimension in M-mode. Following echocardiography,
ventricular tissues were collected and stored at −20°C for 20 min.
The samples were then sliced into 2-mm thick sections and incubated
in 1% 2,3,5-triphenyltetrazolium chloride (Beijing Solarbio Science
& Technology, Beijing, China) at 37°C in 0.2 M Tris buffer (pH
7.4) for 30 min. Then, the sections were placed on clean paper for
imaging. Infarct size was assessed by examining the extracted
hearts; the ratio of average scar size to the average left
ventricular size was calculated. All quantitative evaluations were
performed using ImagePro Plus v6.0 software (Media Cybernetics,
Inc., Rockville, MD, USA).
Cell culture and transfection
The hearts of 1- to 3-day-old neonatal Wistar rats
were isolated and ground in serum-free Dulbecco's modified Eagle's
medium (HyClone; GE Healthcare Life Sciences, Logan, UT, USA), and
then incubated with 0.25% trypsin solution until the tissues were
almost completely digested. After 2 h, non-adherent cardiomyocytes
were collected and re-plated in 6-well plates supplemented with 10%
fetal bovine serum (HyClone; GE Healthcare Life Sciences) and 100
µg/ml penicillin/streptomycin at 5% CO2 and 37°C for 48
h. Prior to all experiments, cardiomyocytes were serum-starved
overnight. Subsequently, cardiomyocytes were treated with 100 µM
H2O2 or 10 µM carvedilol (Bikai
Pharmaceutical Co., Ltd., Haikou, China) for 24 h, or transfected
with 50 nM miR-1 (5′-UGGAAUGUAAAGAAGUAUGUAU-3′), miR-1 inhibitor
(5′-AUACAUACUUCUUUACAUUCCA-3′) or negative control
(5′-UUUGUACUACACAAAAGUACUG-3′), which were synthesized by Guangzhou
RiboBio Co., Ltd. (Guangzhou, China). miRs were transfected using
X-treme GENE siRNA transfection reagent (Roche Diagnostics, Basel,
Switzerland) according to the manufacturer's protocol. After 12 h
of transfection, cardiomyocytes were treated with
H2O2 or carvedilol.
Cell viability assay
An MTT assay was used to determine cell viability.
Cardiomyocytes at a density of 2×104 cells/well were
plated into 96-well plates and transfected, and/or treated with
carvedilol or H2O2. Following incubation with
15 µl MTT for 4 h at 37°C, the culture medium was carefully
removed. Then, 150 µl dimethyl sulfoxide was added to each well.
The plates were agitated to dissolve the formazan crystals.
Finally, the absorbance was measured at 490 nm using an Infinite
M200 Pro microplate spectrophotometer (Tecan Group, Ltd.,
Männedorf, Switzerland).
Identification of target gene
The potential target genes of miR-1 were predicted
using TargetScanHuman (version 7.2; http://www.targetscan.org/vert_72/).
Protein extraction and western blot
analysis
Total protein samples were extracted from the left
ventricular peri-infarct region of rats or cardiomyocytes as
described previously (18).
Briefly, homogenized specimens or cells were lysed in 300 µl of
radioimmunoprecipitation assay buffer (cat. no. R0020; Beijing
Solarbio Science & Technology) containing 1% protease
inhibitor. Then centrifuging at 12,000 × g for 25 min at 4°C. The
protein concentration in the supernatant was examined using a
bicinchoninic acid protein assay (Beyotime Institute of
Biotechnology, Haimen, China). A total of 100 µg protein was loaded
in each lane. Proteins were separated by 10% SDS-PAGE. Following
transfer of the proteins to Pure Nitrocellulose Blotting membranes
(Pall Life Sciences, Port Washington, NY, USA), the blots were
probed with primary antibodies against HSP60 (1:1,000, cat. no.
12165S; Cell Signaling Technology, Inc., Danvers, MA, USA), B-cell
lymphoma 2 (Bcl-2; 1:1,000; cat. no. 26593-1-AP; ProteinTech Group,
Inc., Chicago, IL, USA), Bcl-2-associated X protein (Bax; 1:1,000;
cat. no. 50599-2-Ig; ProteinTech Group, Inc., Chicago, IL, USA) and
GAPDH (1:1,000; cat. no. KC-5G4; Chengdu Kang Cheng Electronic Co.,
Ltd., Chengdu, China), which was the internal control. Membranes
were blocked using 5% skim milk (cat. no. 232100; San Jose, CA,
USA) at 4°C overnight. Membranes were subsequently incubated with
IRDye 800CW-labeled goat anti-rabbit IgG (1:10,000; cat. no.
926-32211; LI-COR Biosciences) and IRDye 800CW-labeled goat
anti-mouse IgG (1:10,000; cat. no. 926-32210; LI-COR Biosciences)
for 1 h at room temperature. Protein expression was analyzed and
quantified using Odyssey Infrared Imaging System (version 3.0;
LI-COR Biosciences).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA samples were extracted from heart tissues
or cardiomyocytes using TRIzol® (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
protocol. The extracted RNA was reverse transcribed into cDNA using
a High-Capacity cDNA RT kit (cat. no. 4368814; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. An RT
primer containing a highly stable stem-loop structure was used to
elongate the target miRNA. The plates were incubated for 15 min at
16°C, for 1 h at 37°C and for 5 min at 85°C. The expression levels
of miR-1 and HSP60 mRNA were determined using a SYBR Green PCR
master mix kit (cat. no. 4309155; Thermo Fisher Scientific, Inc.)
on an ABI 7500 fast Real Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). U6 and GAPDH were employed as the
internal controls for miR-1 and HSP60, respectively. The cDNA
samples were amplified in 96-well plates for 10 min at 95°C,
followed by 40 cycles of 15 sec at 95°C, 30 sec at 60°C and 30 sec
at 72°C. The relative expression of the miRNA and mRNA were
determined using the Cq (2−ΔΔCq) quantification method
(19). The sequences of the
primers used were the following: miR-1 RT primer,
5′-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACATACACAC-3′;
miR-1 forward, 5′-GGCTGGAATGTAAAGAAGTG-3′; miR-1 reverse,
5′-TATCCAGTGCGTGTCGTG-3′; U6 forward,
5′-GCTTCGGCAGCACATATACTAAAAT-3′; U6 reverse,
5′-CGCTTCACGAATTTGCGTGTCAT-3′. HSP60 forward,
5′-GGCTATCGCTACTGGT-3′; HSP60 reverse, 5′-GCAAGTCGCTCGTTCA-3′;
GAPDH forward, 5′-AAGAAGGTGGTGAAGCAGGC-3′; GAPDH reverse,
5′-TCCACCACCCAGTTGCTGTA-3′.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
To evaluate the level of cell apoptosis, a TUNEL
assay was performed using an In Situ Cell Death Detection kit
(Roche Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's protocols. Cardiomyocytes grown on coverslips were
washed with PBS, and fixed in 4% paraformaldehyde solution for 1 h
at 4°C. The cells were permeabilized in a solution containing 0.1%
Triton X-100 for 2 min, followed by incubation in freshly prepared
TUNEL reaction mixture for 1 h at 37°C in the dark. Subsequently,
the TUNEL-stained coverslips were washed with PBS and then
counterstained with DAPI (1:20 dilution; Beyotime Institute of
Biotechnology) for 15 min at room temperature. Nuclei
double-labelled with TUNEL and DAPI were considered to be
TUNEL-positive as detected in ≥5 fields of view under a laser
confocal microscope (Olympus Corporation, Tokyo, Japan;
magnification, ×200).
Statistical analysis
The data are presented as mean ± standard error of
the mean. Statistical comparisons among multiple groups were
performed using a one-way analysis of variance, followed by a
Bonferroni's multiple comparison post-hoc test. Statistical values
were analyzed using GraphPad Prism 5.0 (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Carvedilol ameliorates impaired
cardiac function and decreases infarct size
In our previous study, it was identified that
carvedilol ameliorated impaired cardiac function within infarcted
rats (20). Consistent with a
previous study, echocardiography was performed 24 h post-MI
induction, prior to 2 weeks of treatment with carvedilol. Analysis
demonstrated that MI hearts were significantly dilated, as
evidenced by an increase in the left ventricular systolic internal
dimension, indicating impaired cardiac function; no significant
changes in left ventricular diastolic internal dimension were
observed (Fig. 1A and B).
Carvedilol decreased the left ventricular systolic internal
dimension. The infarct size of MI rats was significantly increased,
whereas the size of infarcted area was decreased by ~20% following
the administration of carvedilol (Fig.
1C and D).
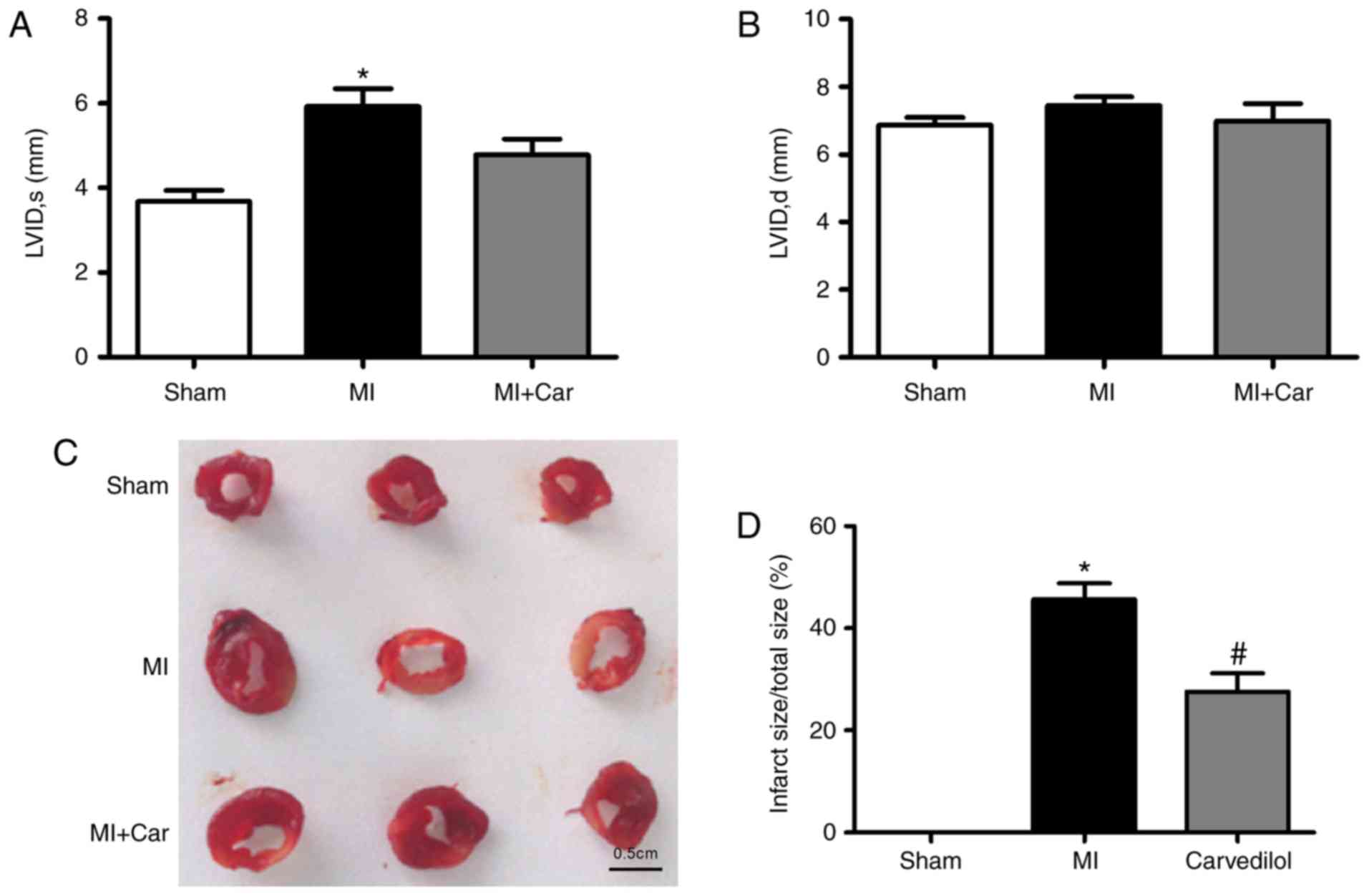 | Figure 1.Effects of carvedilol on cardiac
function and infarct size in a rat model of MI. MI was established
by coronary artery ligation for 24 h. (A) LVID, s, and (B) LVID, d,
values in each model group. (C) Examples of infarcted area of the
left ventricular walls, as indicated by 2,3,5-triphenyltetrazolium
chloride staining. Normal areas were stained brick-red, and the
infarcted areas remained unstained (white). Scale bar=0.5 cm. (D)
Statistical analysis of infarct size from 3 rats. *P<0.05 vs.
Sham; #P<0.05 vs. MI. MI, myocardial infarction;
LVID, s, left ventricular systolic internal dimension; LVID, d,
left ventricular diastolic internal dimension. |
Carvedilol increases cardiomyocyte
viability and inhibits miR-1 expression
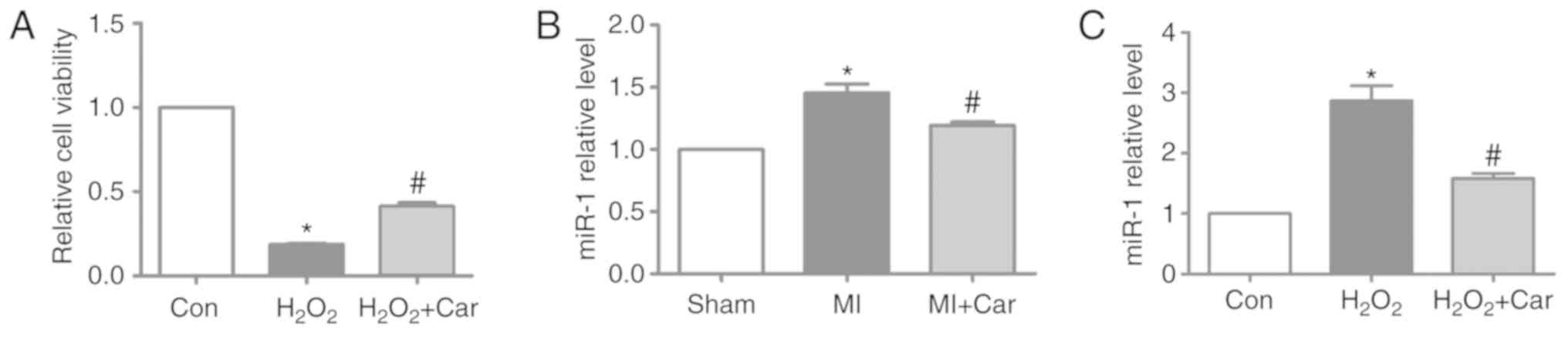
In the present study, 100 µM
H2O2 and 10 µM carvedilol were the treatments
selected for subsequent experiments. It was identified that
H2O2 significantly decreased cardiomyocyte
viability, whereas treatment with carvedilol reversed decreases in
cell viability induced by H2O2 (Fig. 2A). To investigate whether miR-1
expression was altered following carvedilol treatment, RT-qPCR was
performed to detect the miR-1 expression levels in myocardial
tissue and cardiomyocytes. The expression levels of miR-1 were
significantly increased by ~45% in MI myocardial tissues, but were
significantly downregulated in the MI + Car group compared with the
MI group (Fig. 2B). Similarly, in
cultured cardiomyocytes, upregulation of miR-1 expression induced
by H2O2 was significantly reversed following
treatment with carvedilol (Fig.
2C).
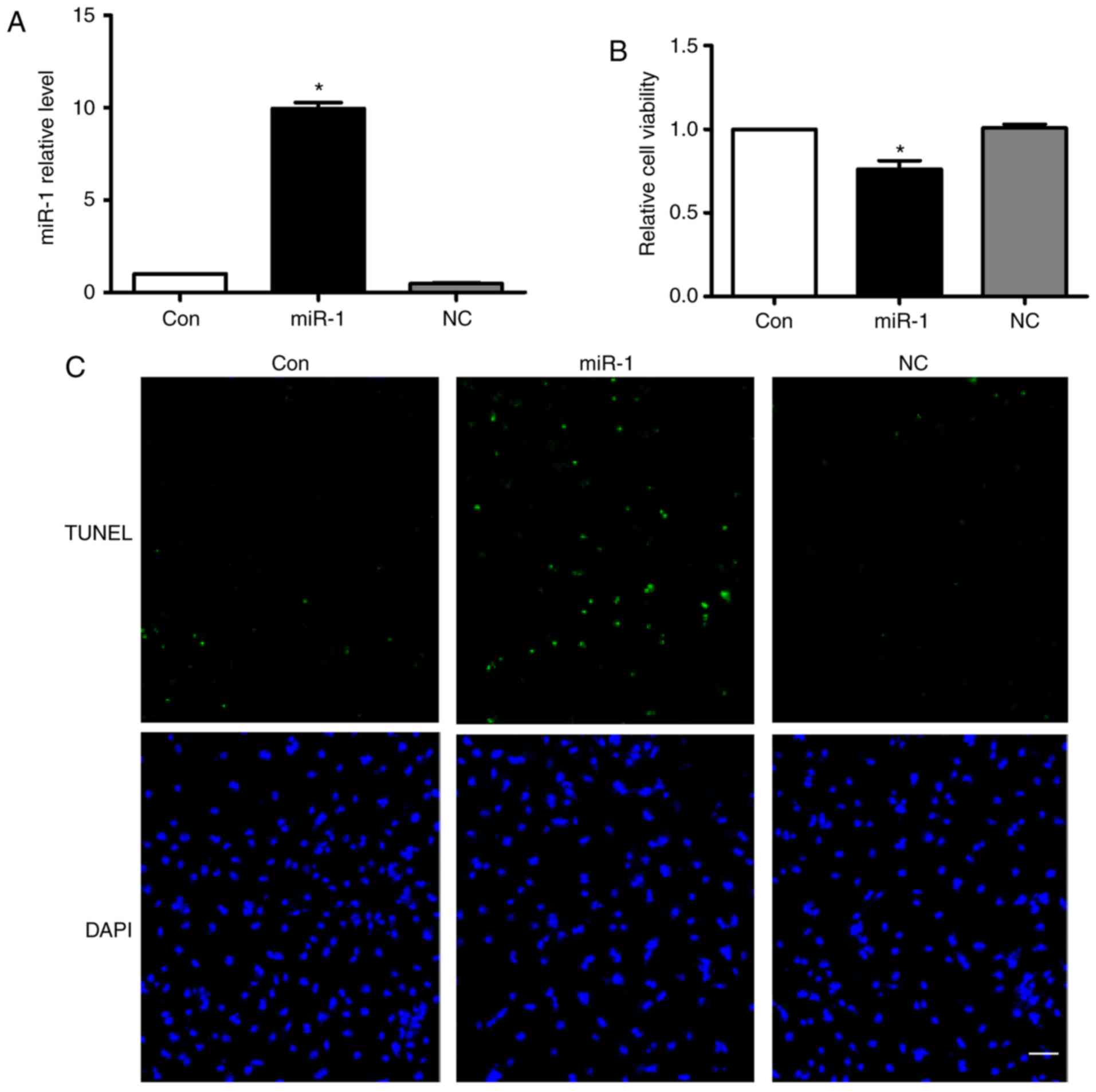
Overexpression of miR-1 induces
cardiomyocyte apoptosis
According to the aforementioned results above, we
hypothesized that the anti-apoptotic effect of carvedilol may be
mediated by downregulating miR-1 expression. Therefore, the effects
of miR-1 on cardiomyocytes were verified. Fig. 3A demonstrated that miR-1 was
successfully transfected into cardiomyocytes. Overexpression of
miR-1 significantly decreased cell viability, while no change was
observed following transfection of the negative control (NC)
(Fig. 3B). Notably, overexpression
of miR-1 substantially induced cardiomyocyte apoptosis, as
determined by TUNEL staining (Fig.
3C). In addition, no significant effects on cardiomyocyte
apoptosis in the NC group were observed (Fig. 3C).
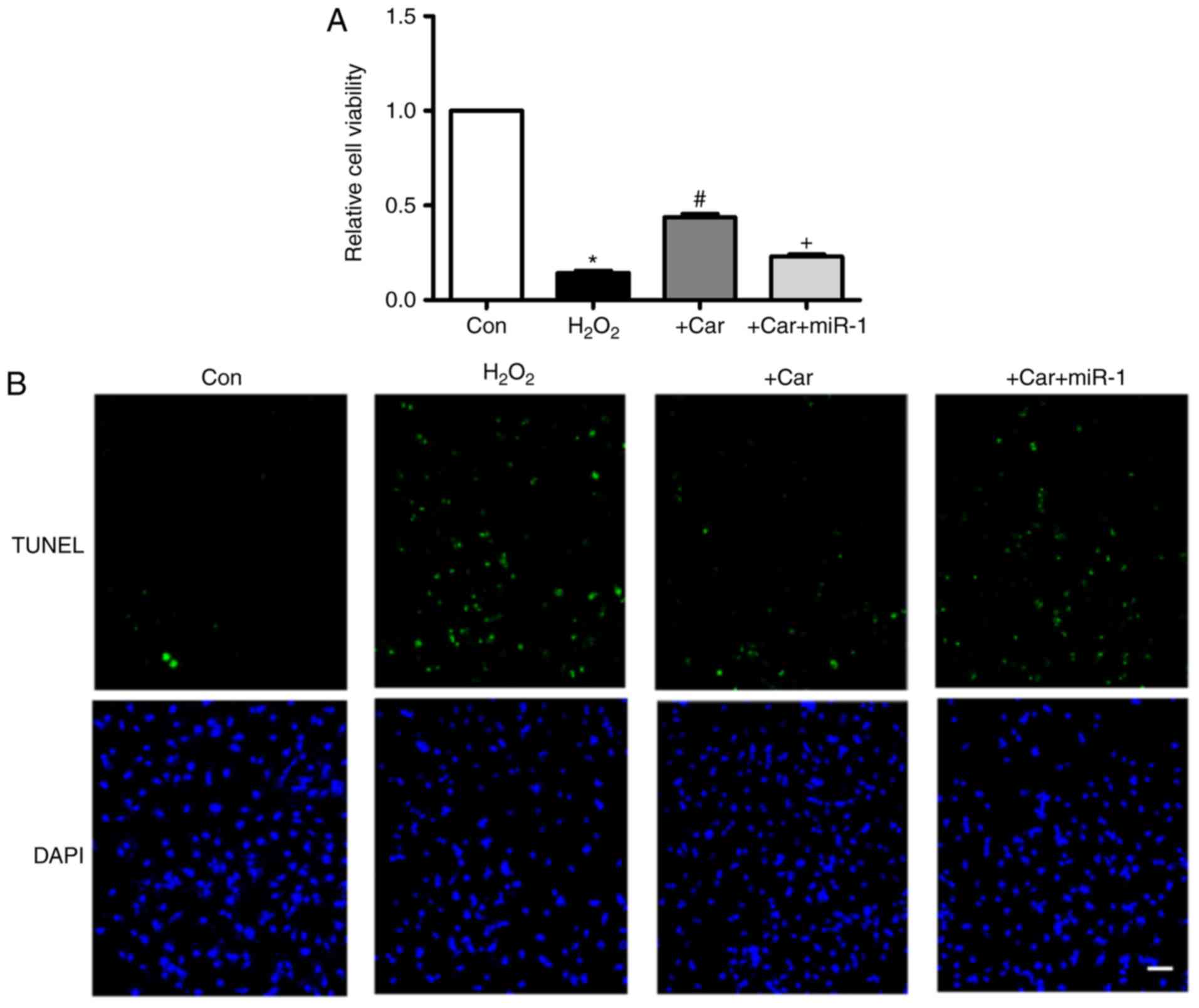
Downregulation of miR-1 is involved in
the anti-apoptotic action of carvedilol
As indicated in Fig.
4A, treatment with carvedilol reversed the decrease in cell
viability induced by H2O2, and the effect was
almost eliminated under the condition of miR-1 transfection.
Additionally, in Fig. 4B, TUNEL
staining indicated that the H2O2-induced
apoptosis of cardiomyocytes was attenuated by carvedilol. As
expected, this was reversed by miR-1, indicating that the observed
anti-apoptotic effect of carvedilol was mediated by down-regulation
of miR-1.
Targeting HSP60 by miR-1 is a
mechanism underlying the cytoprotection of carvedilol
To elucidate the molecular mechanisms by which miR-1
executes its function, HSP60 was bioinformatically predicted to be
the conservative target of miR-1 using the bioinformatics tool
TargetScan. The potential binding sites identified in the 3′UTR of
the human, rat and mouse HSP60 gene for miR-1 are demonstrated in
Fig. 5A. It was identified that
the mRNA and protein expression levels of HSP60 were significantly
decreased under the conditions of MI, while their levels were
increased following pretreatment with carvedilol (Fig. 5B and C). H2O2
treatment markedly inhibited the expression of HSP60 at the mRNA
and protein levels in vitro. These effects were notably
inhibited by carvedilol, whereas cooperation with miR-1 eliminated
the effects of carvedilol (Fig. 5D and
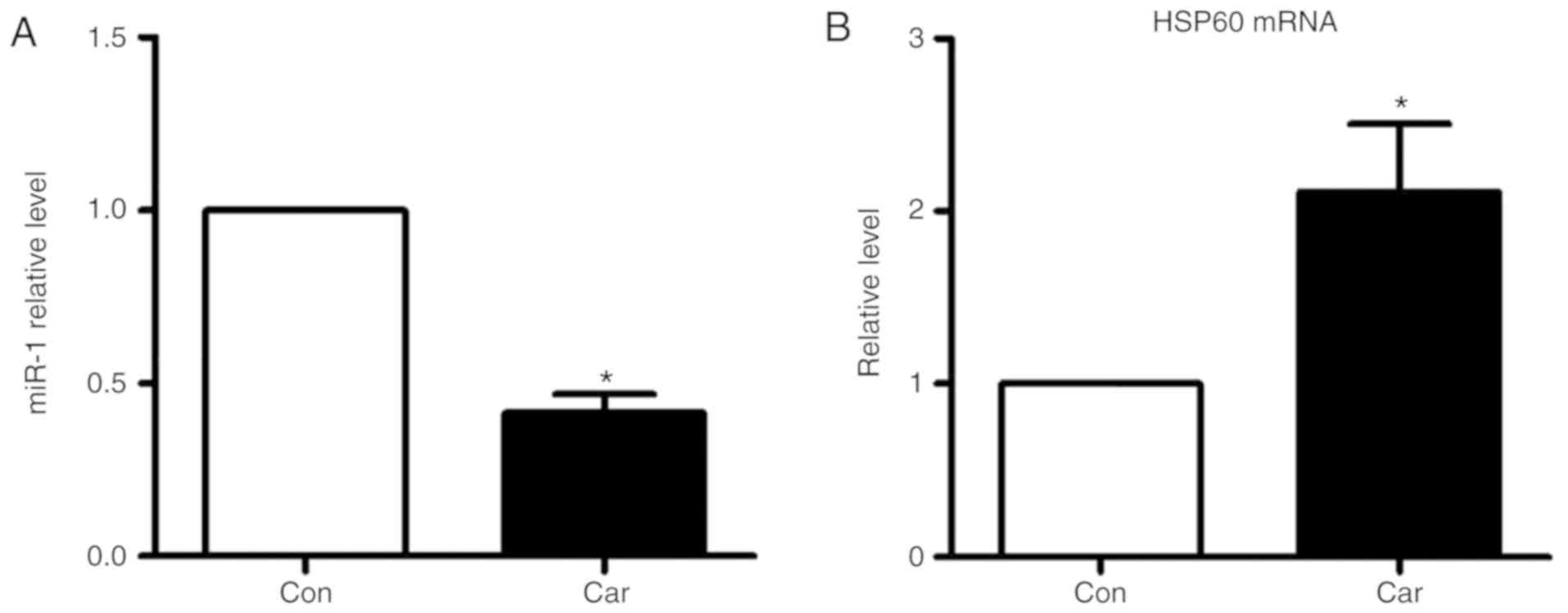
E). Carvedilol alone also decreased the expression of miR-1
(Fig. 6A), and increased the mRNA
expression levels of HSP60 (Fig.
6B).
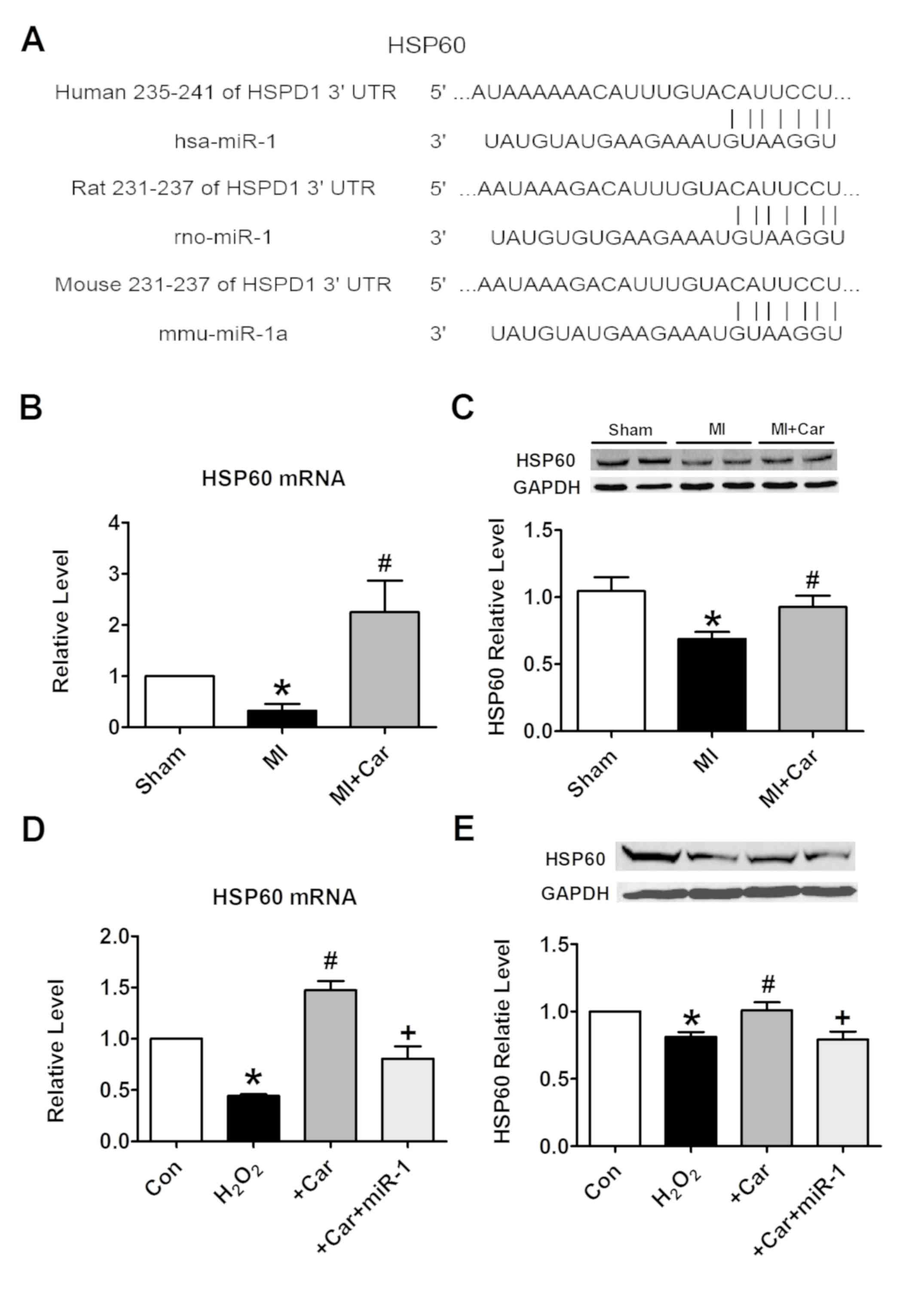 | Figure 5.miR-1 suppresses the expression of
HSP60 by binding to its 3′UTR. (A) Sequence alignment between miR-1
and the 3′UTR of human, rat and mouse HSP60. The matched base pairs
are connected by a vertical line. (B) Effects of carvedilol on
HSP60 mRNA levels in myocardial tissues, as measured by RT-qPCR.
(C) Effects of carvedilol on HSP60 protein expression levels in
myocardial tissues, as measured by western blot analysis. (D)
Effects of carvedilol on HSP60 mRNA levels in cardiomyocytes, as
measured by RT-qPCR. (E) Effects of carvedilol on HSP60 protein
expression levels in cardiomyocytes, as measured by western blot
analysis. Expression levels were normalized to that of GAPDH. All
data are presented as relative levels. *P<0.05 vs. Sham or Con;
#P<0.05 vs. MI or H2O2;
+P<0.05 vs. +Car. Car, carvedilol; Con, control; MI,
myocardial infarction; HSPD1/HSP60, heat shock protein 60; miR,
microRNA; 3′UTR, 3′untranslated region; RT-qPCR, reverse
transcription quantitative polymerase chain reaction. |
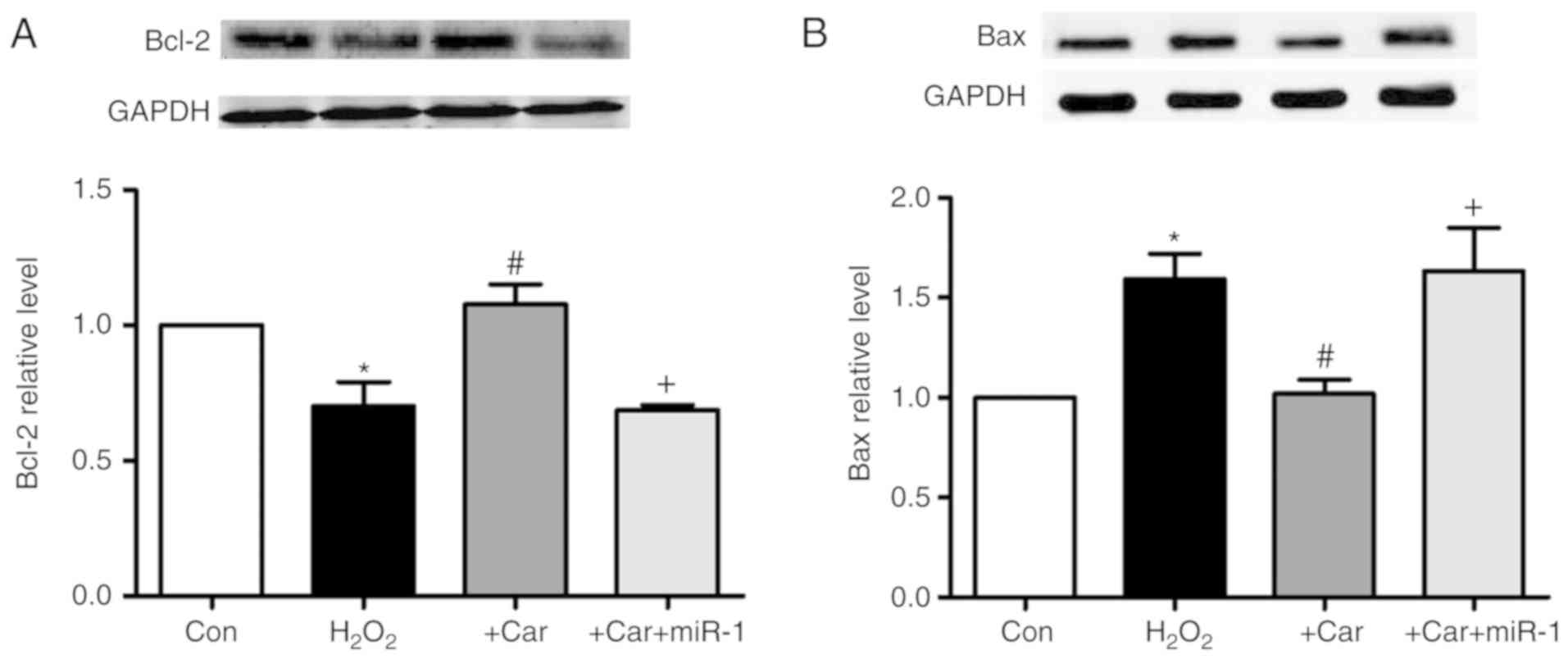
Upregulation of HSP60 by carvedilol is
associated with an increase in Bcl-2 and a decrease in Bax
As demonstrated in Fig.
7A, it was observed that carvedilol abrogated the
H2O2-induced downregulation of Bcl-2 protein
expression. Furthermore, transfection of miR-1 resulted in the
inability of carvedilol to upregulate the expression of Bcl-2.
Conversely, the expression of Bax, a pro-apoptotic factor, was
decreased by carvedilol treatment in the presence of
H2O2, whereas transfection of miR-1 reversed
the effects of carvedilol (Fig.
7B).
Discussion
The mechanism underlying the interaction between
miR-1 and carvedilol in MI may be associated with the decreased
expression of miR-1, which occurred following treatment with
carvedilol in the MI heart model of the present study, which
implies a specific association between carvedilol and miR-1. Based
on the results of the present study, a model of
H2O2-induced cardiomyocyte apoptosis was
established. It was identified that carvedilol protected
cardiomyocytes against H2O2-induced
apoptosis, which was accompanied with the downregulation of miR-1
expression; overexpression of miR-1 reversed the effects of
carvedilol. Bioinformatic analysis revealed that HSP60 was a direct
target of miR-1. Subsequent experiments suggested that the
increased expression of HSP60 may be involved in the mechanism
underlying the effects of carvedilol. Myocardial infarction is a
progressive process, yet the underlying mechanism remains unknown.
These data demonstrated that the downregulation of miR-1, at least
in part, mediated the cytoprotective effects of carvedilol against
cardiomyocyte apoptosis, which provides novel insight into the
roles of miRNAs in the cardioprotective effects against ischemia
exhibited by the β-blocker carvedilol.
It has been suggested that β-blockers exhibit highly
effective roles in the treatment of cardiovascular diseases,
including hypertension, coronary heart disease, angina, myocardial
infarction and heart failure (21,22).
Carvedilol, a third-generation β-blocker, provided improved
protection against vascular events compared with other β-blockers
(6,23,24).
However, the underlying mechanism of this protection is not fully
understood. Previous studies suggested that these protective
effects observed in a number of cardiovascular diseases were due to
the antioxidative properties of carvedilol (25–27).
Carvedilol, a specific agonist of the β1- and β2-ARs, selectively
activates β-arrestin-mediated signaling (28,29).
Similarly, carvedilol has been demonstrated to activate EGFR-ERK
signaling, which protects cardiomyocytes in a cell model system via
β-arrestin 1 (28).
Heat shock proteins, a group of molecular
chaperones, are capable of preventing protein damage and
proteolysis, which may be induced by heat, ischemia, hypothermia
and hypoxia (30). HSP60, an
important member of the heat shock protein family, is an
anti-apoptotic protein expressed in mammalian hearts. HSP60 forms
complexes with Bax, Bcl-2 homologous antagonist killer (Bak) and
Bcl-xS, preventing Bax oligomerization and insertion into the
mitochondrial membrane, thereby circumventing the mitochondrial
apoptotic pathway (12,30,31).
HSP60 served a significant role in the recovery of mitochondrial
function and the protection of cardiomyocytes. HSP60 protected
against the apoptosis of cardiomyocytes by increasing the
activities of complex III and IV in mitochondria, and decreasing
the release and activity of mitochondrial cytochrome c and
caspase-3, respectively, following ischemic stress (32,33).
In the present study, downregulated HSP60 was sufficient to induce
apoptosis, which was associated with a decrease in Bcl-2 and an
increase in Bax expression, as previously demonstrated (31). However, carvedilol reversed the
aforementioned effects to protect cardiomyocytes against
apoptosis.
Several studies have demonstrated that miRNAs serve
important roles in cardiovascular development and its associated
diseases. For example, the cardiac-enriched, muscle-specific miR-1
has been identified for its critical role in myocardial infarction:
Shan et al (34) indicated
that miR-1 and miR-206 participated in cardiomyocyte apoptosis in
MI by suppressing insulin-like growth factor 1. In addition, our
previous study revealed that the upregulation of miR-1 delayed
cardiac conduction and depolarized the cytoplasmic membrane via
potassium voltage-gated channel subfamily J member 2 and gap
junction protein alpha 1 gene downregulation, which encode the
K+ channel subunit Kir2.1 and gap junctional channel
connexin 43, respectively (35).
Overexpression of miR-1 may also contribute to the increased
susceptibility of the heart to atrioventricular block during the
onset of myocardial ischemia (13). In the present study, it was
identified that the β-blocker carvedilol protected against
cardiomyocyte apoptosis by downregulating miR-1 in vivo and
in vitro. This is consistent with the results from a
previous study, which indicated the protective mechanism of β-AR
blockers and tanshinone IIA in cardiomyocytes in ischemia.
Xu et al (20) first proposed that carvedilol
exhibited cardioprotective effects against infarction and oxidative
stress, which were associated with the increased expression levels
of miR-133. These results from previous studies, and those from the
present study, indicated that the combined actions of miR-1 and
miR-133 associated with carvedilol produced beneficial
cardioprotective effects. The two different mechanisms may function
together to protect the heart from myocardial infarction, and may
occur in various pathological conditions. However, how carvedilol
suppresses H2O2-induced increases in miR-1
and decreases in miR-133 expression remains unknown. Additional
studies are required to fully understand the protective mechanisms
of carvedilol under the conditions of ischemic injury.
In addition to miR-1 and miR-133, the association
between carvedilol and associated miRNAs in the negative and
positive regulation of cardiovascular diseases has been described
in several studies (36–40). miR-125a-5p, miR-125b-5p, miR-150,
miR-199a-3p and miR-214 were upregulated by carvedilol stimulation;
this effect was not observed in cells or mice lacking β1-AR, G
protein-coupled receptor kinase 5/6 or β-arrestin 1 (36). miR-125b-5p protected the heart
against acute MI by inhibiting the death of cultured cardiomyocytes
in response to injury, partially through the suppression of the
pro-apoptotic genes Bcl-2 antagonist/killer 1 and Krüppel like
factor 13 (37).
Carvedilol-mediated upregulation of miR-466g or miR-532-5p was
identified to be dependent on β2-ARs, and upregulation of miR-674
by carvedilol was identified to be dependent on β1-ARs (38). β2-AR/β-arrestin-responsive miR-532
conferred cardioprotection against MI though serving as a
gatekeeper of cardiac vascularization by repressing a known
endothelial-to-mesenchymal transition initiator, serine protease 23
(39). The upregulation of
miR-29b, an additional cardioprotective miR, was demonstrated to
contribute to the effects of carvedilol by attenuating post-MI
fibrosis (40). Collectively,
these studies support the hypothesis that the cardioprotective
effects of carvedilol are associated with changes in the expression
levels of carvedilol-responsive miRNAs.
It should be noted that the present study does not
provide insight into the mechanism of action of carvedilol and
miR-1. Previous studies have suggested that β-AR/β-arrestin
signaling is involved in miRNA maturation regulatory network
activated by carvedilol, which may be associated with the
protective effects of this therapeutic agent (36,38).
Therefore, there is an urgent requirement for additional
investigations into the effects of gain or loss-of-function of the
regulatory mechanism proposed in the present study in the
pathophysiology of cardiovascular diseases.
In summary, the present study revealed that
carvedilol protected cardiomyocytes from apoptosis by inhibiting
miR-1 expression in cardiomyocytes; this cardioprotective effect
was associated with increased HSP60 expression within
cardiomyocytes in ischemia. Carvedilol-responsive miRNAs are
increasingly recognized. Therefore, future studies are required to
fully elucidate the potential overlapping/compensatory effects of
known carvedilol-responsive miRNAs and their underlying mechanisms
of action in the pathophysiology of cardiovascular diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the Fund
of Scientific Research Innovation of The First Affiliated Hospital
of Harbin Medical University (grant no., 2016Y005), the National
Natural Science Foundation of China (grant no., 81570399), and the
Program for New Century Excellent Talents in Heilongjiang
Provincial University (grant no., 1254-NCET-012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
YZ, XH and YH conceived and designed the study. YH,
XC, XL, ZL, HD, LL, JZ, JJ, LW, XL, ZP and CX performed data
acquisition and analysis. YH wrote the manuscript. All authors
reviewed and approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the regulations of
Experimental Animal Ethic Committee of Harbin Medical University,
China (Animal Experimental Ethical Inspection Protocol No.
2010102), and conformed to the Guide for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH Publication No. 85-23, revised 1996).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miR-1
|
microRNA-1
|
|
MI
|
myocardial infarction
|
|
HSP60
|
heat shock protein 60
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
Bax
|
Bcl-2-associated X protein
|
References
|
1
|
Writing Group Members, Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Heart disease and stroke
statistics-2016 update: A report from the American Heart
Association. Circulation. 133:e38–e360. 2016.PubMed/NCBI
|
|
2
|
Sun T, Dong YH, Du W, Shi CY, Wang K,
Tariq MA, Wang JX and Li PF: The role of microRNAs in myocardial
infarction: From molecular mechanism to clinical application. Int J
Mol Sci. 18(pii): E7452017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu Q, Zhang J, Xu Y, Huang Y and Wu C:
Effect of carvedilol on cardiomyocyte apoptosis in a rat model of
myocardial infarction: A role for toll-like receptor 4. Indian J
Pharmacol. 45:458–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palojoki E, Saraste A, Eriksson A, Pulkki
K, Kallajoki M, Voipio-Pulkki LM and Tikkanen I: Cardiomyocyte
apoptosis and ventricular remodeling after myocardial infarction in
rats. Am J Physiol Heart Circ Physiol. 280:H2726–H2731. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oliveira PJ, Marques MP, Batista de
Carvalho LA and Moreno AJ: Effects of carvedilol on isolated heart
mitochondria: Evidence for a protonophoretic mechanism. Biochem
Biophys Res Commun. 276:82–87. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang R, Miura T, Harada N, Kametani R,
Shibuya M, Fukagawa Y, Kawamura S, Ikeda Y, Hara M and Matsuzaki M:
Pleiotropic effects of the beta-adrenoceptor blocker carvedilol on
calcium regulation during oxidative stress-induced apoptosis in
cardiomyocytes. J Pharmacol Exp Ther. 318:45–52. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakamura K, Kusano K, Nakamura Y,
Kakishita M, Ohta K, Nagase S, Yamamoto M, Miyaji K, Saito H,
Morita H, et al: Carvedilol decreases elevated oxidative stress in
human failing myocardium. Circulation. 105:2867–2871. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yeh CH, Chen TP, Wang YC, Lin YM and Fang
SW: Carvedilol treatment after myocardial infarct decreases
cardiomyocytic apoptosis in the peri-infarct zone during
cardioplegia-induced cardiac arrest. Shock. 39:343–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Rosa S, Curcio A and Indolfi C:
Emerging role of microRNAs in cardiovascular diseases. Circ J.
78:567–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Latronico MV and Condorelli G: MicroRNAs
and cardiac pathology. Nat Rev Cardiol. 6:419–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pan Z, Sun X, Ren J, Li X, Gao X, Lu C,
Zhang Y, Sun H, Wang Y, Wang H, et al: miR-1 exacerbates cardiac
ischemia-reperfusion injury in mouse models. PLoS One.
7:e505152012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shan ZX, Lin QX, Deng CY, Zhu JN, Mai LP,
Liu JL, Fu YH, Liu XY, Li YX, Zhang YY, et al: miR-1/miR-206
regulate Hsp60 expression contributing to glucose-mediated
apoptosis in cardiomyocytes. FEBS Lett. 584:3592–3600. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Sun L, Zhang Y, Liang H, Li X,
Cai R, Wang L, Du W, Zhang R, Li J, et al: Overexpression of
microRNA-1 causes atrioventricular block in rodents. Int J Biol
Sci. 9:455–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Zhang L, Chu W, Wang B, Zhang J,
Zhao M, Li X, Li B, Lu Y, Yang B and Shan H: Tanshinone IIA
inhibits miR-1 expression through p38 MAPK signal pathway in
post-infarction rat cardiomyocytes. Cell Physiol Biochem.
26:991–998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang Y, Zheng J, Sun Y, Wu Z, Liu Z and
Huang G: MicroRNA-1 regulates cardiomyocyte apoptosis by targeting
Bcl-2. Int Heart J. 50:377–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Y, Zhang Y, Shan H, Pan Z, Li X, Li B,
Xu C, Zhang B, Zhang F, Dong D, et al: MicroRNA-1 downregulation by
propranolol in a rat model of myocardial infarction: A new
mechanism for ischaemic cardioprotection. Cardiovasc Res.
84:434–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
AVMA Quidelines for the Euthanasia of
Animals, . 2013. American Veterinary Medical Association;
Schaumburg, IL: 2013, https://www.avma.org/KB/Policies/Documents/euthanasia.pdfJune
15–2016
|
|
18
|
Zhang Y, Li X, Zhang Q, Li J, Ju J, Du N,
Liu X, Chen X, Cheng F, Yang L, et al: Berberine hydrochloride
prevents postsurgery intestinal adhesion and inflammation in rats.
J Pharmacol Exp Ther. 349:417–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu C, Hu Y, Hou L, Ju J, Li X, Du N, Guan
X, Liu Z, Zhang T, Qin W, et al: β-Blocker carvedilol protects
cardiomyocytes against oxidative stress-induced apoptosis by
up-regulating miR-133 expression. J Mol Cell Cardiol. 75:111–121.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kurdi M and Booz GW: Carvedilol protects
the infarcted heart by upregulating miR-133: First evidence that
disease state affects β-adrenergic arrestin-biased signaling? J Mol
Cell Cardiol. 76:12–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Poirier L and Tobe SW: Contemporary use of
β-blockers: Clinical relevance of subclassification. Can J Cardiol.
30 (5 Suppl):S9–S15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Poole-Wilson PA, Swedberg K, Cleland JG,
Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M,
Remme WJ, et al: Comparison of carvedilol and metoprolol on
clinical outcomes in patients with chronic heart failure in the
carvedilol or metoprolol European trial (COMET): Randomised
controlled trial. Lancet. 362:7–13. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Remme WJ, Torp-Pedersen C, Cleland JG,
Poole-Wilson PA, Metra M, Komajda M, Swedberg K, Di Lenarda A,
Spark P, Scherhag A, et al: Carvedilol protects better against
vascular events than metoprolol in heart failure: Results from
COMET. J Am Coll Cardiol. 49:963–971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Budni P, Pedrosa RC, Garlet TR, Dalmarco
EM, Dalmarco JB, Lino MR, Simionato EL, Amara JA, Frode TS and
Wilhelm Filho D: Carvedilol attenuates oxidative stress in chronic
chagasic cardiomyopathy. Arq Bras Cardiol. 98:218–224. 2012.(In
English). View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li YC, Ge LS, Yang PL, Tang JF, Lin JF,
Chen P and Guan XQ: Carvedilol treatment ameliorates acute
coxsackievirus B3-induced myocarditis associated with oxidative
stress reduction. Eur J Pharmacol. 640:112–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhuang XF, Yin CQ, Wang HY and Sun NL:
Distinctive effects of carvedilol in the non-infarct zone:
Remodelling of the ligated rat heart linked to oxidative stress. J
Int Med Res. 37:1354–1364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim IM, Tilley DG, Chen J, Salazar NC,
Whalen EJ, Violin JD and Rockman HA: Beta-blockers alprenolol and
carvedilol stimulate beta-arrestin-mediated EGFR transactivation.
Proc Natl Acad Sci USA. 105:14555–14560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wisler JW, DeWire SM, Whalen EJ, Violin
JD, Drake MT, Ahn S, Shenoy SK and Lefkowitz RJ: A unique mechanism
of beta-blocker action: Carvedilol stimulates beta-arrestin
signaling. Proc Natl Acad Sci USA. 104:16657–16662. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gupta S and Knowlton AA: HSP60, Bax,
apoptosis and the heart. J Cell Mol Med. 9:51–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kirchhoff SR, Gupta S and Knowlton AA:
Cytosolic heat shock protein 60, apoptosis, and myocardial injury.
Circulation. 105:2899–2904. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hollander JM, Lin KM, Scott BT and
Dillmann WH: Overexpression of PHGPx and HSP60/10 protects against
ischemia/reoxygenation injury. Free Radic Biol Med. 35:742–751.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin KM, Lin B, Lian IY, Mestril R,
Scheffler IE and Dillmann WH: Combined and individual mitochondrial
HSP60 and HSP10 expression in cardiac myocytes protects
mitochondrial function and prevents apoptotic cell deaths induced
by simulated ischemia-reoxygenation. Circulation. 103:1787–1792.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL,
Zhu JN, Liu XY, Zhang YY, Li Y, Lin SG and Yu XY: Upregulated
expression of miR-1/miR-206 in a rat model of myocardial
infarction. Biochem Biophys Res Commun. 381:597–601. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B,
Zhang Y, Xu C, Bai Y, Wang H, et al: The muscle-specific microRNA
miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1
and KCNJ2. Nat Med. 13:486–491. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim IM, Wang Y, Park KM, Tang Y, Teoh JP,
Vinson J, Traynham CJ, Pironti G, Mao L, Su H, et al:
β-arrestin1-biased β1-adrenergic receptor signaling regulates
microRNA processing. Circ Res. 114:833–844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bayoumi AS, Park KM, Wang Y, Teoh JP,
Aonuma T, Tang Y, Su H, Weintraub NL and Kim IM: A
carvedilol-responsive microRNA, miR-125b-5p protects the heart from
acute myocardial infarction by repressing pro-apoptotic bak1 and
klf13 in cardiomyocytes. J Mol Cell Cardiol. 114:72–82. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Teoh JP, Bayoumi AS, Aonuma T, Xu Y,
Johnson JA, Su H, Weintraub NL, Tang Y and Kim IM:
β-arrestin-biased agonism of β-adrenergic receptor regulates
Dicer-mediated microRNA maturation to promote cardioprotective
signaling. J Mol Cell Cardiol. 118:225–236. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bayoumi AS, Teoh JP, Aonuma T, Yuan Z,
Ruan X, Tang Y, Su H, Weintraub NL and Kim IM: MicroRNA-532
protects the heart in acute myocardial infarction, and represses
prss23, a positive regulator of endothelial-to-mesenchymal
transition. Cardiovasc Res. 113:1603–1614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu JN, Chen R, Fu YH, Lin QX, Huang S,
Guo LL, Zhang MZ, Deng CY, Zou X, Zhong SL, et al: Smad3
inactivation and MiR-29b upregulation mediate the effect of
carvedilol on attenuating the acute myocardium infarction-induced
myocardial fibrosis in rat. PLoS One. 8:e755572013. View Article : Google Scholar : PubMed/NCBI
|