Introduction
Osteopetrosis refers to a heterogeneous group of
rare bone disorders characterized by reduced osteoclast activity
(1–3), which results in increased bone mass
and decreased bone strength. The disease is classified into three
major clinical subtypes according to the severity and mode of
inheritance: Autosomal recessive osteopetrosis (ARO); intermediate
ARO (IARO); and autosomal dominant osteopetrosis (ADO) (4). Mutations in the chloride channel 7
(CLCN7) gene locus on chromosome 16p13.3 have been reported
to cause ARO, IARO and ADO type II (ADO-II) (5,6). To
date, all reported cases of ADO-II are associated with mutations in
the CLCN7 gene. The CLCN7 gene encodes the 803-amino
acid chloride channel protein 7 (ClC-7), which provides the
chloride conductance necessary for efficient proton pumping in the
osteoclast ruffled membrane (4)
and is involved in acidification of the resorption lacuna. A low-pH
microenvironment is required during normal bone resorption
(1). Osteoclasts are
multinucleated giant cells responsible for bone resorption, which
serve important roles during bone growth and tissue renewal
(7). With the balance of bone
modeling and remodeling, the normal mineralized matrix erosion
activity of osteoclasts serves to maintain healthy and mechanically
competent bone (8). Conversely,
dysfunctional osteoclasts lead to osteopetrosis. The majority of
patients are diagnosed with ADO, with an incidence of ~1 per 20,000
individuals (2,9).
ADO-II (OMIM 166600; www.omim.org),
also known as Albers-Schönberg disease or marble bone disease, is a
form of ADO comprising a clinical spectrum ranging from very mild
to severe disease phenotypes, with a reported penetrance of 56–90%
in different studies (1,10). The range of phenotypes observed
under the ADO-II spectrum includes asymptomatic disease,
osteosclerosis, a high rate of fractures, osteomyelitis, and
hematological and neural defects (2). IARO is a milder form of ARO; patients
with IARO manifest mandibular prognathism, occasional
osteomyelitis, hepatosplenomegaly and a tendency to develop
fractures (4). Radiographs of
affected individuals reveal diffuse osteosclerosis and
pathognomonic findings of ‘bone-within-bone’ or ‘endobones’ due to
parallel bands of dense bone, which are often prominent in the
pelvis, vertebrae and long bones (11). The thickening of the end plate in
the vertebrae is termed ‘sandwich vertebrae’ or ‘rugger jersey
spine’. Alterations in the metaphysis of long bones are termed
‘Erlenmeyer flask bone deformities’ (12,13).
At present, >23 mutations in CLCN7 have
been identified in Chinese families with ADO-II (11,14–20).
Among them, 14 mutations were reported in our previous studies
(10,14,16,20).
A few cases of IARO in China have been reported by Xue et al
(17), Pang et al (19) and Zhang et al (16). In the present study, the clinical
and molecular characterization of another five patients with ADO-II
and two patients with IARO were reported. Additionally, four novel
mutations were identified.
Materials and methods
Patients
The present study was approved by the Ethics
Committee of the Shanghai Jiao Tong University Affiliated Sixth
People's Hospital. All subjects who participated in the study were
recruited by the Department of Osteoporosis and Bone Disease of the
Shanghai Jiao Tong University Affiliated Sixth People's Hospital
between December 2016 and December 2018, and all signed informed
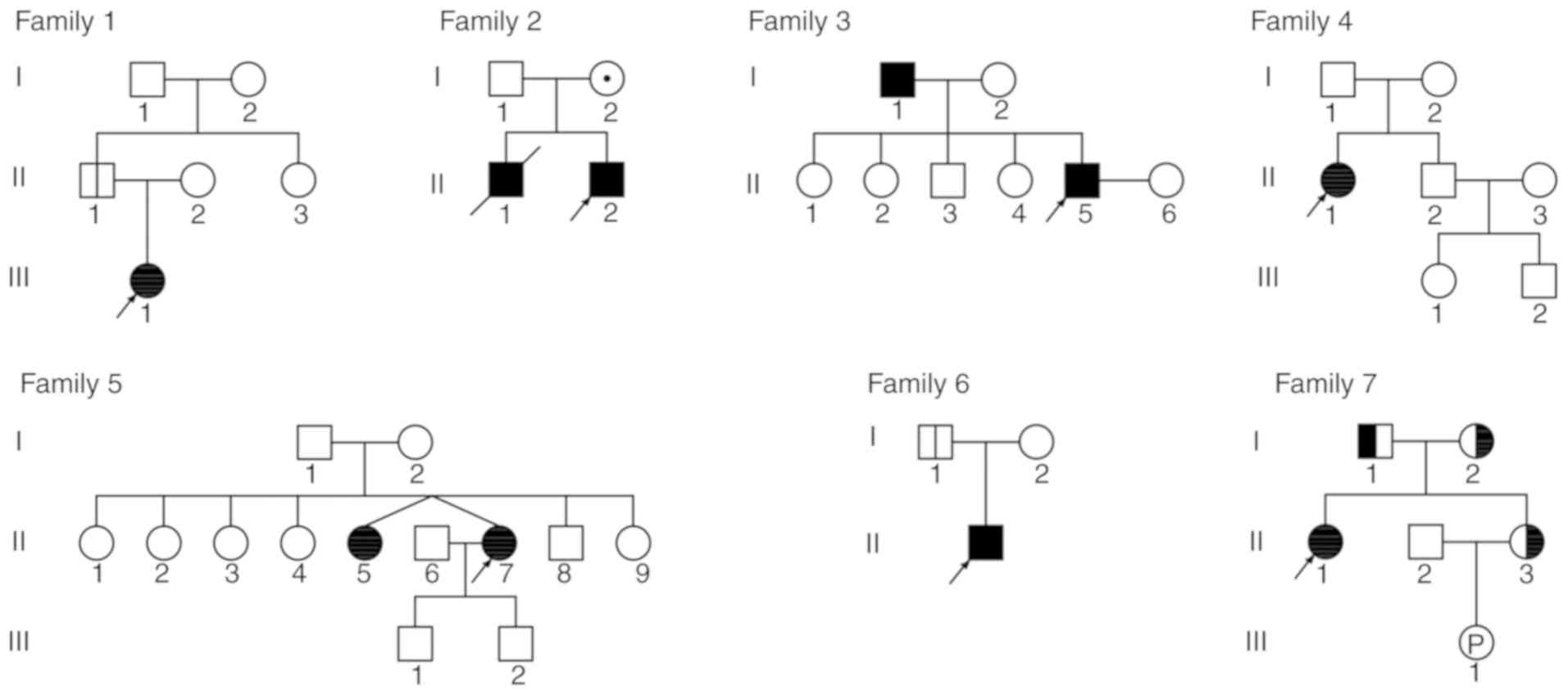
consent documents prior to entering the project. A total of seven
families were included. All subjects were of Han ethnicity and had
non-consanguineous parents. The pedigrees of the families with
ADO-II and IARO are presented in Fig.
1.
Biochemical measurements
Fasting peripheral blood samples were collected from
each subject during a clinical visit and were analyzed in the
central laboratory of Shanghai Jiao Tong University Affiliated
Sixth People's Hospital. Full blood count, including haemoglobin
and platelets, and phosphate, calcium, alkaline phosphatase (ALP),
creatinine, alanine transaminase, aspartate transaminase, lactate
dehydrogenase (LDH), creatine kinase (CK) and CK-MB levels were
measured using a Hitachi 7600 automatic biochemical analyzer
(Hitachi, Ltd., Tokyo, Japan). Serum parathyroid hormone (PTH)
concentrations, and Serum 25-hydroxyvitamin D (25OHD) levels were
measured using an ECLIA Elecsys autoanalyzer (E170; Roche
Diagnostic GmbH, Mannheim, Germany).
Measurement of radiographs and bone
mineral density (BMD)
Radiographic studies were performed at the
Department of Radiology of Shanghai Jiao Tong University Affiliated
Sixth People's Hospital. X-rays of the skull, thoracic and lumbar
vertebrae, distal femoral and proximal tibiae, and pelvis were
acquired to detect bone abnormalities.
The BMD (g/cm2) of the lumbar spine
(L1-4) and the left proximal femur, including the femoral neck and
total hip, were measured using dual-energy X-ray absorptiometry
densitometers at the Department of Osteoporosis and Bone Disease of
Shanghai Jiao Tong University Affiliated Sixth People's
Hospital.
Molecular genetics analyses
All fasting blood samples were collected in the
morning for laboratory tests and DNA analyses. Genomic DNA was
isolated from peripheral white blood cells from 2 ml blood using a
DNA extraction kit (Shanghai Laifeng Biotechnology Co., Ltd.,
Shanghai, China; http://www.lifefeng.com) according to the
manufacturer's protocol. The CLCN7 gene was screened for
mutations in the probands from the seven families. A database was
analysed and additional mutation sites were identified in other
family members and 250 healthy ethnically matched controls (125
males and 125 females) (14). All
25 exons of the CLCN7 gene, including the exon-intron
boundaries, were amplified via polymerase chain reaction (PCR)
using 16 pairs of primers: CLCN7E1 forward (F):
5′-CGACCGGGCCTCGGTGGTT-3′, CLCN7E1 reverse (R):
5′-GCGGCCTCCGAAGACTCCAGACC-3′, CLCN7E2 F:
5′-AGCCGGATCAGTTCTGCTT-3′, CLCN7E2 R: 5′-CACTCCTCCGTCTGAGAGCA-3′,
CLCN7E3_4 F: 5′-CCCCATGTGCAGTTCTCTTG-3′, CLCN7E3_4 R:
5′-AGCAGCCTTCTTGGTTACGG-3′, CLCN7E5_6 F:
5′-CACTGGGCCCTTCATAATCC-3′, CLCN7E5_6 R:
5′-TTCTAAAAGTGCCCGGGTTG-3′, CLCN7E7 F: 5′-GACGTGTGTGCTGCTCTTCC-3′,
CLCN7E7 R: 5′-CAAACTGAAAGCGGGAAACTG-3′, CLCN7E8_9 F:
5′-CCCAGCCACTCTGCCTGATC-3′, CLCN7E8_9 R: 5′-CGCCAGGCTGTCCTCAGAT-3′,
CLCN7E10_11 F: 5′-AGCCCCTTCCCCTGCACAGC-3′, CLCN7E10_11 R:
5′-CGATGGGTGGCCCCAAGGTG-3′, CLCN7E12 F:
5′-CTTCCCCTCTTGCTCTCCACT-3′, CLCN7E12 R:
5′-CTATCGATGGCACGGAGAGTC-3′, CLCN7E13_14 F:
5′-GGTGGTCCTTGAGTTTCAGCA-3′, CLCN7E13_14 R:
5′-TAAACCCCATTCCACCACGTC-3′, CLCN7E15 F:
5′-CAGTGTCCTCCATCAGGGACT-3′, CLCN7E15 R:
5′-CATTTCTCCTGGGTCCACATC-3′, CLCN7E16 F:
5′-GTGTCCTCCTTGCCCTCTGT-3′, CLCN7E16 R:
5′-GATCCTCCTGCCTTGGTCTCT-3′, CLCN7E17 F:
5′-CTCCCCATGGGATCCTTTTAG-3′, CLCN7E17 R:
5′-CCTGGTCCAGACTCCACACAT-3′, CLCN7E18_19 F:
5′-CAGCAGGGTGTACTGTGCTAGG-3′, CLCN7E18_19 R:
5′-CAGAAACCCTGAGCCTACCC-3′, CLCN7E20_21 F:
5′-GGGGTAGGCTCAGGGTTTCT-3′, CLCN7E20_21 R:
5′-ATGGCTGCACACTCAGCTTC-3′, CLCN7E22_23 F:
5′-GAGGCTGGTGTGAGCAGGTAG-3′, CLCN7E22_23 R:
5′-CAGAGTCACCGAGTCCTCTCC-3′, CLCN7E24_25 F:
5′-TCGGTGACTCTGTCTCCTGTG-3′, CLCN7E24_25 R:
5′-ACTGCTGGGGAGCATGGTT-3′. The PCR was performed using the HotStar
Taq polymerase (Takara Bio, Inc., Otsu, Japan). The thermocycling
conditions were the following: 95°C for 2 min, followed by 11
cycles of 94°C for 20 sec, 64°C −0.5°C/cycle for 40 sec and 72°C
for 1 min, followed by 24 cycles of 94°C for 20 sec, 58°C for 30
sec and 72°C for 1 min, the final extension was performed at 72°C
for 2 min for all primer pairs except for the except for
CLCN7E10_11. The thermocycling conditions used to amplify the
genomic region corresponding to CLCN7E10_11 were the following:
Initial denaturation at 95°C for 2 min, followed by 35 cycles of
96°C for 10 sec and 68°C for 1 min. Direct sequencing was performed
using the BigDye Terminator Cycle Sequencing Ready Reaction kit,
v.3.1 (Applied Biosystems; Thermo Fisher Scientific, Inc.), and the
resulting PCR products were directly sequenced using an automated
ABI PRISM 3130 sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Basic Local Alignment Search Tool (www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?link_loc=BlastHomeAd)
was used to perform homology analysis of the R286W, G793R, Y746D,
Y99C, E313K and P470L sites in eight vertebrate species. The
potential causal effects of the R286W, G793R, Y746D, Y99C, E313K
and P470L missense mutations were predicted using PolyPhen-2
software (genetics.bwh.harvard.edu/pph2) (21).
Results
Clinical manifestations of
patients
Table I presents
detailed clinical parameters, biochemical results and BMDs for the
patients. Fig. 2 presents
radiographs from patients with ADO-II and IARO. The patients with
ADO-II were diagnosed at 5–47 years old; five were male (II1 in
family 1, I1 and II5 in family 3, and I1 and II1 in family 6) and
four were female (III1 in family 1, II1 in family 4, and II5 and
II7 in family 5). Patients with ADO-II: One patient exhibited a
notably below-average height (II7 in family 5); one patient
exhibited pigeon chest (II1 in family 6); three patients
experienced fractures of the rib, femur, tibia and radius (II5 in
family 3, II1 in family 4, II1 in family 6); one patient exhibited
dental abnormalities (II5 in family 3); three patients exhibited
anemia (III1 in family 1, II1 in family 4, II7 in family 5); and
one patient, the proband of family 4, was diagnosed with
thrombocytopenia and splenomegaly. Additionally, the proband of
family 4 exhibited a visual impairment from birth.
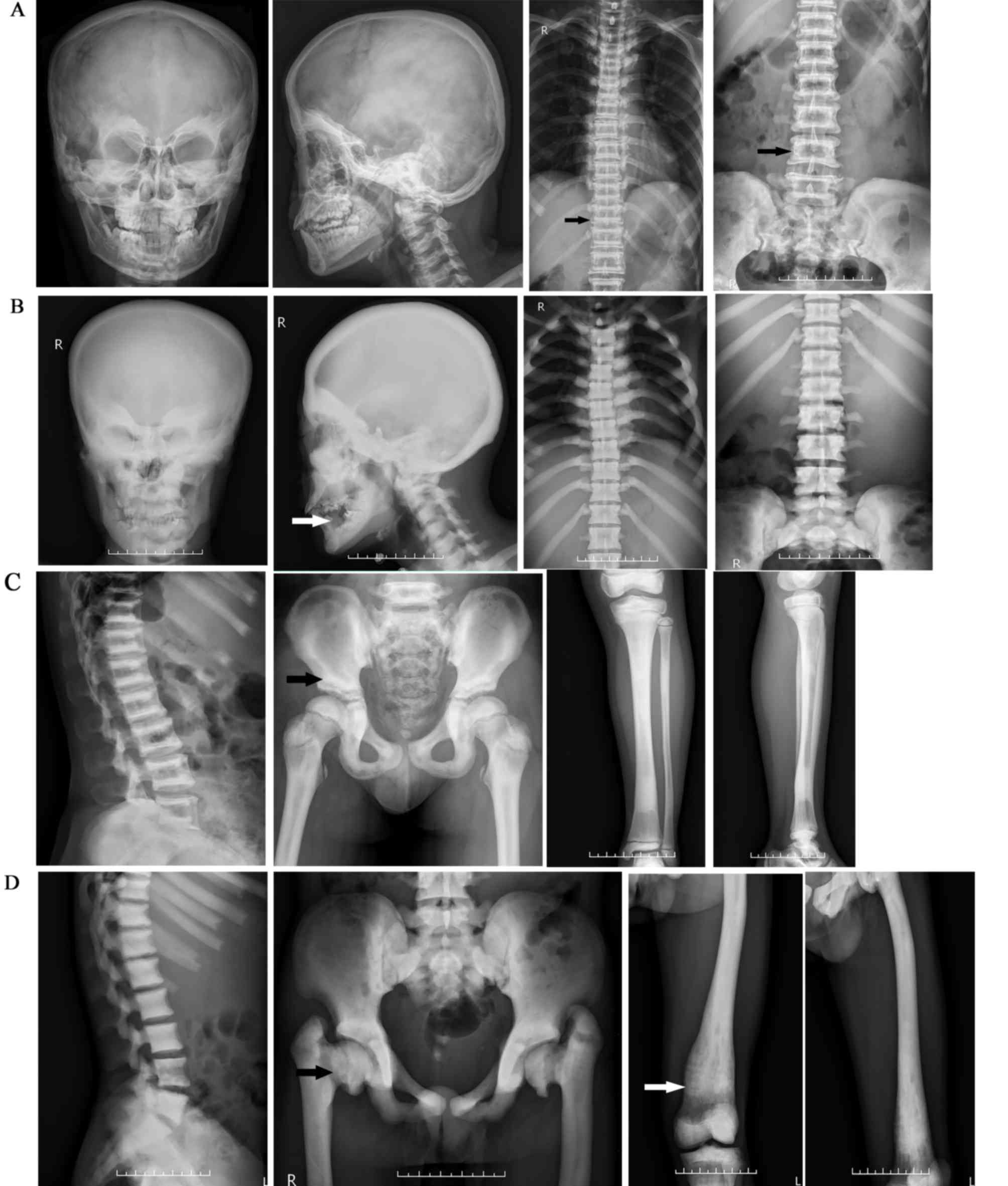 | Figure 2.Radiology results for the patients
(X-rays of skull of A, pelvis, tibia and fibula of C are taken from
the proband of family 6. X-rays of vertebrae of A and C are taken
from the proband of family 5. X-rays of B and D are taken from the
proband of family 2). (A and C) X-rays from a patient with ADO-II.
Note the generalized increase in the bone density of the skull,
vertebrae, pelvis, tibia and fibula. Radiology revealed vertebral
endplate thickening (the black arrow in A indicates a sandwich
vertebrae sign) and typical iliac wings (the black arrow of C
indicates bone-in-bone appearance). (B and D) X-rays from a patient
with IARO. Note the diffuse sclerosis of the skull, vertebrae,
pelvis and femur. Radiology revealed tooth destruction (white arrow
in B), hip deformity (black arrow in D) and the Erlenmeyer flask
deformity (white arrow in D) of the distal femur. ADO-II, autosomal
dominant osteopetrosis type II; IARO, intermediate autosomal
recessive osteopetrosis. |
 | Table I.Clinical characteristics, biochemical
results and BMD values of patients with osteopetrosis and family
members. |
Table I.
Clinical characteristics, biochemical
results and BMD values of patients with osteopetrosis and family
members.
| Family | Member | Sex | Age (years) | Height (cm) | Weight (kg) | LDH (U/l) | CK (U/l) | ALP (U/l) | Hb (g/l) | PLT
(109/l) | L1-4 (Z score) | FN (Z score) | TH (Z score) | Dental problem | Bone fracture | Other clinical
characteristics |
|---|
| 1 | Proband | F | 15 | 153.3 | 61 | 248 | 179 | 79 | 124 | 255 | 7.7 | 4.8 | 6.6 | No | No | No |
|
| Father | M | 42 | 174.7 | 92 | NA | NA | NA | NA | NA | 0.5 | 0.2 | 0.7 | No | No | No |
| 2 | Proband | M | 24 | 153.0 | 45 | 205 | 139 | 150 | 86 | 49 | 14.1 | 14.4 | 14.6 | Yes | Yes | Hepatosplenomegaly,
gallstone, secondary hyperparathyroidism |
| 3 | Proband | M | 28 | 170.5 | 68 | 190 | 156 | 54 | 149 | 217 | 11.1 | 11.1 | 9.1 | Yes | Yes | No |
| 4 | Proband | F | 32 | 153.0 | 67 | 256 | 86 | 100 | 71 | 83 | 11.5 | NA | NA | No | Yes | Splenomegaly,
visual impairment, tinnitus |
| 5 | Proband | F | 42 | 148.5 | 48 | NA | NA | 46 | 108 | 249 | 9.6 | 6.0 | 6.0 | No | No | Secondary
hyperparathyroidism |
| 6 | Proband | M | 10 | 143.7 | 41 | 210 | 207 | 170 | 146 | 273 | 6.8 | 5.4 | 6.0 | No | Yes | Pigeon chest |
|
| Father | M | 41 | 160.5 | 69 | NA | NA | 80 | 165 | 211 | 0.3 | −0.3 | −0.3 | No | No | No |
| 7 | Proband | F | 37 | 148.2 | 49 | 274 | NA | 67 | 62 | 350 | 16.9 | 17.6 | 14.1 | Yes | Yes | Visual and audile
impairment, pigeon chest, cyst, arthralgia, ankylosis,
deformity |
|
| Father | M | 63 | 170.0 | 65 | NA | NA | NA | NA | NA | 0.4 | 0.5 | −0.3 | No | No | No |
|
| Mother | F | 63 | 160.0 | 56 | NA | NA | 77 | 126 | 225 | −0.8 | −1.1 | −1.3 | No | No | No |
In family 1, a 15-year-old female (proband, III1),
the only daughter of nonconsanguineous parents, was born at term
with a normal height and weight. The height and weight of the
proband at the time of assessment were 153.3 cm and 61.0 kg,
respectively. At ~1 month prior to evaluation, the proband
complained of back pain. X-rays revealed an increased bone density
in vertebrae, with the sandwich vertebrae sign. Laboratory data
revealed an elevation in LDH levels and a minor reduction in
hemoglobin (Hb) levels. Subsequently, her parents visited our
department, and presented with normal BMDs.
The proband (II2) of family 2, a 24-year-old male,
was the second child of nonconsanguineous parents. The proband
exhibited growth retardation. The weight and height of the patient
were 45 kg and 153 cm, respectively. He complained of discomfort in
the abdomen and were diagnosed with osteopetrosis with anemia and
hepatosplenomegaly at 3 months. He only had 20 teeth, and exhibited
yellowish skin and a swollen abdomen. Additionally, he experienced
a cementoma 6 years previously; there was no deterioration.
Fractures occurred in the neck of the femur when he was 4 years old
and in the distal radius when he was 16 years old due to a
non-violent etiology. He experienced anemia with thrombocytopenia
for the majority of his life, and received a red cell suspension
>3 times due to severe anemia. X-rays revealed diffuse sclerosis
(Fig. 2). Laboratory data revealed
increased ALP levels, and reduced Hb and PLT levels. The proband's
parents exhibited normal clinical features as assessed by accessory
examinations, including radiographs and BMD. The proband's older
brother succumbed at the age of 3 years and was also diagnosed with
osteopetrosis.
In family 3, the proband (II5), a 28-year-old male,
was the youngest child of nonconsanguineous parents. The proband
came to our department as a result of abnormal skeletal
radiological manifestations. The weight and height of the proband
were 68.0 kg and 170.5 cm, respectively. He complained of shoulder
pain 10 days prior to the visit. A rib fracture with a non-violent
etiology occurred during childhood. Skeletal radiography revealed
an increase in bone density in the vertebrae with the sandwich
vertebrae sign. Laboratory data were normal. Of the seven members
of this family, including the proband's parents and other
relatives, no abnormal clinical manifestations were observed, and
they refused to undergo radiography or BMD assessments.
In family 4, the proband (II1), a 32-year-old
female, was the first child of nonconsanguineous parents. She was
born at term with normal delivery. The height and weight of the
proband at the time of the examination were 153.0 cm and 67.0 kg,
respectively. She was blind from birth. Fractures with a
non-violent etiology occurred in the left femur twice when she was
1 and 20 years old, and in the right femur when she was 10 years
old. Tinnitus and splenomegaly had developed in recent years.
X-rays revealed an increase in the bone density of vertebrae with
the sandwich sign and the pelvis with the bone-in-bone sign.
Laboratory data revealed elevated LDH levels, and reduced Hb and
PLT levels. Of the six members of the proband's family, including
her parents and other relatives, no abnormal clinical
manifestations or BMD were observed.
In family 5, the proband (II7), a 42-year-old
female, was the sixth child of nonconsanguineous parents. She came
to our department as a result of abnormal skeletal radiological
manifestations. Her weight and height were 48 kg and 148.5 cm,
respectively. X-rays revealed a diffuse increase in bone density
with evidence of the sandwich sign in vertebrae and bone-in-bone
sign in the pelvis. Laboratory data revealed reduced Hb levels. The
same signs were observed in X-rays from the proband's twin sister.
The clinical manifestations, laboratory data, radiographs and BMDs
of the proband's sons were normal.
In family 6, the proband (II1), a 10-year-old male,
the only child of nonconsanguineous parents, was born at term with
a normal length and weight. The proband's height and weight at the
time of examination were 143.7 cm and 41.0 kg, respectively. He had
suffered from recurrent influenza since childhood. Fractures with a
non-violent etiology occurred in the left tibia when he was 3 years
old and in the radius when he was 10. Pigeon chest was observed.
Laboratory data revealed elevated CK levels. Their radiograph
exhibited a typical appearance. The clinical manifestations,
laboratory data, radiographs and BMDs of the proband's parents were
normal.
The proband (II1) of family 7, a 37-year-old female,
the first child of nonconsanguineous parents, experienced growth
retardation. Her height and weight at the time of examination were
148.2 cm and 49.0 kg, respectively. She exhibited cysts and
recurrent infections of the right knee joint during the previous 10
years. She suffered arthralgia, ankylosis and deformity of the
right knee with claudication. Amblyopia of the left eye and
amblyacousia of the left ear had occurred since childhood. She also
experienced dental problems, including misalignment, loose teeth,
tooth loss and dental cavities. A fracture of the right radius with
a non-violent etiology occurred when she was 17 years old. She
fainted numerous times as a result of anemia. X-rays revealed a
diffuse increase in bone density. Laboratory data revealed
increased LDH levels and a reduction in Hb levels. The three
members of the proband's family, including her parents and sister,
did not display abnormal clinical manifestations, laboratory data,
radiographs or BMDs.
Genetic analysis
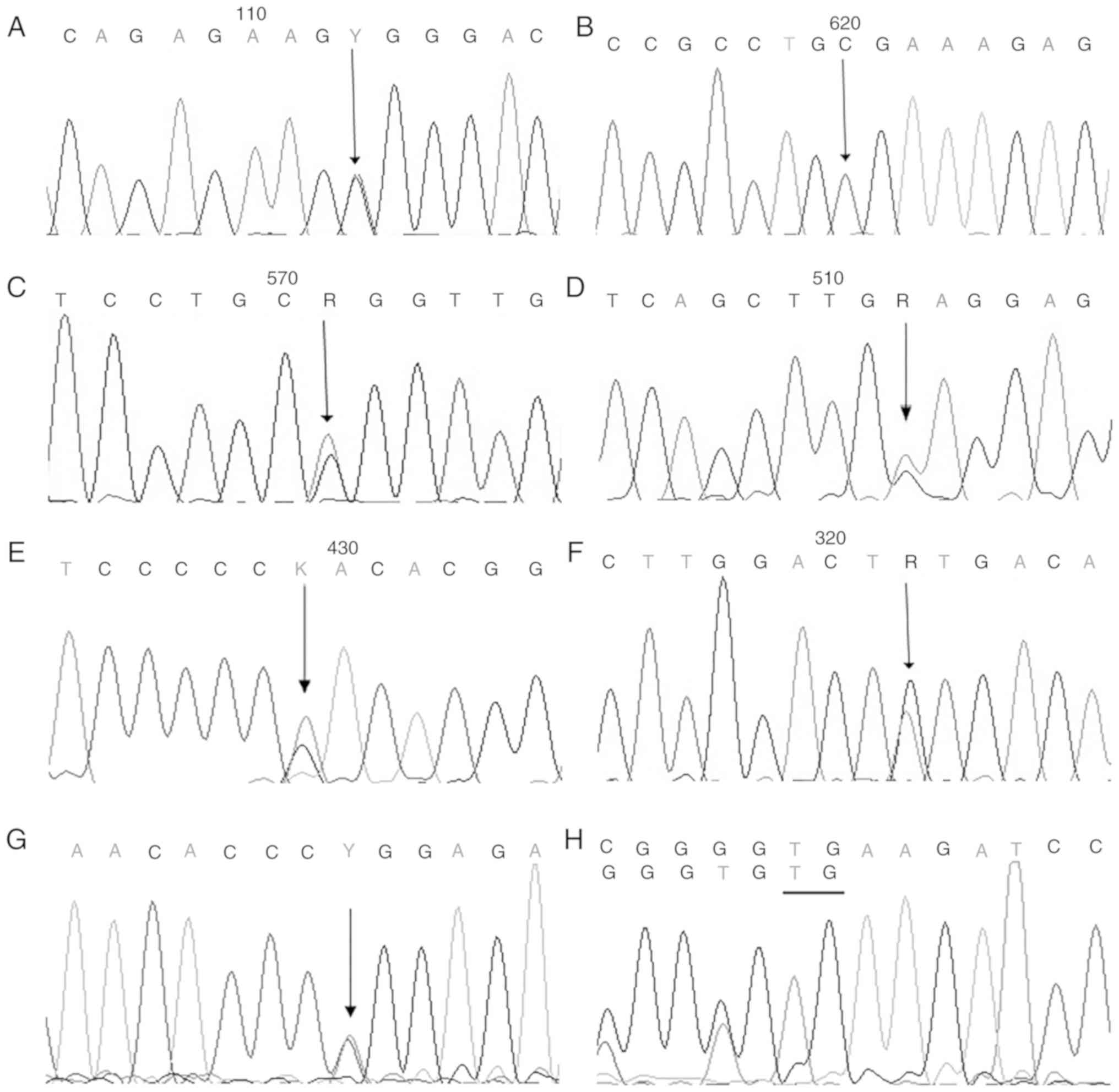
A total of one homozygous and four heterozygous
missense mutations, one splice mutation and one compound
heterozygous mutation in the CLCN7 gene were identified in
the probands (Table II; Fig. 3). The heterozygous mutation
(Y746D), the splice mutation (c.2232-2A>G), the homozygous
mutation (G793R) and the compound heterozygous mutation (P470L and
K217X) were novel. Additionally, six missense mutations,
c.856C>T (R286W), c.2236T>G (Y746D), c.296A>G (Y99C),
c.937G>A (E313K), c.2377G>C (G793R) and c.1409C>T (P470L),
occurred at a highly conserved position, according to a comparison
of the protein sequences from eight vertebrates. The missense
mutations in the CLCN7 gene (R286W, G793R, Y746D, Y99C,
E313K and P470L) were predicted by PolyPhen-2 to exhibit pathogenic
effects (Table II). Based on the
genetic analysis, the probands in families 1, 3, 4, 5 and 6 with
heterozygous mutations were diagnosed with ADO-II, whereas the
proband in family 2 with the homozygous mutation and the proband in
family 7 with the compound heterozygous mutation were diagnosed
with IARO.
| Table II.Summary of the findings of the
molecular analysis of patients with osteopetrosis. |
Table II.
Summary of the findings of the
molecular analysis of patients with osteopetrosis.
| Family no. | Patient/sex | Mutation site | Mutation type | Protein level | DNA level | PolyPhen-2
score | Status |
|---|
| 1 | III1/F | EXON9 | Missense | p.Arg286Trp | c.856C>T | 1.000 | Heterozygous |
|
| II1/M | EXON9 | Missense | p.Arg286Trp | c.856C>T | 1.000 | Heterozygous |
| 2 | II2/M | EXON24 | Missense | p.Gly793Arg | c.2377G>C | 0.751 | Homozygous |
|
| I2/F | EXON24 | Missense | p.Gly793Arg | c.2377G>C | 0.751 | Heterozygous |
| 3 | II5/M | INTRON22 | Putative aberrant
splicing | – | c.2232-2A>G | – | Heterozygous |
|
| I1/M | INTRON22 | Putative aberrant
splicing | – | c.2232-2A>G | – | Heterozygous |
| 4 | II1/F | EXON10 | Missense | p.Glu313Lys | c.937G>A | 1.000 | Heterozygous |
| 5 | II7/F | EXON22 | Missense | p.Tyr746Asp | c.2236T>G | 1.000 | Heterozygous |
| 6 | II1/M | EXON3 | Missense | p.Tyr99Cys | c.296A>G | 1.000 | Heterozygous |
|
| I1/M | EXON3 | Missense | p.Tyr99Cys | c.296A>G | 1.000 | Heterozygous |
| 7 | II1/F | EXON16 | Missense | p.Pro470Leu | c.1409C>T | 1.000 | Compound
heterozygous |
|
|
| EXON7 | Insertion | p.Lys217X | c.647_648dupTG | – |
|
|
| I1/M | EXON16 | Missense | p.Pro470Leu | c.1409C>T | 1.000 | Heterozygous |
|
| I1/F | EXON7 | Insertion | p.Lys217X | c.647_648dupTG | – | Heterozygous |
|
| II3/F | EXON7 | Insertion | p.Lys217X | c.647_648dupTG | – | Heterozygous |
In family 1, a known heterozygous missense mutation,
c.856C>T, in exon 9 of the CLCN7 gene was identified in
the proband (III1) and her father (II1), resulting in an
arginine-to-tryptophan substitution at p.286. In family 2, a novel
homozygous missense mutation, c.2377G>C, in exon 24 of the
CLCN7 gene was identified in the proband (II2), and a
heterozygous mutation was detected in his mother (I2), resulting in
a glycine-to-arginine substitution at p.793. In family 3, a novel
heterozygous splice mutation, c.2232-2A>G, leading to truncated
protein in intron 22 was identified in the proband (II5) and his
father (I1). In family 4, a known heterozygous missense mutation,
c.937G>A, in exon 10 of the CLCN7 gene was identified in
the proband (II1), resulting in a glutamate-to-lysine substitution
at p.313. In family 5, the novel heterozygous missense mutation
c.2236T>G in exon 22 was identified in the proband (II7) and her
twin sister (II5), resulting in a tyrosine-to-aspartate
substitution at p.746. In family 6, a known heterozygous missense
mutation, c.296A>G, in exon 3 of the CLCN7 gene was
identified in the proband (II1) and his father (I1), resulting in a
tyrosine-to-cysteine substitution at p.99. In family 7, a novel
insertion mutation (c.647_648 dupTG) in exon 7 was identified in
the proband (II1), and her mother (I2) and sister (II3), resulting
in a lysine-to-stop codon substitution at p.217 that produced a
truncated protein. Additionally, a missense c.1409C>T mutation
in exon 16 of the CLCN7 gene was identified in the proband
(II1) and her father (I1), resulting in a proline-to-leucine
substitution at p.470. The eight mutations were not detected in the
250 healthy controls.
Discussion
In the present study, direct sequencing of the
CLCN7 gene in the seven families revealed four heterozygous
missense mutations (R286W, Y746D, Y99C and E313K), one homozygous
missense mutations (G793R), one splice mutation (c.2232-2A>G)
and one compound heterozygous mutation (P470L, K217X) in the
probands. The heterozygous missense mutations (R286W, Y99C and
E313K) and the compound heterozygous mutation (P470L) have been
reported previously (10,15,18,19).
The mutations (R286W, Y746D, Y99C, G793R, E313K and P470L) all
occurred at highly conserved positions among eight vertebrate
species and were predicted to exert a pathogenic effect by
PolyPhen-2. The CLCN7 gene is expressed at high levels in
the osteoclast ruffled membrane; ClC-7 provides the chloride
conductance required for efficient proton pumping (1,4–5). CLC
chloride channels are homodimers composed of two subunits, each
containing 18 intramembranous α-helices, four highly conserved
Cl− binding sites and two cystathionine β synthase (CBS)
domains (CBS1 and CBS2) located near the carboxyl terminus of the
topological domain (19). Cleiren
et al (5) first identified
mutations in the CLCN7 gene as causing ADO-II. The mutations
(R286W, Y99C, E313K and P470L) in the CLCN7 gene have been
hypothesized to be located in the intramembrane α-helices, creating
a positive electrical potential to prevent the fast flux of
Cl− at the binding site (19). The Y746D, G793R and c.2232-2A>G
mutations were hypothesized to be located in CBS2. Y746D and G793R
are hypothesized to disturb protein sorting and interfere with the
localization of the protein in the ruffled-border formation, which
is required for normal lysosomal function and bone resorption
(22,23). The novel splicing mutation
(c.2232-2A>G) in the CLCN7 gene occurred at the splice
acceptor site, potentially interfering with the splicing of exon 23
with exon 24 and thereby truncating the functional domains. The
novel insertion mutation (c.647_648 dupTG) in exon 7 resulted in a
lysine-to-stop codon substitution at p.217, leading to the
synthesis of a truncated protein. The novel CLCN7 mutations
identified in the present study may increase understanding of the
spectrum of CLCN7 mutations in this disorder.
Osteopetrosis is a rare inherited metabolic disease.
ADO-II was first described by Albers-Schönberg in 1904 (5). The onset of ADO-II may occur in
adulthood or childhood (16,23).
Patients with ADO-II generally experience symptoms including
osteosclerosis, fractures, bone pain, osteomyelitis,
osteoarthritis, rickets, deafness and blindness (19). CLCN7-dependent IARO exhibits
an autosomal recessive inheritance profile and is milder than
CLCN7-associated ARO (1). A
few cases of IARO have been previously reported (16–17,19).
The onset usually occurs in the first year of life; patients
frequently reach adulthood, compared with the <3-year life
expectancy of patients with ARO (24).
In the present study, five families with ADO-II and
two families with IARO were reported. The proband of family 4 (II1)
diverged from other patients with ADO-II due to her impaired
vision, increased tinnitus, bone marrow failure and splenomegaly
secondary to extramedullary hematopoiesis. Waguespack et al
(25) reported the rate of severe
visual loss in subjects with ADO, with an overall prevalence of
19%, and a rare rate (~2%) of bone marrow failure. The proband
suffered from vision loss from birth, consistent with previous
reports (19,25). The mechanism by which visual loss
occurs remains unclear.
In family 2, the proband (II2), a 24-year-old male
from nonconsanguineous parents, was diagnosed with IARO caused by a
homozygous mutation in the CLCN7 gene, and presented with
early-onset bone marrow failure (anemia and thrombocytopenia) and
splenomegaly. The proband inherited one mutation from his mother;
the peripheral blood DNA of his father was normal. Therefore, two
possibilities existed to explain the presence of the other
mutation: A germ cell mutation or germ cell mosaicism in the gonads
of the proband's father, or a mutation in the fertilized egg
itself. The proband's older brother (II1), who was diagnosed with
osteopetrosis, succumbed at 3 years of age due to bone marrow
failure and massive hepatosplenomegaly; the genotype of the brother
is not known.
In family 7, the proband (II1), a 37-year-old female
from nonconsanguineous parents, was diagnosed with IARO caused by a
compound heterozygous mutation in the CLCN7 gene, and
presented with early-onset hearing impairments, unilateral vision
impairments and pigeon chest. Notably, the P470L mutation was
previously reported by Xue et al (17) to cause IARO when homozygous;
however, the heterozygous mutation P470L, inherited from the
patient's father, was not predicted to cause disease. Conversely,
the inheritance of an insertion mutation (resulting in a
lysine-to-stop codon substitution at p.217 and truncated protein)
from the patient's mother resulted in another type of IARO due to
the lack of compensatory ClC-7 protein. The patient manifested
notably more severe symptoms than the patients with ADO-II.
In the present study, seven symptomatic patients
(including family members) with ADO-II presented classical clinical
manifestations and radiographic findings. The father (II1) in
family 1 and the father (I1) in family 6 were unaffected gene
carriers with normal biochemical measurements, radiographs and
BMDs. The penetrance of ADO-II was ~66%, with a highly variable
phenotype. The association between genotype and phenotype was
difficult to determine in the present study. Modifier genes, DNA
methylation or intrinsic osteoclast factors may serve roles in the
manifestation of this disease. Further studies should be performed
to determine the mechanism of incomplete penetrance.
There were a number of limitations in our study. The
relatively small sample size, including only two cases of IARO,
were insufficient to characterize all the properties of
CLCN7-dependent osteopetrosis or determine
phenotype-genotype associations. In addition, investigations into
the mechanisms underlying the effects of these novel mutations were
not determined, and further in-depth research is required.
In conclusion, various mutations (R286W, Y746D,
Y99C, G793R, E313K, c.2232-2A>G, P470L and K217X) in the
CLCN7 gene were identified in six patients (the probands in
family 1, 2, 3, 5, 6, 7) with familial osteopetrosis and one
patient (the proband in family 4) with sporadic osteopetrosis.
Furthermore, the phenotypes of patients with ADO-II and IARO were
characterized. The findings from the present study increase the
spectrum of reported CLCN7 gene mutations and improve the
present understanding of osteopetrosis. Additionally, it may aid
further studies aiming to dissect the heterogeneity of ADO-II and
IARO.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Basic Research Program of China (grant no. 2014CB942903),
the National Natural Science Foundation of China (grant nos.
81570794 and 81770871 to Z.-L.Z.) and the Shanghai Municipal
Commission of Health and Family Planning (grant no.
20164Y0062).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
ZZ and HY designed and conceived the present study
and analyzed the genetic test results. LL and SL analyzed the
clinical information, analyzed the genetic test results and drafted
the manuscript. CW collected and analyzed data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Shanghai Jiao Tong University Affiliated Sixth
People's Hospital. All participants provided written informed
consent.
Patient consent for publication
All patients within this study provided consent for
the publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Del Fattore A, Cappariello A and Teti A:
Genetics, pathogenesis and complications of osteopetrosis. Bone.
42:19–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coudert AE, de Vernejoul MC, Muraca M and
Del Fattore A: Osteopetrosis and its relevance for the discovery of
new functions associated with the skeleton. Int J Endocrinol.
2015:3721562015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Palagano E, Blair HC, Pangrazio A,
Tourkova I, Strina D, Angius A, Cuccuru G, Oppo M, Uva P, Van Hul
W, et al: Buried in the middle but guilty: Intronic mutations in
the TCIRG1 gene cause human autosomal recessive osteopetrosis. J
Bone Miner Res. 30:1814–1821. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tolar J, Teitelbaum S and Orchard P:
Osteopetrosis. N Engl J Med. 351:2839–2849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cleiren E, Bénichou O, Van Hul E, Gram J,
Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama
T, et al: Albers-Schönberg disease (autosomal dominant
osteopetrosis, type II) results from mutations in the ClCN7
chloride channel gene. Hum Mol Genet. 10:2861–2867. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bénichou O, Cleiren E, Gram J, Bollerslev
J, de Vernejoul MC and Van Hul W: Mapping of autosomal dominant
osteopetrosis type II (Albers-Schönberg disease) to chromosome
16p13.3. Am J Hum Genet. 69:647–654. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cappariello A, Maurizi A, Veeriah V and
Teti A: Reprint of: The great beauty of the osteoclast. Arch
Biochem Biophys. 561:13–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Teti A: Bone development: Overview of bone
cells and signaling. Curr Osteoporos Rep. 9:264–273. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuroyanagi Y, Kawasaki H, Noda Y, Ohmachi
T, Sekiya S, Yoshimura K, Ohe C, Michigami T, Ozono K and Kaneko K:
A fatal case of infantile malignant osteopetrosis complicated by
pulmonary arterial hypertension after hematopoietic stem cell
transplantation. Tohoku J Exp Med. 234:309–312. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng H, Shao C, Zheng Y, He JW, Fu WZ,
Wang C and Zhang ZL: Two novel mutations of CLCN7 gene in Chinese
families with autosomal dominant osteopetrosis (type II). J Bone
Miner Metab. 34:440–446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng H, He D, Rong P, Xu H, Yuan L, Li L,
Lu Q and Guo Y: Novel CLCN7 mutation identified in a Han Chinese
family with autosomal dominant osteopetrosis-2. Mol Pain.
12:17448069166526282016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Faden MA, Krakow D, Ezgu F, Rimoin DL and
Lachman RS: The Erlenmeyer flask bone deformity in the skeletal
dysplasias. Am J Med Genet A 149A. 1334–1345. 2009. View Article : Google Scholar
|
|
13
|
Boudin E and Van Hul W: Sclerosing bone
dysplasias. Best Pract Res Clin Endocrinol Metab. 32:707–723. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang ZL, He JW, Zhang H, Hu WW, Fu WZ, Gu
JM, Yu JB, Gao G, Hu YQ, Li M and Liu YJ: Identification of the
CLCN7 gene mutations in two Chinese families with autosomal
dominant osteopetrosis (type II). J Bone Miner Metab. 27:444–451.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang C, Zhang H, He JW, Gu JM, Hu WW, Hu
YQ, Li M, Liu YJ, Fu WZ, Yue H, et al: The virulence gene and
clinical phenotypes of osteopetrosis in the Chinese population: Six
novel mutations of the CLCN7 gene in twelve osteopetrosis families.
J Bone Miner Metab. 30:338–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang X, Wei Z, He J, Wang C and Zhang Z:
Novel mutations of CLCN7 cause autosomal dominant osteopetrosis
type II (ADOII) and intermediate autosomal recessive osteopetrosis
(ARO) in seven Chinese families. Postgrad Med. 129:934–942. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xue Y, Wang W, Mao T and Duan X: Report of
two Chinese patients suffering from CLCN7-related osteopetrosis and
root dysplasia. J Craniomaxillofac Surg. 40:416–420. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng B, Li R, Hu Y, Hu B, Zhao Q, Liu H,
Yuan P and Wang Y: A novel mutation and a known mutation in the
CLCN7 gene associated with relatively stable infantile malignant
osteopetrosis in a Chinese patient. Gene. 576:176–181. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pang Q, Chi Y, Zhao Z, Xing X, Li M, Wang
O, Jiang Y, Liao R, Sun Y, Dong J and Xia W: Novel mutations of
CLCN7 cause autosomal dominant osteopetrosis type II (ADO-II) and
intermediate autosomal recessive osteopetrosis (IARO) in Chinese
patients. Osteoporos Int. 27:1047–1055. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng H, Zhang Z, He JW, Fu WZ, Wang C and
Zhang ZL: Identification of two novel CLCN7 gene mutations in three
Chinese families with autosomal dominant osteopetrosis type II.
Joint Bone Spine. 81:188–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lange PF, Wartosch L, Jentsch TJ and
Fuhrmann JC: ClC-7 requires Ostm1 as a beta-subunit to support bone
resorption and lysosomal function. Nature. 440:220–223. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sobacchi C, Schulz A, Coxon FP, Villa A
and Helfrich MH: Osteopetrosis: Genetics, treatment and new
insights into osteoclast function. Nat Rev Endocrinol. 9:522–536.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Villa A, Guerrini MM, Cassani B, Pangrazio
A and Sobacchi C: Infantile malignant, autosomal recessive
osteopetrosis: The rich and the poor. Calcif Tissue Int. 84:1–12.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Waguespack SG, Hui SL, Dimeglio LA and
Econs MJ: Autosomal dominant osteopetrosis: Clinical severity and
natural history of 94 subjects with a chloride channel 7 gene
mutation. J Clin Endocrinol Metab. 92:771–778. 2007. View Article : Google Scholar : PubMed/NCBI
|