Introduction
Cerebrovascular disease (CVD) is a common disease
affecting the nervous system and may be caused by various types of
cerebrovascular injuries (1).
Notably, CVD morbidity increased over the past years (2,3). In
addition, CVD may affect the quality of life, and ~40% of patients
with CVD present severe disabilities (4). Ischemic cerebrovascular disease
accounts for ~75% of all CVD cases and presents high disability,
mortality and recurrence rates (5).
Treatments aimed to re-establish blood flow and
blood perfusion in the ischemic penumbra following cerebral
ischemia are necessary to improve the outcomes of patients with CVD
(6,7). Previous studies demonstrated that
metabolic disorders, oxidative stress, neurotoxicity of the
excitatory amino acids, inflammatory cell infiltration, cell
apoptosis, microvascular neovascularization and additional factors
are involved in the development of cerebral ischemia-reperfusion
(I/R) injury (8,9). The structure and the function of the
vasculature may be severely impaired following I/R injury,
influencing the permeability of the blood-brain barrier and the
mechanisms underlying vascular homeostasis; these processes may
aggravate cerebral edema, leading to a decline in the clinical
outcomes of the patients (6,10,11).
Therefore, reducing reperfusion injury following cerebral
infarction is an important topic of research.
Cytochrome c oxidase subunit 6B1 (COX6B1) is one of
the subunits of the cytochrome c oxidase (COX) and is expressed in
numerous cell types, including yeast and HeLa cells (12). The human COX6B1 gene is located on
chromosome 19q13; the coding sequence comprises 261 base pairs and
the protein contains 87 amino acids (12–15).
The principal role of COX6B1 is to connect two COX monomers to form
a dimer, serving a role in the cell respiratory chain (16). Alterations in the dimeric structure
of the COX complex affects its function, and defects in the
assembly of a functional dimer may cause severe diseases (17,18).
A previous study demonstrated that genetic mutations in the
conserved regions of COX6B1 may lead to mitochondrial
encephalopathy (16).
Mitochondrial diseases are frequently associated with mutations in
or an absence of COX (19).
Additionally, previous studies reported that abnormal alternations
of COX6B1 markedly affect the function of COX, potentially leading
to the occurrence of cerebromyopathy, hydrocephalus and other
diseases (16,17). COX6B1 protein may be associated
with the development of the central nervous system (16). Previous studies reported that
COX6B1 may protect the myocardium against I/R injury by regulating
mitochondrial function (20,21);
however, whether COX6B1 protects hippocampal neurons against I/R
injury has not been reported, and the molecular mechanisms
underlying the role of COX6B1 in the development of the nervous
system remains unclear.
In the present study, a model of I/R injury was
constructed using rat hippocampal neurons. Additionally, the
molecular mechanisms underlying the role of COX6B1 following I/R
injury in hippocampal neurons was investigated.
Materials and methods
Animals
A total of 6 pregnant Sprague-Dawley (SD) rats at
gestational day 18 (age, 2 months; weight, 180–200 g) were obtained
from Guangdong Medical Laboratory Animal Center (Foshan, China) and
maintained at 22±1°C, 40–70% humidity, with free access to food and
water, under a 12-h light/dark cycle. The animal experiments were
approved by The Ethics Committee of The Nanchuan People's Hospital
Affiliated to Chongqing Medical University (Chongqing, China).
Extraction of hippocampal neurons
Pregnant SD rats were anesthetized by ether, and 12
fetal rats were extracted. The fetal rats were transferred to Petri
dishes containing 75% ethanol and the heads were removed using a
guillotine (cat. no. 7950; Ugo Basile SRL, Gemonio, Italy).
Subsequently, the hippocampi were dissected and digested with 2.5
g/l trypsin (Beyotime Institute of Biotechnology, Haimen, China)
for 15 min at 37°C. Dulbecco's modified Eagle's medium/Ham's F-12
nutrient mixture (DMEM/F12; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) was added to the tissues and
gently agitated. Cell suspension was filtered through a 400-µm
nylon mesh sieve, centrifuged at 1,000 × g for 5 min and
resuspended in DMEM/F12 at a density of 1×106 cells/ml.
The neurons obtained were cultured in an incubator under a
humidified atmosphere containing 5% CO2 at 37°C for 24
h. Following a 24-h incubation, DMEM/F12 was replaced with
neurobasal medium containing 2% B-27 supplement (Thermo Fisher
Scientific, Inc.) and 5 µmol/l cytosine arabinoside (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) was added to inhibit glial cell
proliferation; cells were subsequently incubated for 48 h at 37°C.
Following a 48-h incubation, the medium was replaced, and cells
were cultured until further experimentation; 50% of the culture
medium was replaced every 3 days.
Immunofluorescence assay
Following culturing, hippocampal neurons were fixed
with 4% paraformaldehyde at room temperature for 20 min on glass
coverslips. PBS containing 0.2% Triton X-100 (Thermo Fisher
Scientific, Inc.) was used for permeabilizing cells for 10 min.
Cells were blocked with 5% bovine serum albumin (Bovogen
Biologicals Pty Ltd., Victoria, Australia) at room temperature for
25 min. The cells were incubated with anti-microtubule-associated
protein 2 (MAP2) antibody (1:100; cat. no. ab32454; Abcam,
Cambridge, UK) at 4°C for 24 h, and washed with PBS three times.
Subsequently, the cells were incubated with goat anti-rabbit
fluorescein isothiocyanate (FITC)-conjugated immunoglobulin G (IgG;
1:5,000; cat. no. ab6717; Abcam) at 37°C for 1 h. Following
incubation, the cells were washed with PBS three times, and
subsequently incubated with DAPI (Thermo Fisher Scientific, Inc.)
at room temperature for 10 min. Stained samples were observed under
an MF43 fluorescence microscope (magnification, ×200 and ×400;
Micro-shot Technology Limited, Guangzhou, China).
Oxygen-glucose
deprivation/reoxygenation (OGD/R) in vitro model establishment
Hippocampal neurons (1×106 cells/ml) were
cultured in neurobasal medium containing 2% B-27 supplement at 37°C
with 5% CO2 for 10 days and cells were exposed to OGD by
replacing the neurobasal medium with Earle's balanced salt solution
(EBSS) without glucose. The cells were maintained in an anaerobic
chamber (YQX–II; Shanghai CIMO Medical Instrument Co., Ltd.,
Shanghai, China) with 95% N2 and 5% CO2 for
60 min at 37°C. Subsequently, cells were subjected to reoxygenation
treatment (incubation in neurobasal medium in an incubator with 5%
CO2 at 37°C) for 10, 30 or 60 min prior to further
experimentation.
Cell viability analysis
Following the aforementioned OGD/R treatment, a Cell
Counting Kit-8 (CCK-8) was performed to determine the viability of
cells following OGD and OGD/R treatment. The hippocampal neurons
(2×103 cells/well) were seeded into 96-well plates and
maintained in an incubator for 24 h at 37°C. Following incubation,
10 µl CCK-8 reagent (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) was added into each well. Cells were
transferred into the incubator and maintained for 2 h at 37°C. A
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
was used to detect the absorbance at 450 nm.
Cell transfection
The pcDNA3.1(+)-empty vector (EV) and
pcDNA3.1(+)-COX6B1 vector were purchased from GenomeDitech Co.,
Ltd., (Shanghai, China). Cells were seeded (2×105
cells/well) into 6-well plates, serum-starved overnight, and
subsequently transfected with COX6B1 vector (20 nM) using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells were transfected for 36 h at 37°C prior to
subsequent experimentation. Cells were transfected with the
plasmids prior to OGD/R.
Cell grouping
To assess the effects of OGD/R treatment on neurons,
cells were divided into five experimental groups: i) Control group,
in which untreated neurons were exposed to PBS; ii) OGD group, in
which cells were exposed to OGD for 60 min, as aforementioned,
without reoxygenation; iii) OGD-10 group, OGD cells exposed to
reoxygenation treatment for 10 min; iv) OGD-30 group, OGD cells
exposed to reoxygenation treatment for 30 min; v) OGD-60 group, OGD
cells exposed to reoxygenation treatment for 60 min. Based upon the
results of this experiment, OGD-60 treatment was selected to be the
I/R injury model in subsequent experiments. To investigate the role
of COX6B1 in OGD/R-induced damage, cells were divided into six
groups: i) Control group, untreated cells; ii) EV group, cells
transfected with pcDNA3.1(+)-EV; iii) COX6B1 group, cells
transfected with pcDNA3.1(+)-COX6B1 overexpression vector; iv) EV +
I/R group, OGD-60 cells transfected with pcDNA3.1(+)-EV; v) I/R
group, untransfected OGD-60 cells; and vi) COX6B1 + I/R group,
OGD-60 cells transfected with pcDNA3.1(+)-COX6B1. All experiments
were performed at least three times.
Cytosolic Ca2+ levels
analysis
The cytosolic Ca2+ levels of cells in the
control, EV, COX6B1, I/R, EV + I/R and COX6B1 + I/R groups were
determined by flow cytometry. Following transfection, hippocampal
neurons were seeded (2×104 cells/well) into 6-well
plates and cultured in an incubator for 24 h 37°C prior to exposure
to the aforementioned OGD/R protocol. The staining reagent Fluo-3,
AM (Sigma-Aldrich; Merck KGaA) was added into the wells and the
cells were incubated at 37°C for 45 min. Subsequently, the cells
were incubated with Hank's Balanced Salt Solution (HBSS; Thermo
Fisher Scientific, Inc.) containing 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) for 40 min at 37°C. Following incubation, the
cells were washed with HBSS for 3 times. The cells were resuspended
in HBSS and incubated at 37°C for 10 min. The concentration of
intracellular Ca2+ was assessed by flow cytometry (BD
Biosciences, San Jose, CA, USA) and FSC Express version 3 software
(De Novo Software, Glendale, CA, USA).
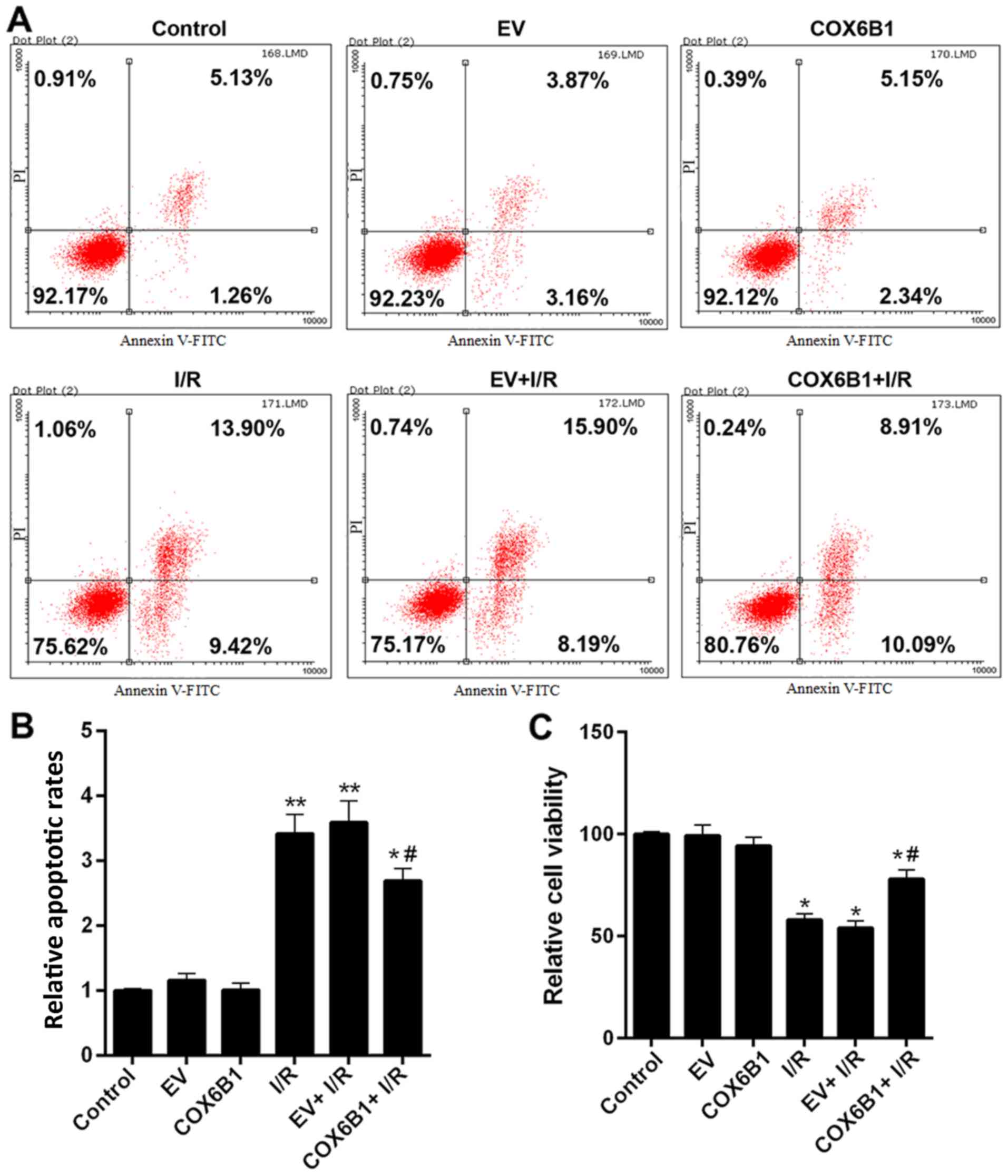
Cell apoptosis analysis
The cell apoptosis of cells in the control, EV,
COX6B1, I/R, EV + I/R and COX6B1 + I/R groups were determined by
flow cytometry. Following transfection, hippocampal neurons were
seeded (2×104 cells/well) into 6-well plates and
cultured in an incubator for 24 h prior to exposure to the
aforementioned OGD/R protocol. The cells were then incubated with
Annexin V-FITC and propidium iodide (Beijing Solarbio Science &
Technology Co., Ltd.) in the dark for 20 min at room temperature. A
flow cytometer and FSC Express version 3 software were used to
measure the level of apoptosis. Advanced apoptotic cells were
presented in upper right quadrants, and early apoptotic cells in
lower right quadrants. The relative apoptosis rate of total
apoptotic cells (early and advanced) was calculated.
Western blotting
A Mitochondria/Cytosol Fractionation kit (AmyJet
Scientific Inc., Wuhan, China) was used to separate the
mitochondrial and cytosolic fractions. Total protein was extracted
from neurons (2×104 cells/well in 6-well plates) using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology). The protein concentration was determined using
Bradford method (Beyotime Institute of Biotechnology). A total of
30 µg protein from each sample was separated by 12% SDS-PAGE and
transferred to a polyvinylidene fluoride (PVDF) membrane (EMD
Millipore, Billerica, MA, USA). The PVDF membrane was blocked using
5% skimmed milk at 37°C for 60 min. Subsequently, the membranes
were incubated at 4°C for 24 h with the following primary
antibodies: Anti-COX6B1 (1:600; cat. no. 11425-1-AP; ProteinTech
Group, Inc., Chicago, IL, USA), apoptosis regulator BCL-2 (1:1,000;
cat. no. ab194583; Abcam), anti-BCL2-associated X, apoptosis
regulator (BAX; 1:800; cat. no. ab53154; Abcam), anti-cytochrome c
(cyt c; 1:600; cat. no. ab13575; Abcam), anti-cytochrome c oxidase
subunit 4I1 (COX4I1; 1:1,000; cat. no. ab33985; Abcam) and
anti-β-actin (1:800; cat. no. ab8226; Abcam). Subsequently, the
PVDF membranes were incubated at room temperature for 1.5 h with
one of the following secondary antibodies: Rabbit anti-mouse IgG
(1:7,000; cat. no. 58802; Cell Signaling Technology, Inc., Danvers,
MA, USA) or goat anti-rabbit IgG (1:700; cat. no. ab6721; Abcam).
Protein bands were visualized with Enhanced Chemiluminescent
reagents (EMD Millipore) and the protein expression level was
detected using a Molecular Imager® Gel Doc™ XR+ System
(cat. no. 1708195; Bio-Rad Laboratories, Inc.) and ImageJ version
1.46 software (National Institutes of Health, Bethesda, MD, USA).
β-actin was used as the loading control for total proteins and
cytosolic fractions, whereas COX4I1 was used as the loading control
for the mitochondrial fraction; protein expression was normalized
to β-actin and COX4I1.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
A Mitochondria/Cytosol Fractionation kit was used to
separate the mitochondrial and cytosolic fractions. Total RNA was
extracted from hippocampal neurons (2×104 cells/well in
6-well plates) using TRIzol® reagent (Thermo Fisher
Scientific, Inc.). RNA was reverse transcribed to cDNA using
TIANScript cDNA Synthesis kit (Tiangen Biotech Co., Ltd., Beijing,
China) according to the manufacturer's protocol. cDNA was amplified
using TeloPrime Full-Length cDNA amplification kit (Lexogen GmbH,
Vienna, Austria), according to the manufacturer's protocol. qPCR
experiments were performed using a SYBR Premix Ex Taq™ Real-Time
PCR kit (Takara Bio, Inc., Otsu, Japan). qPCR thermocycling
conditions were as follows: Initial denaturation at 95°C for 1 min,
followed by 45 cycles at 96°C for 15 sec and at 63°C for 45 sec,
with a final extension step at 75°C for 10 min. β-actin was used as
the reference gene, except for mitochondrial fraction, for which
COX4I1 was used as the reference gene. The primers were purchased
from Sigma-Aldrich (Merck KGaA) and are listed in Table I. The relative expression of genes
was calculated using the 2−ΔΔCq quantification method
(22) and normalized to β-actin
and COX4I1 expression.
 | Table I.Primer sequences used in reverse
transcription-quantitative polymerase chain reactions. |
Table I.
Primer sequences used in reverse
transcription-quantitative polymerase chain reactions.
| Gene | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| COX6B1 | F:
AAGAACTACAAAACCGCCCC | 195 |
|
| R:
ATCCCAGGCTGAGACCCAT |
|
| BAX | F:
GAGACACCTGAGCTGACCTT | 187 |
|
| R:
CGTCTGCAAACATGTCAGCT |
|
| BCL-2 | F:
GCCTTCTTTGAGTTCGGTGG | 221 |
|
| R:
CTGAGCAGCGTCTTCAGAG |
|
| Cyt c | F:
GGAGGCAAGCATAAGACTGG | 210 |
|
| R:
TGCCCTTTCTCCCTTCTTCT |
|
| β-actin | F:
TGTGTTGTCCCTGTATGCC | 232 |
|
| R:
AATGTCACGCACGATTTCCC |
|
Statistical analysis
All experiments were performed at least three times.
GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) was
used to perform statistical analysis. Data are presented as the
mean ± standard deviation. Statistical comparisons were performed
using one-way analysis of variance followed by Tukey's test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cell viability and COX6B1 expression
levels are decreased in hippocampal neurons exposed to OGD/R
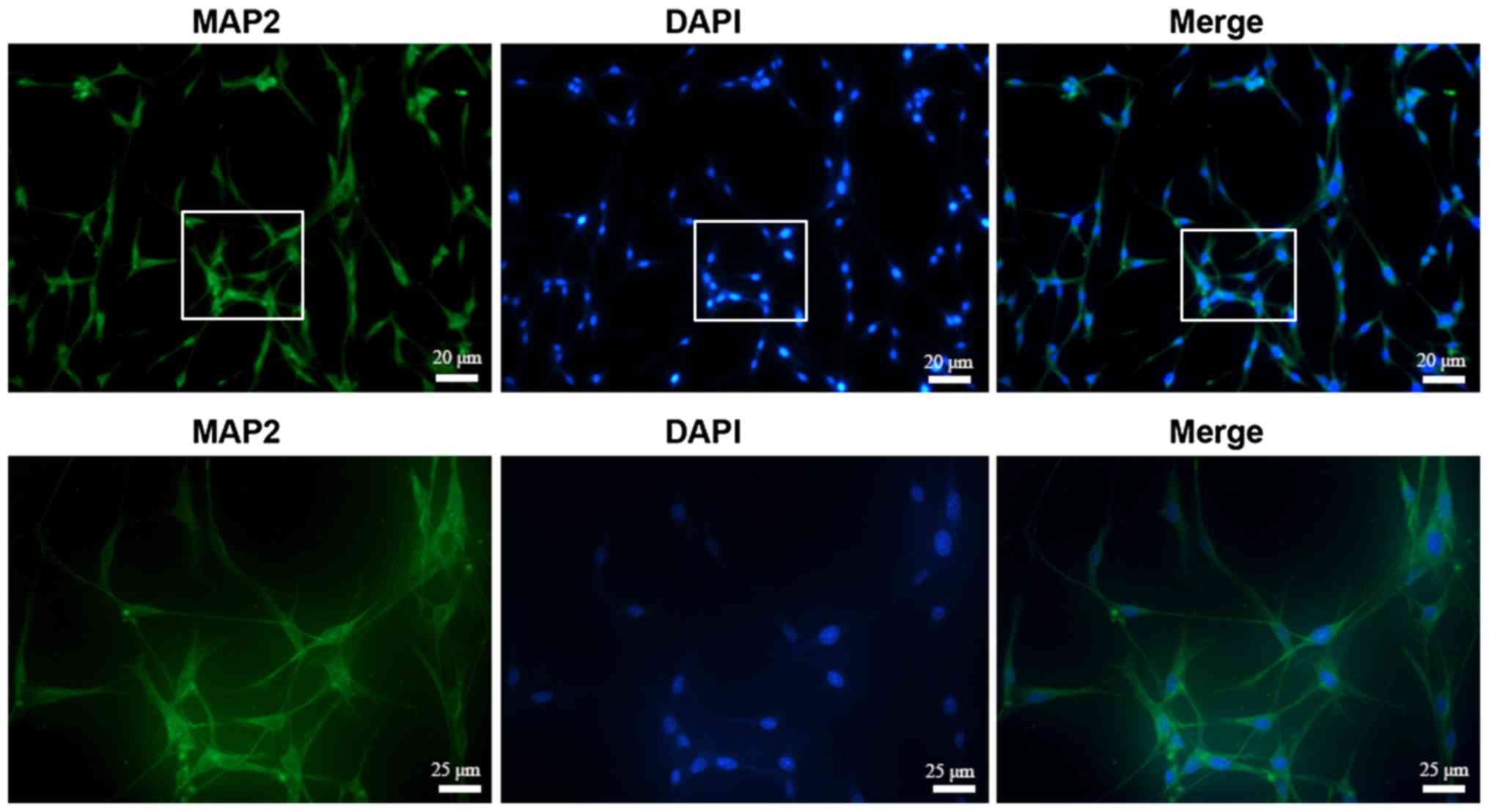
The cellular identity of the extracted hippocampal
neurons was tested by immunofluorescence assay and >95% of cells
were identified to be positive for MAP2 (Fig. 1). The effects of OGD and OGD/R on
hippocampal neurons were investigated, and cell viability and
COX6B1 mRNA and protein expression levels were examined by CCK-8
assay, RT-qPCR analysis and western blotting, respectively.
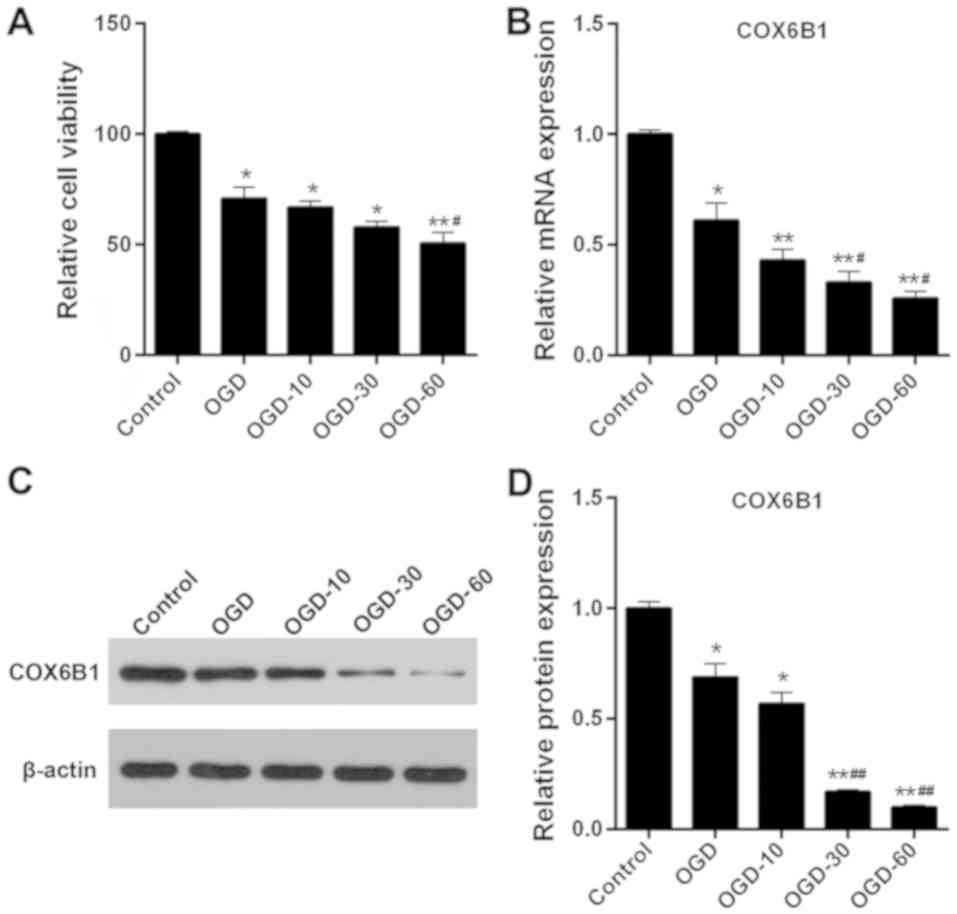
Following exposure to OGD, cell viability decreased significantly
in hippocampal neurons compared with the Control group (Fig. 2A). Furthermore, the viability of
cells in the OGD-10 and OGD-30 groups was markedly decreased
compared with in OGD group, and significantly decreased in the
OGD-60 group. The mRNA and protein expression levels of COX6B1 were
significantly reduced in the OGD group, compared with the Control
group (Fig. 2B); additionally, the
mRNA and protein expression levels of COX6B1 were significantly in
OGD-30 and −60 groups compared with in the OGD group (Fig. 2C and D). As OGD-60 treatment
exhibited the most pronounced effects on cell viability and COX6B1
expression, it was selected as the I/R injury model in subsequent
experiments.
Overexpression of COX6B1 decreases the
cytosolic levels of Ca2+ in hippocampal neurons
increased following I/R
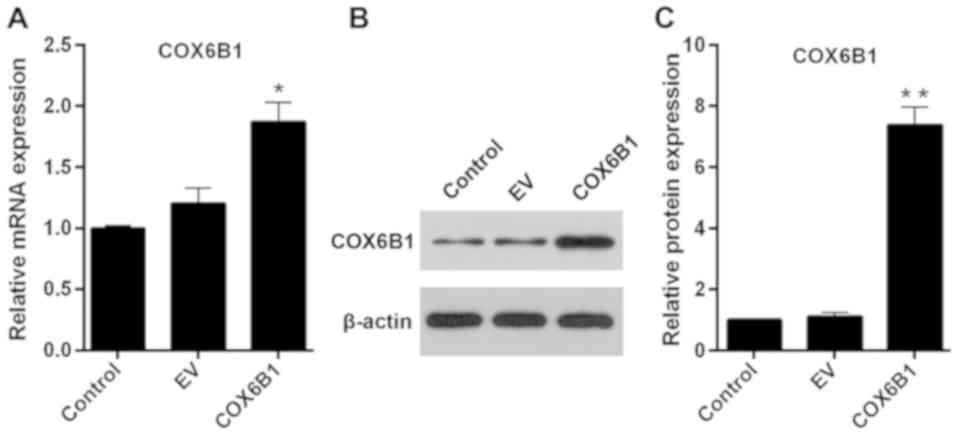
The transfection efficiency of COX6B1 overexpression
vectors was detected using RT-qPCR and western blotting.
Hippocampal neurons transfected with pcDNA3.1(+)-COX6B1 exhibited
an increase in the mRNA and protein expression levels of COX6B1
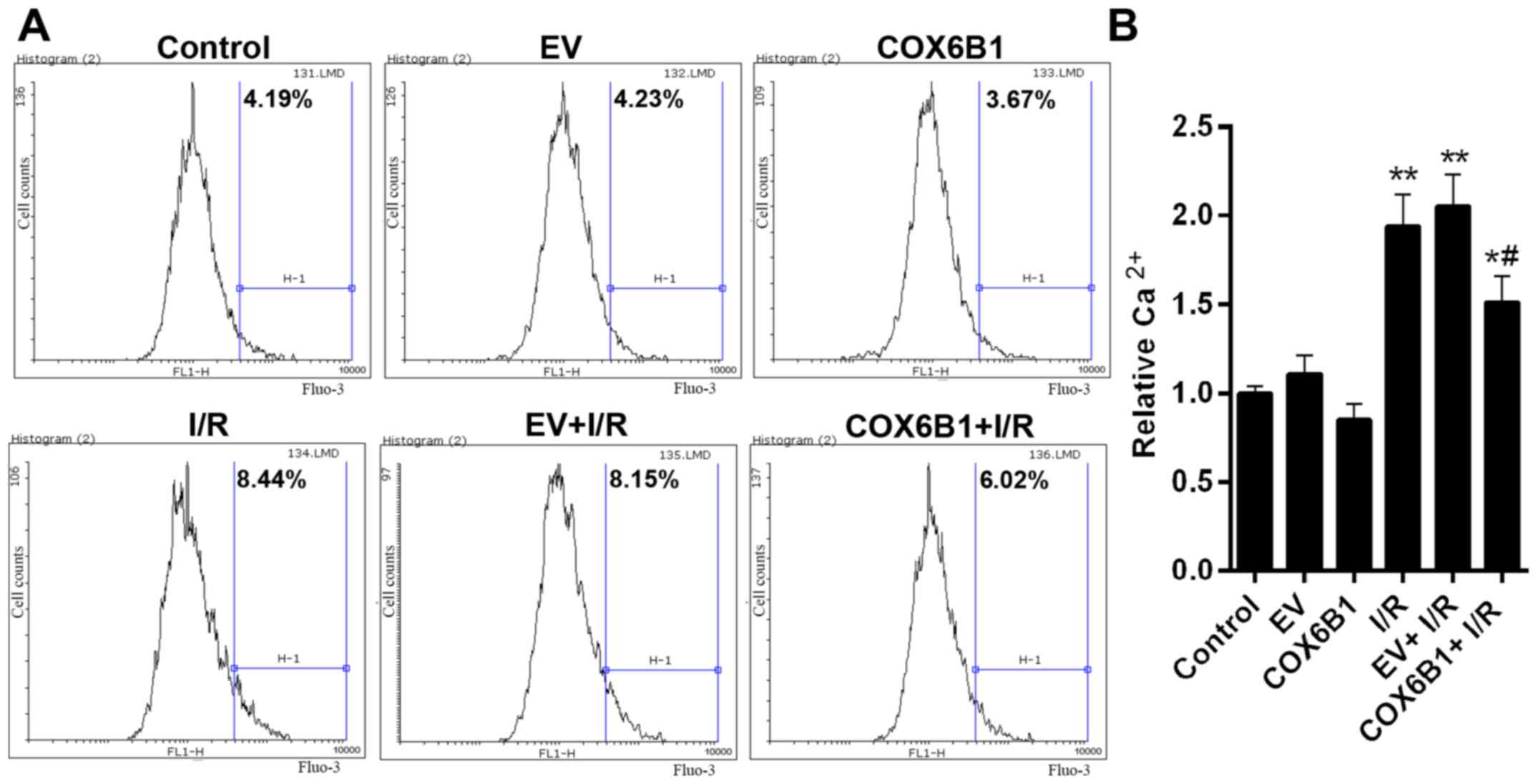
compared with the control EV-transfected cells (Fig. 3). To investigate the role of COX6B1
in the intracellular levels of Ca2+ in hippocampal
neurons, flow cytometry was conducted. The cytosolic levels of
Ca2+ were 4.19% in the Control group, 4.23 in EV, 3.67
in COX6B1, 8.44 in I/R, 8.15 in EV + I/R and 6.02% in COX6B1 + I/R
treated cells (Fig. 4A). The
relative concentration of Ca2+ was significantly
increased in the I/R group compared with the EV group (Fig. 4B); however, COX6B1 overexpression
decreased the concentration of Ca2+ compared with the EV
+ I/R group.
Overexpression of COX6B1 represses
I/R-induced apoptosis of hippocampal neurons and increases cell
viability
To investigate the effects of COX6B1 on apoptosis
and viability of hippocampal neurons, flow cytometry and CCK-8
assays were conducted, respectively. Cells exposed to I/R exhibited
an increased total apoptotic rate (early + advanced) compared with
the EV group (Fig. 5A and B).
Nevertheless, COX6B1 overexpression in hippocampal neurons
decreased the apoptosis induced by I/R injury (Fig. 5A and B). In addition, cell
viability was decreased following I/R injury, whereas
overexpression of COX6B1 increased the viability of cells following
I/R injury (Fig. 5C).
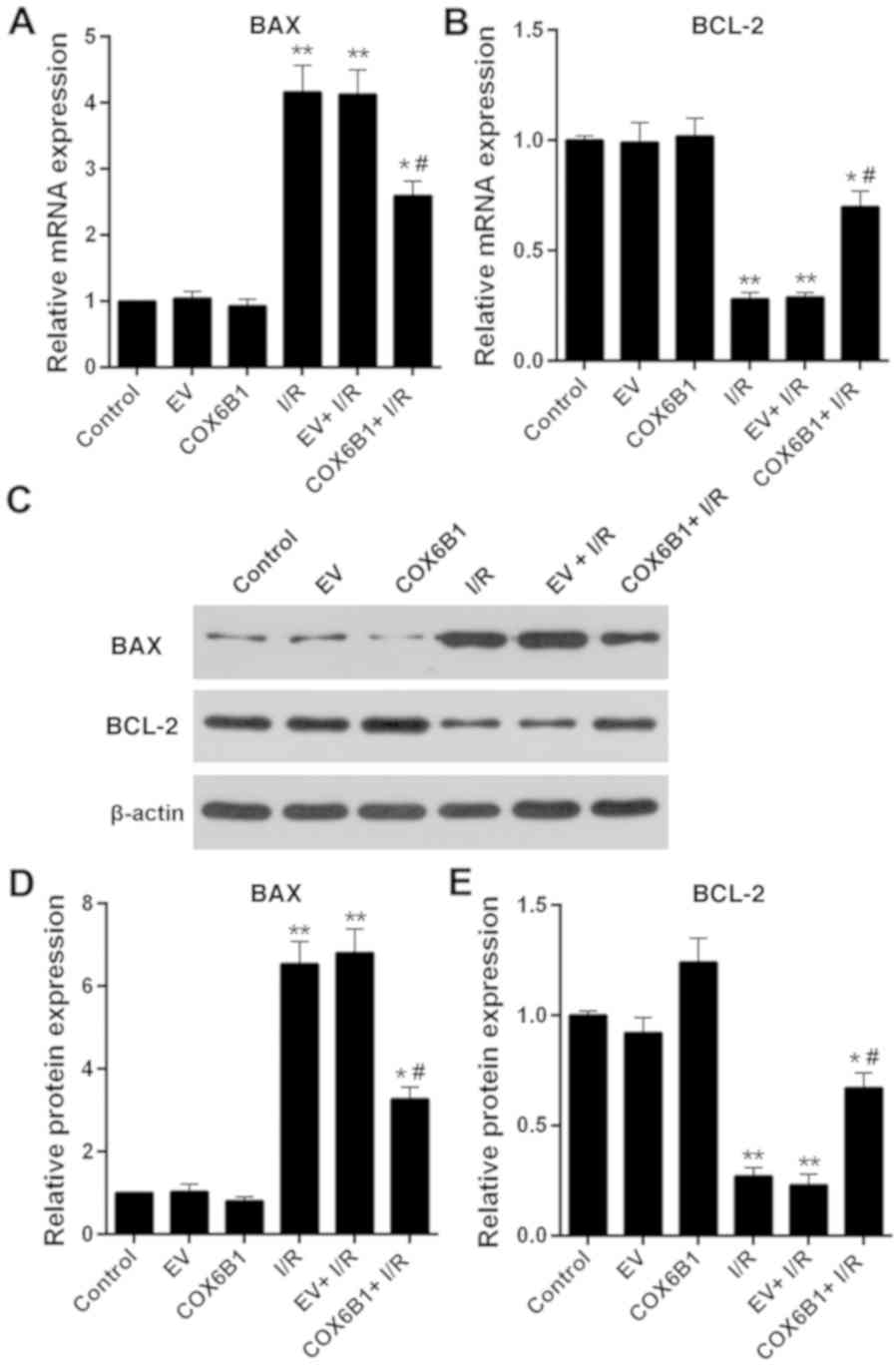
Overexpression of COX6B1 increases the
expression levels of BCL-2 and decreases the expression levels of
BAX in hippocampal neurons following I/R injury
To investigate apoptotic pathway components
downstream of COX6B1 in hippocampal neurons, the expression levels
of apoptosis-associated factors BCL-2 and BAX were investigated by
RT-qPCR and western blotting. The mRNA expression levels of BAX in
the I/R, EV + I/R and COX6B1 + I/R groups were significantly
increased compared with those in the EV group, whereas the
expression levels of BCL-2 were significantly decreased compared
with in the EV group (Fig. 6A and
B, respectively). Overexpression of COX6B1 following I/R injury
decreased the mRNA expression levels of BAX and increased the
expression levels of BCL-2 compared with the EV + I/R group.
Similar alterations in the expression of BAX and BCL-2 were
observed at the protein level, as assessed by western blotting
(Fig. 6C-E).
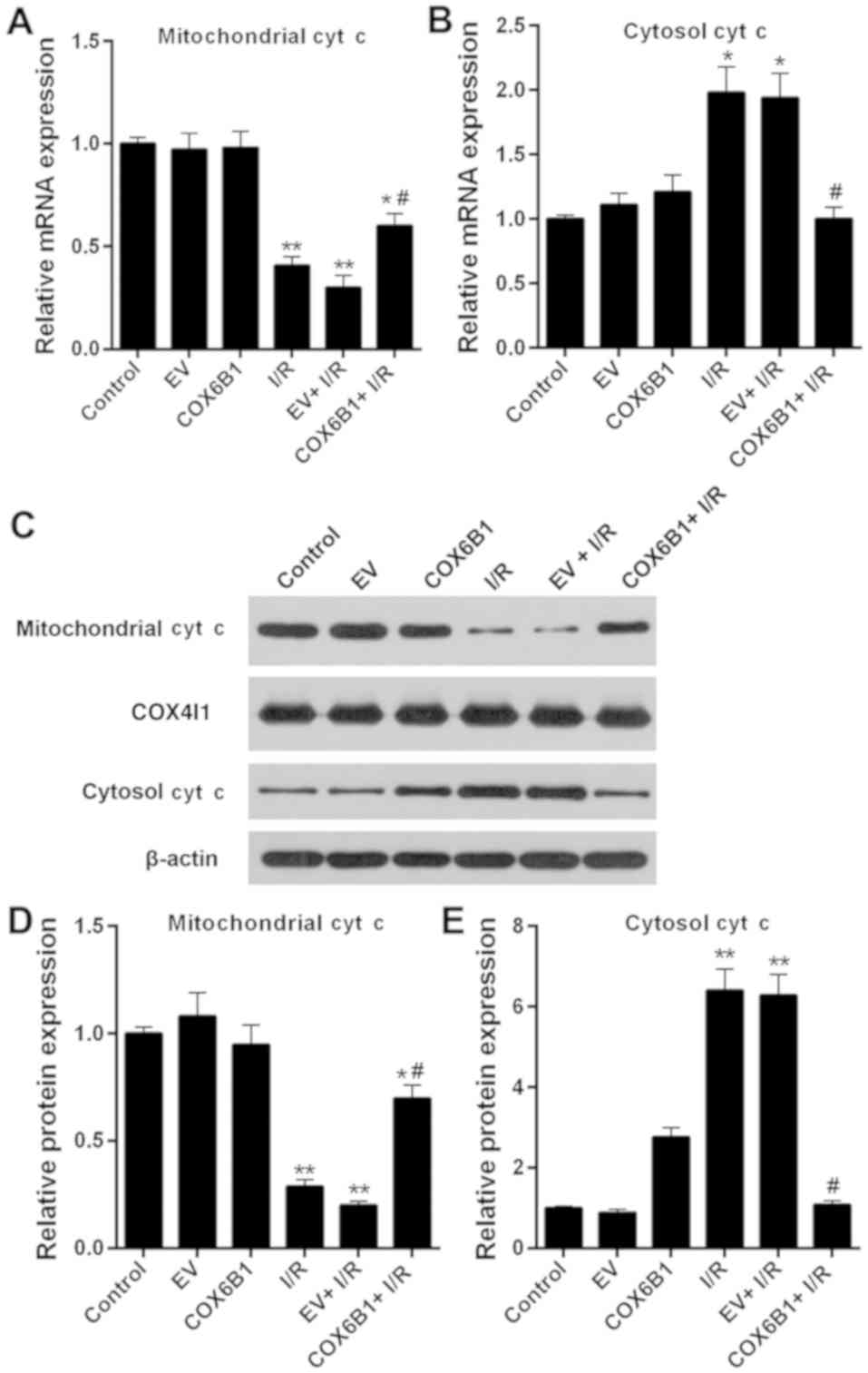
Overexpression of COX6B1 increases the
expression levels of mitochondrial cyt c and decreases the
expression levels of cytosolic cyt c in hippocampal neurons
following I/R
To investigate the role of COX6B1 on the expression
levels of cyt c in hippocampal neurons, RT-qPCR and western
blotting were performed. The expression levels of mitochondrial cyt
c decreased, whereas the expression levels of cytosolic cyt c
increased following I/R injury; COX6B1 overexpression reversed the
effects of I/R injury (Fig.
7).
Discussion
Primary cultured hippocampal neurons are frequently
harvested from hippocampi of newborn (<24-h-old) rodents, owing
to their localization and the amount of available material
(23,24), and these cells have been used to
establish experimental models to study the function of neurons
in vitro. Therefore, in the present study, the hippocampal
neurons from fetal rats were selected as a model to study I/R
injury. MAP2 is a specific marker of hippocampal neurons (25). MAP2 protein expression was used to
confirm the identity of the extracted cells used in the present
study; therefore, the extracted cells were identified as
hippocampal neurons. OGD exposure has been used by various previous
studies to investigate neuronal ischemia in vitro (26,27).
Therefore, in the present study, this method was selected to
establish an I/R model in hippocampal neurons. Cell viability
decreased by ~50% in cells exposed to OGD for 60 min followed by
reoxygenation treatment for 60 min, as assessed by CCK-8 assay.
Massa et al (16) demonstrated that genetic mutations
affecting the 20th amino acid residue of COX6B1 protein may cause
severe infantile encephalomyopathies. In addition, previous studies
reported that knockout of COX5A and COX6A may lead to a decline in
the number of neurons in animal models (28,29).
Nevertheless, the molecular mechanism underlying COX6B1 function in
hippocampal neurons remains unclear. In the present study, the
expression levels of COX6B1 decreased following ischemic or I/R
injury. Therefore, it was hypothesized that COX6B1 may serve a role
in hippocampal neurons following I/R injury. Subsequently, in the
present study, COX6B1 was overexpressed in hippocampal neurons
exposed to OGD/R, which reversed the I/R-induced decrease in
viability of hippocampal neurons.
Dysregulation of intracellular Ca2+
homeostasis is a mechanism of cell death caused by various factors
(30,31). Cerebral ischemia leads to membrane
depolarization and causes the release of presynaptic excitatory
transmitters, including glutamic acid and aspartic acid, and
extracellular Ca2+ ions enter the cells via
voltage-gated channels and N-methyl-D-aspartate receptor-gated
channels (32). An increase in
intracellular Ca2+ may lead to the activation of
Ca2+-dependent enzymes, including cytoplasmic
phospholipases and proteases, which may cause the degradation of
cell membranes and the disruption of the neuronal cytoskeleton,
damaging the structure and function of neurons (33,34).
In the present study, the levels of cytosolic Ca2+ were
increased following I/R neuronal injury, whereas overexpression of
COX6B1 significantly decreased I/R-induced cytosolic
Ca2+ levels in hippocampal neurons. The present results
suggested that COX6B1 overexpression protected against I/R-induced
neuronal damage by inhibiting the cytosolic levels of
Ca2+.
A number of previous studies demonstrated that
neuronal apoptosis served a principal role in neuronal death
following cerebral I/R (26,35,36).
In accordance with these previous studies, the present study
demonstrated that apoptosis was promoted following I/R injury.
Additionally, overexpression of COX6B1 was able to suppress
I/R-induced neuronal apoptosis. Subsequently, the molecular
mechanism underlying neuronal apoptosis was investigated in the
present study. The proteins belonging to the BCL-2 family are
involved in the apoptotic pathway, and regulate the permeability of
the outer membranes of mitochondria (37). BCL-2 and BAX are antagonistic
proteins. BCL-2 serves anti-apoptotic roles, whereas BAX serves a
role in promoting apoptosis (38).
Aboutaleb et al (39)
demonstrated that the expression level of BCL-2 decreased, whereas
BAX expression increased in hippocampal neurons following I/R
injury (39). In accordance with
that previous study, the present results revealed that the
expression level of BCL-2 decreased following I/R injury, whereas
the expression level of BAX was increased. However, overexpression
of COX6B1 significantly reversed the expression levels of BCL-2 and
BAX in hippocampal neurons following I/R injury. The present
results suggested that COX6B1 overexpression was able to reduce
I/R-induced neuronal apoptosis by downregulating the expression
level of BAX and by upregulating the expression level of BCL-2.
In addition, Wang et al (40) reported that cyt c was detectable in
the cytoplasm of neurons following I/R injury, and appropriate
ischemic post-conditioning significantly decreased the cytoplasmic
levels of cyt c in neurons. The release of cyt c from mitochondria
to the cytoplasm is an important step of apoptosis (41–43).
Therefore, in the present study, the levels of mitochondrial and
cytoplasmic cyt c in hippocampal neurons were investigated. The
results suggested that the expression level of mitochondrial cyt c
decreased; however, the expression level of cytosolic cyt c
increased in hippocampal neurons following I/R injury. This may be
a result of the destruction of the mitochondrial outer membrane
induced by I/R injury, leading to the release of cyt c from the
mitochondrial membrane space. Conversely, overexpression of COX6B1
reversed the effects of I/R injury; the expression levels of
mitochondrial cyt c in the COX6B1 + I/R group were significantly
increased compared with in the EV + I/R group, whereas the
expression levels of cytoplasmic cyt c were significantly decreased
in the COX6B1 + I/R group compared with in the EV + I/R group. The
overexpression of COX6B1 may reduce the IR-induced damage to the
mitochondrial outer membrane. Of note, there was no clear
difference in the mRNA expression of cytosolic cyt c between the EV
and the COX6B1 group, whereas the protein expression of cytosolic
cyt c in the COX6B1 group was markedly (but not significantly)
increased compared with in the EV group. It may be that the
overexpression of COX6B1 promoted a small release of cyt c from the
mitochondria to the cytoplasm; however, this would require further
investigation. The present results suggested that COX6B1
overexpression reversed I/R-induced neuronal apoptosis by
inhibiting the release of cyt c from mitochondria to the cytoplasm.
However, the present study presents some limitations; the specific
molecular mechanism underlying the function of COX6B1 in I/R injury
was not investigated. Furthermore, the present experiments were
performed in vitro and further in vivo analyses are
required to confirm the present observations.
Collectively, the present study results indicated
that COX6B1 overexpression was able to partially protect neurons
following I/R-induced damage by increasing cell viability and by
decreasing the levels of cytosolic Ca2+ and apoptosis.
The results may facilitate the development of novel strategies for
CVD prevention and treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SY made substantial contributions to the conception
and design of the study. PW, JX and LJ were involved in the
acquisition, analysis and interpretation of data. All authors
approved of the final version of the manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by The Ethics
Committee of The Nanchuan People's Hospital Affiliated to Chongqing
Medical University (Chongqing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Leira Y, Blanco M, Blanco J and Castillo
J: Association between periodontal disease and cerebrovascular
disease. A review of the literature. Rev Neurol. 61:29–38. 2015.(In
Spanish). PubMed/NCBI
|
|
2
|
Wang Q, Yu H, Jiang C, Sun R, Qi M, Sun S,
Xu G, Cai H, Zhang Z, Zhao F, et al: Cerebral infarction as initial
presentation in stress cardiomyopathy: Case report and literature
review. Medicine (Baltimore). 97:e108042018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang MY, Wu HW, Xu LP and Yang HJ:
Pharmacological effect of Schisandrae Chinensis Fructus and
relative active components on cardiovascular and cerebrovascular
diseases. Zhongguo Zhong Yao Za Zhi. 43:1536–1546. 2018.(In
Chinese). PubMed/NCBI
|
|
4
|
Dai L, Song L, Li X, Yang Y, Zheng X, Wu
Y, Li C, Zhao H and Wang Y, Wu S and Wang Y: Association of
visit-to-visit blood pressure variability with the risk of
all-cause mortality and cardiovascular events in general
population. J Clin Hypertens (Greenwich). 20:280–288. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Griffin RL, Falatko SR, Aslibekyan S,
Strickland V and Harrigan MR: Aspirin for primary prevention of
stroke in traumatic cerebrovascular injury: Association with
increased risk of transfusion. J Neurosurg. 1–8. 2018.doi:
10.3171/2017.12.JNS172284. (Epub ahead of print). PubMed/NCBI
|
|
6
|
Endepols H, Mertgens H, Backes H,
Himmelreich U, Neumaier B, Graf R and Mies G: Longitudinal
assessment of infarct progression, brain metabolism and behavior
following anterior cerebral artery occlusion in rats. J Neurosci
Methods. 253:279–291. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Struys T, Govaerts K, Oosterlinck W,
Casteels C, Bronckaers A, Koole M, Van Laere K, Herijgers P,
Lambrichts I, Himmelreich U and Dresselaers T: In vivo evidence for
long-term vascular remodeling resulting from chronic cerebral
hypoperfusion in mice. J Cereb Blood Flow Metab. 37:726–739. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan Y, Zhang C, Peng W, Li T, Yin J, Kong
Y, Lan C, Li X, Wang R and Hu Z: Retraction notice to ‘Secretory
pathway Ca2+-ATPase isoform 1 knockdown promotes Golgi apparatus
stress injury in a mouse model of focal cerebral
ischemia-reperfusion: In vivo and in vitro study’ [Brain Res. 1642
(2016) 189–196]. Brain Res. 1670:2532017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang ML, Tao T, Xu J, Liu Z and Xu D:
Antiapoptotic effect of gene therapy with recombinant adenovirus
vector containing hypoxia-inducible factor-1α after cerebral
ischemia and reperfusion in rats. Chin Med J (Engl). 130:1700–1706.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hossmann KA: The two pathophysiologies of
focal brain ischemia: Implications for translational stroke
research. J Cereb Blood Flow Metab. 32:1310–1316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mui K, Yoo AJ, Verduzco L, Copen WA,
Hirsch JA, González RG and Schaefer PW: Cerebral blood flow
thresholds for tissue infarction in patients with acute ischemic
stroke treated with intra-arterial revascularization therapy depend
on timing of reperfusion. AJNR Am J Neuroradiol. 32:846–851. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lenka N, Vijayasarathy C, Mullick J and
Avadhani NG: Structural organization and transcription regulation
of nuclear genes encoding the mammalian cytochrome c oxidase
complex. Prog Nucleic Acid Res Mol Biol. 61:309–344. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barrientos A, Barros MH, Valnot I, Rötig
A, Rustin P and Tzagoloff A: Cytochrome oxidase in health and
disease. Gene. 286:53–63. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huttemann M, Jaradat S and Grossman LI:
Cytochrome c oxidase of mammals contains a testes-specific isoform
of subunit VIb-the counterpart to testes-specific cytochrome c? Mol
Reprod Dev. 66:8–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu J, Wang K, Rodova M, Esteves R, Berry
D, E L, Crafter A, Barrett M, Cardoso SM, Onyango I, et al:
Polymorphic variation in cytochrome oxidase subunit genes. J
Alzheimers Dis. 21:141–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Massa V, Fernandez-Vizarra E, Alshahwan S,
Bakhsh E, Goffrini P, Ferrero I, Mereghetti P, D'Adamo P, Gasparini
P and Zeviani M: Severe infantile encephalomyopathy caused by a
mutation in COX6B1, a nucleus-encoded subunit of cytochrome c
oxidase. Am J Hum Genet. 82:1281–1289. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abdulhag UN, Soiferman D, Schueler-Furman
O, Miller C, Shaag A, Elpeleg O, Edvardson S and Saada A:
Mitochondrial complex IV deficiency, caused by mutated COX6B1, is
associated with encephalomyopathy, hydrocephalus and
cardiomyopathy. Eur J Hum Genet. 23:159–164. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SE, Mori R, Komatsu T, Chiba T,
Hayashi H, Park S, Sugawa MD, Dencher NA and Shimokawa I:
Upregulation of cytochrome c oxidase subunit 6b1 (Cox6b1) and
formation of mitochondrial supercomplexes: Implication of Cox6b1 in
the effect of calorie restriction. Age (Dordr). 37:97872015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Popovic DM: Current advances in research
of cytochrome c oxidase. Amino Acids. 45:1073–1087. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng Y, Madungwe NB, da Cruz Junho CV and
Bopassa JC: Activation of G protein-coupled oestrogen receptor 1 at
the onset of reperfusion protects the myocardium against
ischemia/reperfusion injury by reducing mitochondrial dysfunction
and mitophagy. Br J Pharmacol. 174:4329–4344. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang W, Wang Y, Wan J, Zhang P and Pei F:
COX6B1 relieves hypoxia/reoxygenation injury of neonatal rat
cardiomyocytes by regulating mitochondrial function. Biotechnol
Lett. 41:59–68. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Facci L and Skaper SD: Culture of rodent
cortical, hippocampal, and striatal neurons. Methods Mol Biol.
1727:39–47. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rivera-Carvantes MC, Jarero-Basulto JJ,
Feria-Velasco AI, Beas-Zarate C, Navarro-Meza M, Gonzalez-Lopez MB,
Gudino-Cabrera G and Garcia-Rodriguez JC: Changes in the expression
level of MAPK pathway components induced by monosodium
glutamate-administration produce neuronal death in the hippocampus
from neonatal rats. Neuroscience. 365:57–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caceres A, Banker G, Steward O, Binder L
and Payne M: MAP2 is localized to the dendrites of hippocampal
neurons which develop in culture. Brain Res. 315:314–318. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lei X, Lei L, Zhang Z and Cheng Y:
Diazoxide inhibits of ER stressmediated apoptosis during
oxygenglucose deprivation in vitro and cerebral
ischemiareperfusion in vivo. Mol Med Rep. 17:8039–8046.
2018.PubMed/NCBI
|
|
27
|
Lin YW, Chen TY, Hung CY, Tai SH, Huang
SY, Chang CC, Hung HY and Lee EJ: Melatonin protects brain against
ischemia/reperfusion injury by attenuating endoplasmic reticulum
stress. Int J Mol Med. 42:182–192. 2018.PubMed/NCBI
|
|
28
|
Baden KN, Murray J, Capaldi RA and
Guillemin K: Early developmental pathology due to cytochrome c
oxidase deficiency is revealed by a new zebrafish model. J Biol
Chem. 282:34839–34849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu W, Gnanasambandam R, Benjamin J, Kaur
G, Getman PB, Siegel AJ, Shortridge RD and Singh S: Mutations in
cytochrome c oxidase subunit VIa cause neurodegeneration and motor
dysfunction in Drosophila. Genetics. 176:937–946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su F, Guo AC, Li WW, Zhao YL, Qu ZY, Wang
YJ, Wang Q and Zhu YL: Low-dose ethanol preconditioning protects
against oxygen-glucose deprivation/reoxygenation-induced neuronal
injury by activating large conductance, Ca2+-Activated
K+ channels in vitro. Neurosci Bull. 33:28–40. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Shen Y, Lin HP, Li Z, Chen YY and
Wang S: Large-conductance Ca(2+)-activated K(+) channel involvement
in suppression of cerebral ischemia/reperfusion injury after
electroacupuncture at Shuigou (GV26) acupoint in rats. Neural Regen
Res. 11:957–962. 2016.PubMed/NCBI
|
|
32
|
Savigni DL, O'Hare Doig RL, Szymanski CR,
Bartlett CA, Lozic I, Smith NM and Fitzgerald M: Three Ca2+ channel
inhibitors in combination limit chronic secondary degeneration
following neurotrauma. Neuropharmacology. 75:380–390. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zolezzi JM, Carvajal FJ, Rios JA, Ordenes
D, Silva-Alvarez C, Godoy JA and Inestrosa NC: Tetrahydrohyperforin
induces mitochondrial dynamics and prevents mitochondrial Ca2+
overload after Aβ and Aβ-AChE complex challenge in rat hippocampal
neurons. J Alzheimers Dis. 37:735–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma YY, Li KY, Wang JJ, Huang YL, Huang Y
and Sun FY: Vascular endothelial growth factor acutely reduces
calcium influx via inhibition of the Ca2+ channels in rat
hippocampal neurons. J Neurosci Res. 87:393–402. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li J, Yan D, Liu X, Wang Y, Zhao X, Zhang
Y and Zhang C: U0126 protects hippocampal CA1 neurons against
forebrain ischemia-induced apoptosis via the ERK1/2 signaling
pathway and NMDA receptors. Neurol Res. 40:318–323. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu Z, Cai M, Li X, Zhang J, Wu T, Yang F,
Zhu W, Xiang Y, Zhang W, Xiang J and Cai D: Neuroprotective effects
of Tongxinluo on focal cerebral ischemia and reperfusion injury in
rats associated with the activation of the MEK1/2/ERK1/2/p90RSK
signaling pathway. Brain Res. 1685:9–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouyang YB and Giffard RG: MicroRNAs affect
BCL-2 family proteins in the setting of cerebral ischemia.
Neurochem Int. 77:2–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aboutaleb N, Shamsaei N, Rajabi H,
Khaksari M, Erfani S, Nikbakht F, Motamedi P and Shahbazi A:
Protection of hippocampal CA1 neurons against ischemia/reperfusion
injury by exercise preconditioning via modulation of Bax/Bcl-2
ratio and prevention of caspase-3 activation. Basic Clin Neurosci.
7:21–29. 2016.PubMed/NCBI
|
|
40
|
Wang JY, Shen J, Gao Q, Ye ZG, Yang SY,
Liang HW, Bruce IC, Luo BY and Xia Q: Ischemic postconditioning
protects against global cerebral ischemia/reperfusion-induced
injury in rats. Stroke. 39:983–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Antonawich FJ: Translocation of cytochrome
c following transient global ischemia in the gerbil. Neurosci Lett.
274:123–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Muranyi M and Li PA: Bongkrekic acid
ameliorates ischemic neuronal death in the cortex by preventing
cytochrome c release and inhibiting astrocyte activation. Neurosci
Lett. 384:277–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao H, Yenari MA, Cheng D, Sapolsky RM
and Steinberg GK: Biphasic cytochrome c release after transient
global ischemia and its inhibition by hypothermia. J Cereb Blood
Flow Metab. 25:1119–1129. 2005. View Article : Google Scholar : PubMed/NCBI
|