Introduction
Endothelial dysfunction has been identified as one
of the most important pathogenetic causes of human cerebrovascular
disease (CVD) (1,2). Endothelial dysfunction can cause
damage to the blood-brain barrier and can result in a range of
neurological disorders, including multiple sclerosis, vascular
dementia and subsequent complications of the extremities (3–5).
Cerebral small vessel disease is a condition that involves the
formation of white matter lesions and cerebral microbleeds, and has
been associated with endothelial dysfunction (6).
Elevated serum levels of homocysteine (Hcy) is an
independent risk factor that can damage vascular endothelial cells
and can cause endothelial dysfunction, which in turn contributes to
the occurrence and development of CVDs (7–9).
Several studies have focused on the ability of Hcy to lower the
severity of numerous human diseases (10–12).
Hcy-induced apoptosis of endothelial cells has been reported to
account for Hcy-dependent vascular injury (13). Accumulated evidence suggests that
Hcy can cause endothelial dysfunction. For example, Hcy can inhibit
endothelial nitric oxide (NO) synthase signaling (14) and cell migration by targeting key
angiogenic factors (15).
Furthermore, it can reduce the expression levels of vascular
endothelial growth factor (VEGF)-A and vascular endothelial growth
factor receptor (VEGFR)-2 (16,17).
Hcy can inhibit microvascular endothelial cell formation by
disrupting cell migration via an inducible NO synthase-dependent
mechanism (18,19). Hcy can decrease the invasive
potential of endothelial cells by inhibiting matrix
metalloproteinase (MMP)-2 and urokinase (19); however, the mechanism of
cytotoxicity of Hcy on endothelial cells remains unclear.
Furthermore, to the best of our knowledge, the role of reactive
oxygen species (ROS) in endothelial dysfunction has not been
investigated previously.
Astaxanthin (ATX) is a potent antioxidant that
undertakes a novel mechanism of action. Our previous study revealed
that ATX can attenuate Hcy-induced cardiotoxicity in vitro
and in vivo by inhibiting mitochondrial dysfunction and
oxidative damage (20). It was
reported that ATX could attenuate the astrocyte apoptosis and
reduce traumatic brain injury by inhibiting Na-K-Cl co-transporter
(NKCC1) and the secretion of proinflammatory cytokines (21). These effects were caused by the
suppression of oxidative stress and the upregulation of
brain-derived neurotrophic factor and nerve growth factor mRNA
(22,23). ATX exerted neuroprotective effects
against subarachnoid hemorrhage damage that involved the inhibition
of MMP-9 expression, the upregulation of Akt/glycogen synthase
kinase-3β and the activation of the nuclear factor-like
2-antioxidant responsive element pathway (24–32);
however, the protective effects of ATX against Hcy-induced
endothelial dysfunction and the underlying mechanism require
further investigation.
Materials and methods
Materials
Dulbecco's Modified Eagles medium (DMEM)/F-12 and
fetal bovine serum (FBS) were purchased from Gibco (Thermo Fisher
Scientific, Inc.). ATX (purity, 97%), Hcy (purity, 98%), MTT and
propidium iodide were obtained from Sigma-Aldrich (Merck KGaA). All
primary antibodies used in the present study, including anti-VEGF
(cat. no. 2463), VEGFR2 (cat. no. 9698), phosphorylated (p)-VEGFR2
(cat. no. 2478), Tyr397-focal adhesion kinase (FAK; cat. no. 3283),
FAK (cat. no. 3285) and β-actin (cat. no. 8457) were purchased from
Cell Signaling Technology, Inc. A horseradish peroxidase-linked
goat anti-rabbit immunoglobulin G (cat. no. 7074; Cell Signaling
Technology, Inc.) was used as the secondary antibody. PF-562271 was
purchased from Selleck Chemicals. All solvents used were of
high-performance liquid chromatography grade.
Cell viability assay
Human umbilical vein endothelial cells (HUVECs) were
obtained from the American Type Culture Collection. HUVECs were
cultured in DMEM-F12 containing 10% FBS at 5% CO2 and
37°C in an incubator. Cells (8×103 cells/well) were
seeded in a 96-well plate and treated with Hcy (1, 2, 5, 10 and 20
mM) at 37°C for 72 h. In addition, cells were pre-treated with 1,
2, 5 and 10 µM ATX at 37°C for 6 h and then incubated with 10 mM
Hcy at 37°C for 72 h. Following treatment, 20 µl MTT solution was
added and the cells were incubated at 37°C for another 5 h.
Subsequently, the medium was removed and 150 µl of dimethyl
sulfoxide was added. Cell viability was analyzed at room
temperature (25°C) by detecting the absorbance at 570 nm. The
morphology of HUVECs was observed under a phase contrast-microscope
(magnification, ×400; Nikon Corporation). Five randomly-selected
fields of view per sample were imaged.
Cell migration assay
HUVEC migration was measured by a wound-healing
migration assay. Briefly, HUVECs were seeded in a 6-well tissue
culture plate and cultured at 37°C for 24 h. Scratched wounds were
created by scraping the cell monolayer with a sterile 10 µl pipette
tip. Subsequently, the cells were cultured with DMEM/F-12 medium
(containing 1% FBS). Subsequently, the cells were pre-treated with
5 µM ATX for 6 h and/or 10 mM Hcy or 10 nM PF562271 at 37°C for 48
h. Untreated cells were used as control. The migrated cells were
imaged in five randomly-selected fields of view with a
phase-contrast microscope (magnification, ×200) and the percentage
of migration was quantified by manual counting (% of control).
Cell invasion assay
HUVECs were pre-treated with 5 µM ATX for 6 h and/or
co-incubated with 10 mM Hcy at 37°C for 72 h. Following treatment,
HUVECs (4×104 cells/well) were suspended in 100 µl
DMEM/F-12 medium (FBS-free) and were seeded in the upper layer of a
Matrigel pre-coated Transwell chamber. Complete DMEM/F12 (600 µl,
10% FBS) was added into the lower chamber. Following a 24 h
incubation period at 37°C, the non-invaded cells on the Transwell
were removed using a cotton swab; invaded cells were washed with
PBS, fixed with 10% ethanol for 10 min at room temperature (25°C)
and stained with 0.1% crystal violet for 15 min at room temperature
(25°C). Invaded cells were measured by manual counting with a Nikon
Ti-S inverted microscope (magnification, ×200). In total, five
randomly-selected fields of view per sample were imaged and
analyzed.
Tube formation
In vitro tube formation was examined by a
Transwell assay. Briefly, HUVECs were pre-treated with 5 µM ATX for
6 h and/or co-incubated with 10 mM Hcy at 37°C for 72 h. Following
treatment, HUVECs (104 cells/well) were seeded in
Matrigel pre-coated 48-well plates and incubated at 37°C for 24 h.
In total, five randomly-selected fields of view per sample were
imaged, and the number of tubes formed manually counted using a
Nikon inverted microscope (magnification, ×100).
ROS measurement
The levels of intracellular ROS in HUVECs were
detected by the 2′7′-dichlorfluorescein diacetate (DCFH-DA).
Briefly, HUVECs were incubated with 10 µM DCFH-DA for 20 min at
37°C in the dark. Subsequently, the cells were washed with PBS and
treated with 10 mM Hcy at 37°C for 10, 30, 60 and 120 min. On the
contrary, cells were treated with 5 µM ATX for 60 min and/or
co-treated with 10 mM Hcy at 37°C for 120 min to analyze the
protective effects of ATX. For ROS inhibition, cells were
pre-treated with 5 mM glutathione (GSH) at 37°C for 2 h prior to
ATX/Hcy treatment. The production of ROS was quantified using a
microplate reader by measuring the fluorescence intensity at an
excitation wavelength of 488 nm and an emission wavelength of 525
nm.
Western blotting
Protein expression was detected by western blotting.
Briefly, HUVECs were pre-treated with 5 µM of ATX for 6 h and/or
co-incubated with 10 mM Hcy at 37°C for 72 h. Following treatment,
the cells were collected and lysed on ice for 1 h at 4°C in RIPA
lysis buffer (Nanjing KeyGen Biotech Co., Ltd.). Total protein was
quantified with a Bicinchoninic Acid detection kit. A total of 40
µg of protein was added and separated on a 10% SDS gel at 110 V for
75 min. Following electrophoresis, the proteins were transferred
from the gel onto the nitrocellulose membrane. The membrane was
blocked with 5% non-fat milk at room temperature (25°C) for 1 h and
incubated overnight with a primary antibody (1:1,000) at 4°C,
followed by incubation with the secondary antibody (1:2,000) for 1
h at room temperature (25°C). The target protein was scanned with
X-ray film using an enhanced chemiluminescence system (Kodak).
β-actin was used as the reference protein.
Statistical analysis
The experiments were repeated three times.
Statistical analysis was conducted with the SPSS software (version
13.0; SPSS, Inc.). Data are presented as the mean ± SD. Statistical
evaluation was analyzed by one-way ANOVA followed by a Dunnett's or
Tukey's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ATX inhibits Hcy-induced cytotoxicity
in HUVECs
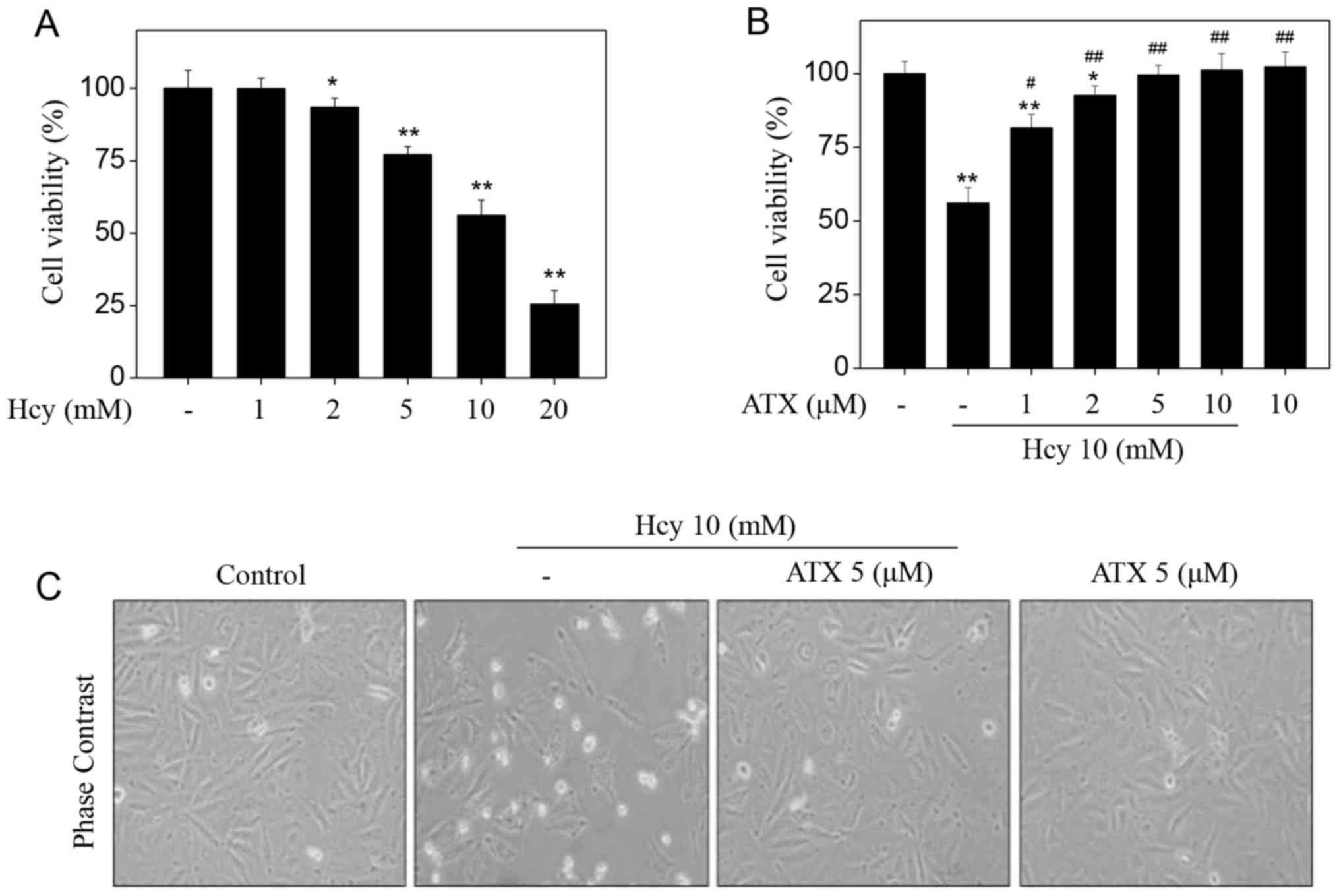
Initially, the toxicity of Hcy towards HUVECs was
examined by an MTT assay. Hcy alone apparently suppressed HUVEC
viability in a dose-dependent manner (Fig. 1A). Treatment of HUVECs with 5, 10
and 20 mM Hcy significantly suppressed the cell viability from 100%
(control) to 77.1, 56.2 and 25.5%, respectively. On the contrary,
pre-treatment of HUVECs with ATX could restore the cell viability
inhibited by Hcy. Pre-treatment of HUVECs with 1, 2 and 5 µM ATX
significantly increased cell viability from 56.2% (Hcy, 10 mM) to
86.1, 92.7 and 99.6%, respectively (Fig. 1B). ATX (10 µM) alone indicated no
cytotoxicity towards HUVECs. In addition, ATX pre-treatment
ameliorated morphological changes induced by Hcy in HUVECs. Hcy
treatment notably decreased cell number, and induced cell shrinkage
(Fig. 1C). These results suggested
that ATX could inhibit Hcy-induced cytotoxicity in HUVECs.
ATX increases cell migration, invasion
and tube formation in Hcy-treated HUVECs
To examine the effects on the functions of
endothelial cells, we examined HUVEC migration, invasion and tube
formation, which are considered indices of angiogenesis. Initially,
Hcy-treated HUVEC migration was analyzed by a wound-healing assay.
Hcy treatment alone significantly inhibited the migration of HUVECs
compared with untreated cells (Fig.
2A), which was demonstrated by the distance between the edges
of the wounded region following 48 h. On the contrary, ATX
pre-treatment appeared to improve the migration of Hcy-treated
cells. Hcy treatment (10 mM) significantly inhibited the migration
rate from 100% (control) to 17.9%; however, ATX pre-treatment (5
µM) significantly improved the migration rate to 77.8% (Fig. 2B). ATX treatment alone indicated no
significant effect on HUVEC migration compared with untreated
cells. The potency of ATX was further examined using cell invasion
and tube formation assays. Hcy treatment alone (10 mM)
significantly inhibited cell invasion and tube formation compared
with the untreated control, whereas ATX pre-treatment (5 µM)
significantly improved cell invasion and tube formation in
Hcy-treated cells (Fig. 2C-F).
Collectively, these results indicated that ATX could improve cell
migration, invasion and tube formation in Hcy-treated HUVECs.
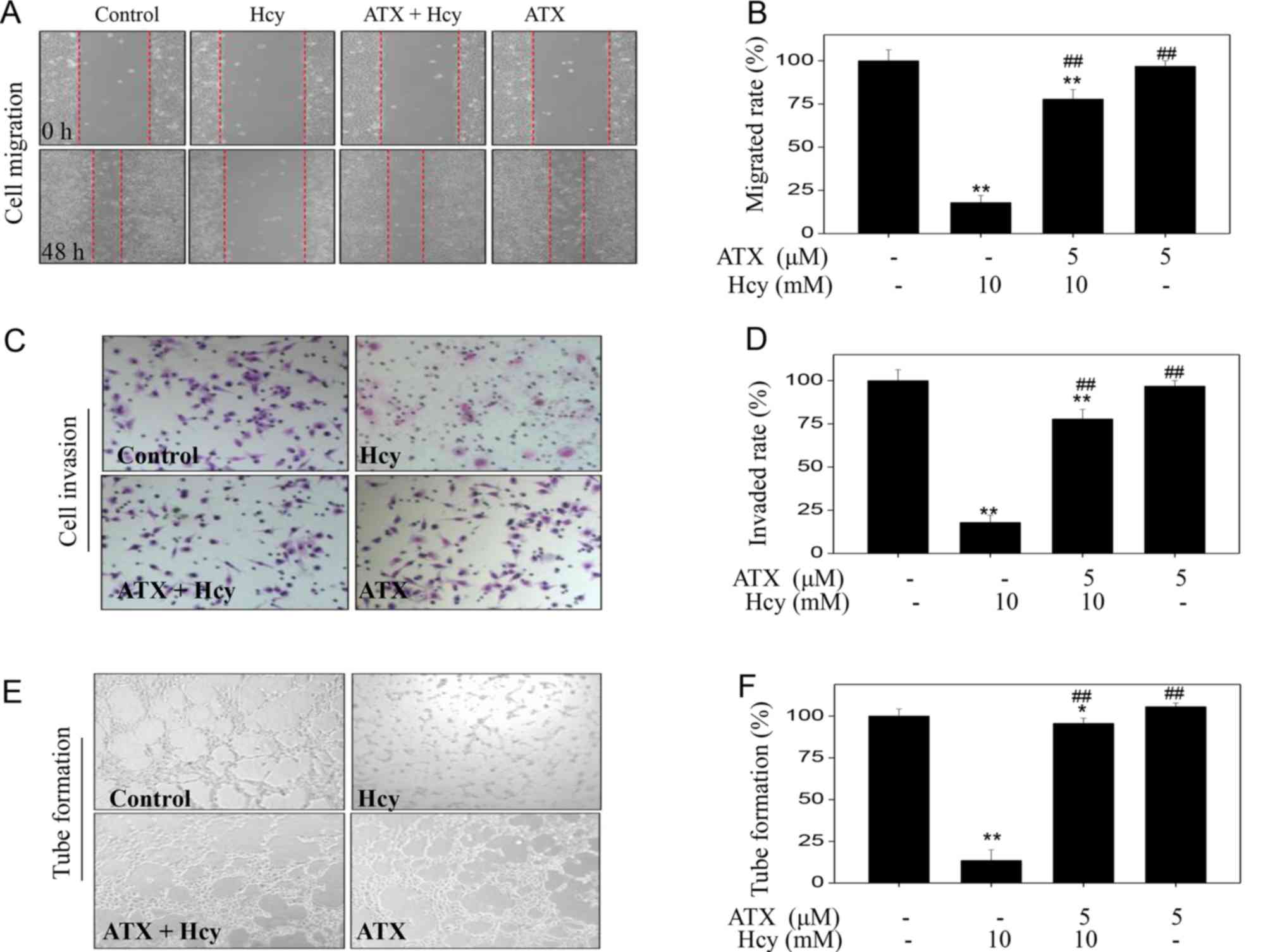 | Figure 2.ATX improves cell migration, invasion
and tube formation in Hcy-treated HUVECs. (A) ATX improved HUVECs
migration. Cells were seeded in a 6-well plate and cultured until
confluent. Cells were scraped by with a pipette tip and treated
with ATX for 6 h or/and co-treated with Hcy 48 h (magnification,
×100). (B) Statistical analysis of the rate of migration. (C) ATX
improved HUVEC invasion. The invasive potential of cells was
analyzed by a Transwell assay (magnification, ×200). (D)
Statistical analysis of the rate of invasion. (E) ATX improved
HUVEC tube formation. (F) Statistical analysis of tube formation.
The migrated cells, invaded cells and the number of tubes formed
were all calculated by manual counting, and expressed as a
percentage of the control (magnification, ×200). All data and
images were obtained from three independent experiments.
*P<0.05, **P<0.01 vs. control; ##P<0.01 vs.
Hcy-treated group. ATX, astaxanthin; Hcy, homocysteine; HUVECs,
human umbilical vascular endothelial cells. |
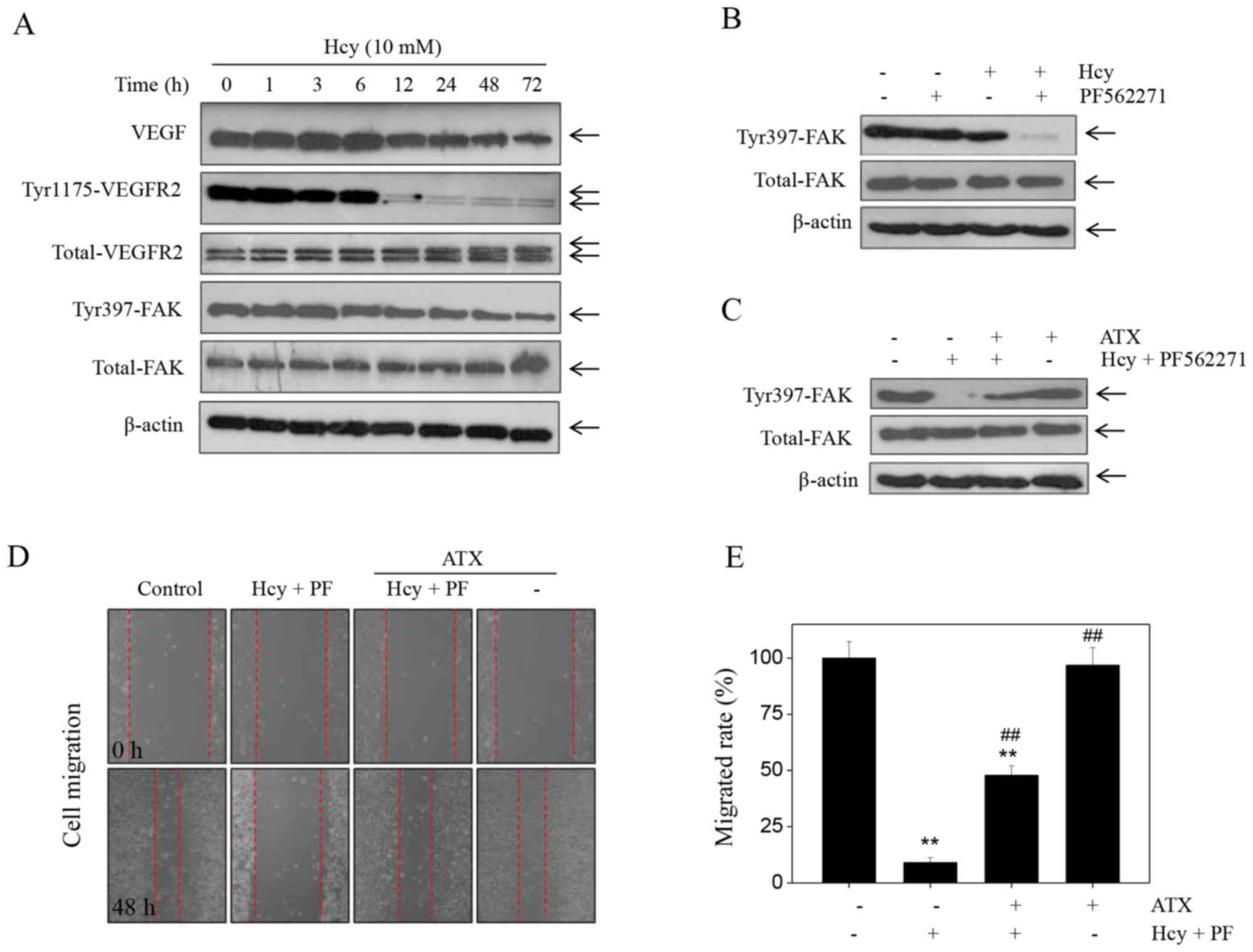
ATX inhibits Hcy-induced effects on
the VEGF-VEGFR2- FAK signaling pathway
Accumulating evidence has suggested that the
VEGF-VEGFR2-FAK is one of the most important pro-angiogenenic
signaling pathways that serve a key role in regulating cell
migration, invasion and tube formation (33). This pathway can be potentially
targeted for therapeutic intervention. Therefore, in the present
study, the expression levels of proteins involved in the
VEGF-VEGFR2-FAK pathway were detected by western blotting.
Treatment of cells with Hcy induced a significant time-dependent
decrease in the expression of VEGF, p-Tyr-VEGFR2 and p-Tyr397-FAK
(Fig. 3A). Notable changes were
noted in the expression levels of total-FAK and total-VEGFR2 in
Hcy-treated cells. To further evaluate the role of FAK, we used the
FAK inhibitor, PF562271. The results indicated that treatment with
PF562271 markedly enhanced the Hcy-induced inhibition of
p-Tyr397-FAK expression (Fig. 3B).
Additionally, PF562271 and Hcy significantly inhibited of HUVEC
migration compared with the control (Fig. 3D), which suggested that Hcy
inhibited HUVEC migration in a FAK-dependent manner. However, ATX
pre-treatment markedly recovered the expression of p-Tyr397-FAK in
HUVECs that were induced by the combined treatment of the FAK
inhibitor and Hcy (PF562271 + Hcy). ATX pre-treatment (5 µM)
reversed the effects of combined treatment of PF562271 and Hcy on
FAK phosphorylation (Fig. 3C). In
addition, ATX pre-treatment significantly increased the rate of
migration of HUVECs (47.8%) compared with the combined treatment
(8.99%; Fig. 3E). Collectively,
these findings indicated that ATX could inhibit Hcy-induced
dysfunction of the VEGF-VEGFR2-FAK signaling pathway.
ATX inhibits ROS-dependent FAK
phosphorylation
Accumulating evidence has shown that Hcy can induce
ROS accumulation, which can further cause cytotoxicity (20–22).
Therefore, the intracellular accumulation of ROS in Hcy-treated
HUVECs was examined. Hcy treatment induced ROS production in a
time-dependent manner, as demonstrated by the enhanced green
fluorescence (Fig. 4A); however,
ATX pre-treatment effectively inhibited Hcy-induced ROS production
(Fig. 4B). In addition, ATX
recovered the levels of Tyr397-FAK phosphorylation and improved
HUVEC viability (Fig. 4C-E), which
indicated similar protective effects to those of GSH, a ROS
scavenger. The results suggested that Hcy induced ROS-dependent FAK
phosphorylation; inhibition of ROS formation by ATX or GSH may
increase FAK phosphorylation. Collectively, these results suggested
that ATX could inhibit ROS-dependent FAK phosphorylation in
Hcy-treated HUVECs.
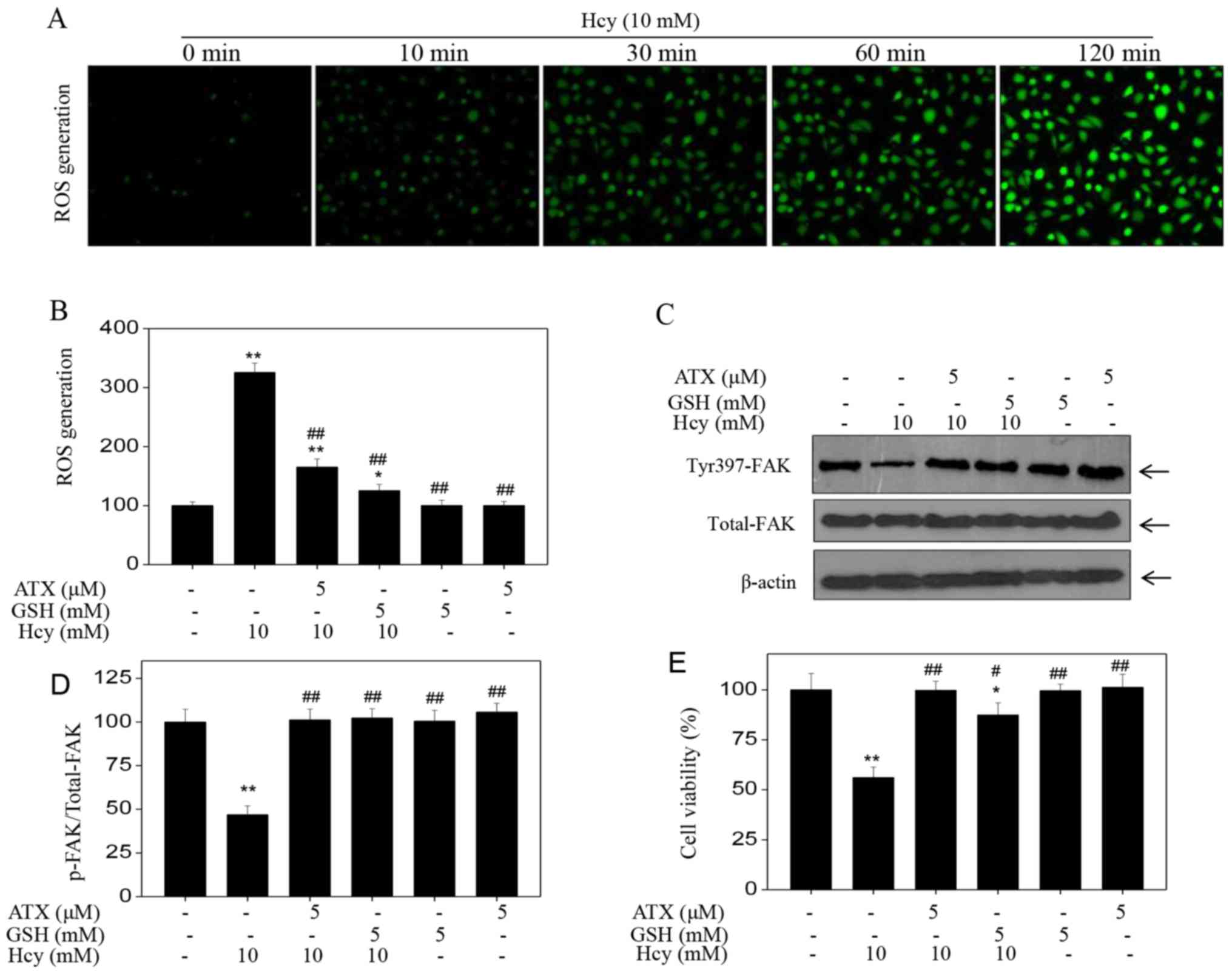 | Figure 4.ATX inhibits ROS-dependent FAK
phosphorylation. (A) Time-dependent ROS generation in Hcy-treated
HUVECs. HUVECs were labeled with 2′7′-dichlorfluorescein diacetate
for 20 min, and cells were washed and treated with 10 mM Hcy for
various durations. ROS generation was analyzed with a fluorescence
microscope (magnification, ×200). (B) ROS generation was quantified
by a microreader. (C) Effects of GSH or ATX pretreatment on
Hcy-induced FAK phosphorylation. HUVECs were pre-treated with 5 mM
GSH for 2 h prior to Hcy treatment. (D) Statistical analysis of
p-FAK expression. (E) Effects of GSH or ATX pre-treatment on
Hcy-induced HUVECs viability. All data and images were obtained
from three independent experiments. *P<0.05, **P<0.01 vs.
control; #P<0.05, ##P<0.01 vs.
Hcy-treated group ATX, astaxanthin; FAK, focal adhesion kinase;
Hcy, homocysteine; HUVECs, human umbilical vascular endothelial
cells; p, phosphorylated; ROS, reactive oxygen species. |
Discussion
Numerous studies have supported the notion that
hyperhomocysteinemia can induce endothelial cell apoptosis and
promote the development of vascular diseases (10–17).
This condition has therefore emerged as an independent risk factor
for human CVD (34). The
pathogenesis of hyperhomocysteinemia-associated human CVD is
remains unclear, but may be due to dysregulated endothelial cell
migration and invasion. Angiogenesis is a critical process required
for physiological processes in the body, such as the regeneration
of the damaged vascular tissues. The process of angiogenesis
includes capillary or posterior venous endothelial cell activation,
proliferation and migration. In addition, endothelial cell
migration is one of the most important processes of angiogenesis.
Endothelial cells can invade surrounding tissues, a prerequisite
for the development of angiogenesis in response to migration
signaling (2,3). Hyperhomocystinemia may cause damage
to vascular endothelial cells and consequently inhibit cell
migration. The morphology of viable cells following Hcy treatment
was notably altered than that of the control group, as determined
by phase-contrast microscopy. These findings indicated that Hcy
affected the normal function of endothelial cells. Atherosclerosis
and cerebral hemorrhages are complex processes initiated at sites
of endothelial cell injury. Injured endothelial cells can cause the
endothelium-dependent relaxation of blood vessels, thereby
resulting in the development of CVDs (35). In the present study, Hcy treatment
significantly inhibited the migration and invasive potentials of
HUVECs compared with the control group. Thus, inhibiting
endothelial cell migration and invasion may suppress the process of
angiogenesis.
The formation of a mature vascular network is
inhibited with vessel destabilization, followed by endothelial cell
re-organization. This process is completed by vessel maturation
(10–17). Angiogenesis requires the
simultaneous precise regulation of a large number of angiogenic
factors, including VEGF and VEGFR2, and their downstream signaling
proteins, namely ERK, AKT and FAK (36). The VEGF-VEGFR2 axis aids
endothelial cell recruitment and vascular permeability, whereas ERK
activates endothelial cell proliferation; FAK promotes cell
migration and invasion. VEGF and VEGFR2 have been considered to be
the most important factors in this pathway, and serve key roles in
regulating angiogenesis via the modulation of the degradation,
differentiation, proliferation and migration of vascular
endothelial cells (36–40). The VEGF-VEGFR axis eventually
promotes the formation of new blood vessels (36–39).
In clinical settings, patients with hyperhomocysteinemia usually
possess endothelial cells with impaired endothelial activities,
including cellular proliferation, migration and adhesion, which can
harm human heart health (41–43).
The present study revealed that Hcy induced endothelial cell
dysfunction, and these effects were reversed by ATX
pre-pretreatment, possibly via the regulation of FAK activation and
increased cell migration in Hcy-treated HUVECs. Our findings
provide insight into the potential therapeutic role of ATX in the
prevention and chemotherapy of Hcy-mediated human CVDs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Undergraduate Training Program for Innovation and Entrepreneurship
(grant no. 201610439043 to FW Wang and grant no. 201510005001 to MH
Zhang), the Sci-Tech Development Project of Taian in Shandong
(grant no. 2016NS1058 to XY Fu) and The Japan Society for the
Promotion of Science (JSPS) Postdoctoral Fellowship (grant no.
P17751 to JK Ma).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
JKM designed the experiments. XJW, DCT, FWW, XYF and
CDF performed the experiments. MHW and XYF analyzed the data and
prepared the images. JKM and XJW wrote the manuscript. All authors
reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fateeva VV and Vorobyova OV: Nitric oxide:
From the mechanism of action to pharmacological effects in
cerebrovascular diseases. Zh Nevrol Psikhiatr Im S S Korsakova.
117:131–135. 2017.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Poggesi A, Pasi M, Pescini F, Pantoni L
and Inzitari D: Circulating biologic markers of endothelial
dysfunction in cerebral small vessel disease: A review. J Cereb
Blood Flow Metab. 36:72–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holm H, Nägga K, Nilsson ED, Ricci F,
Melander O, Hansson O, Bachus E, Magnusson M and Fedorowski A:
Biomarkers of microvascular endothelial dysfunction predict
incident dementia: A population-based prospective study. J Intern
Med. 282:94–101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Michinaga S and Koyama Y: Protection of
the Blood-Brain barrier as a therapeutic strategy for brain damage.
Biol Pharm Bull. 40:569–575. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spencer JI, Bell JS and DeLuca GC:
Vascular pathology in multiple sclerosis: Reframing pathogenesis
around the blood-brain barrier. J Neurol Neurosurg Psychiatry.
89:42–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nezu T, Hosomi N, Aoki S, Kubo S, Araki M,
Mukai T, Takahashi T, Maruyama H, Higashi Y and Matsumoto M:
Endothelial dysfunction is associated with the severity of cerebral
small vessel disease. Hypertens Res. 38:291–297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dayal S, Baumbach GL, Arning E,
Bottiglieri T, Faraci FM and Lentz SR: Deficiency of superoxide
dismutase promotes cerebral vascular hypertrophy and vascular
dysfunction in hyperhomocysteinemia. PLoS One. 12:e01757322017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hatefi M, Behzadi S, Dastjerdi MM,
Ghahnavieh AA, Rahmani A, Mahdizadeh F, Hafezi Ahmadi MR and
Asadollahi K: Correlation of homocysteine with cerebral hemodynamic
abnormality, endothelial dysfunction markers, and cognition
impairment in patients with traumatic brain injury. World
Neurosurg. 97:70–79. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Škovierová H, Vidomanová E, Mahmood S,
Sopková J, Drgová A, Červeňová T, Halašová E and Lehotský J: The
molecular and cellular effect of homocysteine metabolism imbalance
on human health. Int J Mol Sci. 17(pii): E17332016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Catena C, Colussi G, Url-Michitsch M, Nait
F and Sechi LA: Subclinical carotid artery disease and plasma
homocysteine levels in patients with hypertension. J Am Soc
Hypertens. 9:167–175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang BR, Ou Z, Jiang T, Zhang YD, Zhao HD,
Tian YY, Shi JQ and Zhou JS: Independent correlation of serum
homocysteine with cerebral microbleeds in patients with acute
ischemic stroke due to large-artery atherosclerosis. J Stroke
Cerebrovasc Dis. 25:2746–2751. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu GH, Kong FZ, Dong XF, Wu DF, Guo QZ,
Shen AR, Cheng QZ and Luo WF: Association between
hyperhomocysteinemia and stroke with atherosclerosis and small
artery occlusion depends on homocysteine metabolism-related vitamin
levels in Chinese patients with normal renal function. Metab Brain
Dis. 32:859–865. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Z, Wei C, Zhou Y, Yan T, Wang Z, Li
W and Zhao L: Homocysteine induces apoptosis of human umbilical
vein endothelial cells via mitochondrial dysfunction and
endoplasmic reticulum stress. Oxid Med Cell Longev.
2017:57365062017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan TT, Li Q, Zhang XH, Wu WK, Sun J, Li
L, Zhang Q and Tan HM: Homocysteine impaired endothelial function
through compromised vascular endothelial growth
factor/Akt/endothelial nitric oxide synthase signalling. Clin Exp
Pharmacol Physiol. 37:1071–1077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pan L, Yu G, Huang J, Zheng X and Xu Y:
Homocysteine inhibits angiogenesis through cytoskeleton remodeling.
Biosci Rep. 37(pii): BSR201708602017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oosterbaan AM, Steegers EA and Ursem NT:
The effects of homocysteine and folic acid on angiogenesis and VEGF
expression during chicken vascular development. Microvasc Res.
83:98–104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Q, Li Q, Chen Y, Huang X, Yang IH,
Cao L, Wu WK and Tan HM: Homocysteine-impaired angiogenesis is
associated with VEGF/VEGFR inhibition. Front Biosci (Elite Ed).
4:2525–2535. 2012.PubMed/NCBI
|
|
18
|
Chen CH, Beard RS and Bearden SE:
Homocysteine impairs endothelial wound healing by activating
metabotropic glutamate receptor 5. Microcirculation. 19:285–295.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rodrıguez-Nieto S, Chavarrıa T,
Martınez-Poveda B, Sánchez-Jiménez F, Rodríguez Quesada A and
Medina MA: Anti-angiogenic effects of homocysteine on cultured
endothelial cells. Biochem Biophys Res Commun. 293:497–500. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fan CD, Sun JY, Fu XT, Hou YJ, Li Y, Yang
MF, Fu XY and Sun BL: Astaxanthin attenuates homocysteine-induced
cardiotoxicity in vitro and in vivo by inhibiting mitochondrial
dysfunction and oxidative damage. Front Physiol. 8:10412017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pang X, Si J, Xu S, Li Y and Liu J:
Simvastatin inhibits homocysteine-induced CRP generation via
interfering with the ROS-p38/ERK1/2 signal pathway in rat vascular
smooth muscle cells. Vascul Pharmacol. 88:42–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian X, Zhao L, Song X, Yan Y, Liu N, Li
T, Yan B and Liu B: HSP27 inhibits homocysteine-induced endothelial
apoptosis by modulation of ROS production and mitochondrial
Caspase-dependent apoptotic pathway. Biomed Res Int.
2016:48478742016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang M, Cui Z, Cui H, Wang Y and Zhong C:
Astaxanthin protects astrocytes against trauma-induced apoptosis
through inhibition of NKCC1 expression via the NF-κB signaling
pathway. BMC Neurosci. 18:422017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nai Y, Liu H, Bi X, Gao H and Ren C:
Protective effect of astaxanthin on acute cerebral infarction in
rats. Hum Exp Toxicol. 37:929–936. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang M, Cui Z, Cui H, Cao Y, Zhong C and
Wang Y: Astaxanthin alleviates cerebral edema by modulating NKCC1
and AQP4 expression after traumatic brain injury in mice. BMC
Neurosci. 17:602016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee DH, Lee YJ and Kwon KH:
Neuroprotective effects of astaxanthin in oxygen-glucose
deprivation in SH-SY5Y cells and global cerebral ischemia in rat. J
Clin Biochem Nutr. 47:121–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X and Osawa T: Astaxanthin protects
neuronal cells against oxidative damage and is a potent candidate
for brain food. Forum Nutr. 61:129–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu YP, Liu SY, Sun H, Wu XM, Li JJ and Zhu
L: Neuroprotective effect of astaxanthin on H(2)O(2)-induced
neurotoxicity in vitro and on focal cerebral ischemia in vivo.
Brain Res. 1360:40–48. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shen H, Kuo CC, Chou J, Delvolve A,
Jackson SN, Post J, Woods AS, Hoffer BJ, Wang Y and Harvey BK:
Astaxanthin reduces ischemic brain injury in adult rats. FASEB J.
23:1958–1968. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wen X, Huang A, Hu J, Zhong Z, Liu Y, Li
Z, Pan X and Liu Z: Neuroprotective effect of astaxanthin against
glutamate-induced cytotoxicity in HT22 cells: Involvement of the
Akt/GSK-3β pathway. Neuroscience. 303:558–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu Q, Zhang XS, Wang HD, Zhang X, Yu Q, Li
W, Zhou ML and Wang XL: Astaxanthin activates nuclear factor
erythroid-related factor 2 and the antioxidant responsive element
(Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in
rats and attenuates early brain injury. Mar Drugs. 12:6125–6141.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang XS, Zhang X, Wu Q, Li W, Wang CX,
Xie GB, Zhou XM, Shi JX and Zhou ML: Astaxanthin offers
neuroprotection and reduces neuroinflammation in experimental
subarachnoid hemorrhage. J Surg Res. 192:206–213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang XS, Zhang X, Wu Q, Li W, Zhang QR,
Wang CX, Zhou XM, Li H, Shi JX and Zhou ML: Astaxanthin alleviates
early brain injury following subarachnoid hemorrhage in rats:
Possible involvement of Akt/bad signaling. Mar Drugs. 12:4291–4310.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang XS, Zhang X, Zhou ML, Zhou XM, Li N,
Li W, Cong ZX, Sun Q, Zhuang Z, Wang CX and Shi JX: Amelioration of
oxidative stress and protection against early brain injury by
astaxanthin after experimental subarachnoid hemorrhage. J
Neurosurg. 121:42–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bi YL, Mi PY, Zhao SJ, Pan HM, Li HJ, Liu
F, Shao LR, Zhang HF, Zhang P and Jiang SL: Salinomycin exhibits
anti-angiogenic activity against human glioma in vitro and
in vivo by suppressing the VEGF-VEGFR2-AKT/FAK signaling
axis. Int J Mol Med. 39:1255–1261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lai WK and Kan MY: Homocysteine-induced
endothelial dysfunction. Ann Nutr Metab. 67:1–12. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tiwari A, Pattanaik N, Mohanty Jaiswal A
and Dixit M: Increased FRG1 expression reduces in vitro cell
migration, invasion and angiogenesis, ex vivo supported by
reduced expression in tumors. Biosci Rep. 37(pii): BSR201710622017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bai Y, Bai L, Zhou J, Chen H and Zhang L:
Sequential delivery of VEGF, FGF-2 and PDGF from the polymeric
system enhance HUVECs angiogenesis in vitro and CAM angiogenesis.
Cell Immunol. 323:19–32. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Heldin J, O'Callaghan P, Hernández Vera R,
Fuchs PF, Gerwins P and Kreuger J: FGD5 sustains vascular
endothelial growth factor A (VEGFA) signaling through inhibition of
proteasome-mediated VEGF receptor 2 degradation. Cell Signal.
40:125–132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lv J, Sun B, Mai Z, Jiang M and Du J:
STAT3 potentiates the ability of airway smooth muscle cells to
promote angiogenesis by regulating VEGF signalling. Exp Physiol.
102:598–606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mahecha AM and Wang H: The influence of
vascular endothelial growth factor-A and matrix metalloproteinase-2
and-9 in angiogenesis, metastasis, and prognosis of endometrial
cancer. Onco Targets Ther. 10:4617–4624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang F, Pacheco MTF, Chen P, Liang D and
Li W: Secretogranin III promotes angiogenesis through MEK/ERK
signaling pathway. Biochem Biophys Res Commun. 495:781–786. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Abdelzaher LA, Imaizumi T, Suzuki T,
Tomita K, Takashina M and Hattori Y: Astaxanthin alleviates
oxidative stress insults-related derangements in human vascular
endothelial cells exposed to glucose fluctuations. Life Sci.
150:24–31. 2016. View Article : Google Scholar : PubMed/NCBI
|