Introduction
Ischemic heart disease is a leading cause of
mortality in a number of countries, especially the United States
(1,2). Despite the advances in therapeutic
strategy over the past decade, such as percutaneous coronary
intervention, antiplatelet and antithrombotic therapies and
angioplasty, ischemic heart disease remains prevalent and a major
culprit for heart failure (3,4).
One therapeutic strategy for this disease is
controlling ischemia/reperfusion (I/R) injury (5,6),
that can activate autophagy and consequent cell apoptosis (7). When the ischemic myocardium is
re-perfused with oxygen and substrate-rich blood, I/R injury
develops to further damage the heart (4). Therefore, an increasing number of
studies have been carried out to investigate the myocytic autophagy
and apoptosis induced by I/R injury (8–10).
CD47 signal controls second messengers, such as
calcium, cAMP and cGMP. In vascular cells, CD47 inhibits the
production and effector pathways of nitric oxide (NO). Therefore,
CD47 signaling can regulate blood flow, platelet homeostasis and
angiogenesis (11,12). Studies have demonstrated that
reducing or blocking CD47 can profoundly protect cells and tissues
from apoptosis induced by I/R injury (11–19)
or radiation injury through activating autophagic flux (20–22).
But so far, the effect of CD47 downregulation on myocytic autophagy
induced by I/R injury remains unclear.
A hypoxia/reoxygenation (H/R) in vitro model
is appropriate for exploring the molecular mechanisms and functions
of autophagy during myocardial ischemia/reperfusion (I/R) (23). In this study, using the H9c2 cell
line to model I/R injury induced cardiomyocyte apoptosis, the
effect of CD47 on cellular I/R injury and autophagy was
investigated.
Materials and methods
H9c2 cell culture
The H9c2 cells (ventricular myocardiocyte, rat in
origin; Cell Bank of the Chinese Academy of Sciences, Shanghai,
China) were seeded in 6-well plates (2×104
cells/cm2) and cultured in Dulbecco's modified Eagle's
medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) containing
10% (v/v) fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and 100 mmol/l penicillin-streptomycin in a
humidified atmosphere (95% air and 5% CO2, 37°C). The
medium was replenished every two days.
H/R in H9c2
Cardiomyocyte hypoxia was induced by exposing the
cells to 1% O2, 94% N2 and 5% CO2
for 24 h in a modular incubator (Model 3131; Forma Scientific;
Thermo Fisher Scientific, Inc.). Then the cells were reoxygenated
(95% air, 5% CO2, 37°C) for 12 h (24). Cells under normoxia were used as a
control throughout the experiments. All experiments were repeated
three times.
Transfection
H9c2 cells were digested to form a single-cell
suspension and plated in 6-well plates until the cells entered the
logarithmic growth phase. When the cell density reached ~80%, 20 nM
short interfering (Si)-CD47 or a scrambled Si-RNA (Si-NC) was
transfected into the cells using Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
sequences of siRNAs are: Si-CD47, 5′-AGAUUUGACUUUACUAAGCAG-3′ and
Si-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′. After 24 h of transfection, the
mRNA expression of CD47 was determined by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Experiment design
A total of two experiments were performed. In
experiment 1, H9c2 cells were assigned to 5 groups. i) Control
group: H9c2 cells were maintained under normoxic conditions without
the CD47 antibody (cat. no. sc-53050; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) or CQ treatment; ii) H/R group: H9c2 cells
were subjected to 24 h of hypoxia followed by 12 h of reoxygenation
(24); iii) CD47 group: H9c2 cells
were treated with the CD47 antibody (7 µg/ml for 2 h) and incubated
under normoxic conditions (25);
iv) H/R+CD47 group: H9c2 cells treated with the CD47 antibody were
subjected to 24 h of hypoxia followed by 12 h of reoxygenation; v)
H/R+CD47+CQ group: H9c2 cells treated with CD47 antibody (7 µg/ml
for 2 h) and CQ (10 mmol/l for 1 h; C6628; Sigma-Aldrich; Merck
KGaA) were subjected to 24 h of hypoxia followed by 12 h of
reoxygenation. CQ was used to inhibit lysosomal acidification and
autophagosome-lysosome fusion (26). H9c2 cells were treated with CD47
and CQ for the indicated times as described previously (25,26).
In experiment 2, H9c2 cells were assigned to 3 groups: i)
Si-NC+H/R: H9c2 cells treated with Si-NC were subjected to 24 h of
hypoxia followed by 12 h of reoxygenation; ii) Si-CD47: H9c2 cells
treated with SiCD47; iii) Si-CD47+H/R: H9c2 cells treated with
Si-CD47 were subjected to 24 h of hypoxia followed by 12 h of
reoxygenation. All experiments were repeated three times.
Determination of cell injury
Using commercially available kits for
malondialdehyde (MDA; cat. no. A003-1), lactate dehydrogenase (LDH;
cat. no. A020-2), creatinine kinase-muscle/brain (CK-MB; cat. no.
H197) and superoxide dismutase (SOD; cat. no. A001-1; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China), the
biochemical parameters of the medium supernatant and H9c2 cells
were measured.
Reactive oxygen species (ROS)
staining
To evaluate cell production of ROS, the slides of
cells were incubated with 10 µmol/l dihydroethidium (DHE; D-23107;
Invitrogen, Thermo Fisher Scientific, Inc.) in PBS in the dark for
exactly 30 min at room temperature. Then, the sections were rinsed
twice with cold PBS and imaged at 490–560 nm using an Olympus
microscope (1–71, Olympus Corporation) immediately.
Determination of cellular
apoptosis
To detect the apoptosis rate of the H9C2 cells, we
used an Annexin V-fluorescein isothiocyanate (FITC) Assay kit (cat.
no. 556547; BD Biosciences; Becton, Dickinson and Company, Franklin
Lakes, NJ, USA). Briefly, the granulosa cells were washed twice
with cold PBS which was removed afterwards from the cell pellet.
The cells were resuspended in 500 µl of binding buffer. Then 5 µl
of Annexin V-FITC and 5 µl of PI staining solution was added. The
cells were vortexed, incubated for 15 min in the dark and detected
by flow cytometer (BD FACSCalibur; BD Biosciences; Becton,
Dickinson and Company). The mean fluorescent intensity of the
annexin V/PI double staining in the myocytes was analyzed using
FlowJo software version 10.4.2 (FlowJo LLC). The proportion of
apoptotic cells (mortality) was defined as the percentage of
Annexin V+-FITC+ cells (n=9 per group).
Electron microscopy
For transmission electron microscope (TEM)
examination, cells were digested and centrifuged (800 × g for 5 min
at 25°C) to form cell pellets, and fixed in 2.5% glutaraldehyde for
48 h at 4°C, postfixed in 0.5% osmium tetroxide, dehydrated and
embedded in epoxy resin. Ultrathin sections (90 nm thick) were made
and examined using transmission electron microscope (Tecnai G2
Spirit Bio TWIN; FEI Ltd.) at accelerating voltage 80 kV and
×10,000 magnification. Electron micrographs (five fields of view
per cell) were randomly examined (five cells at four corners and in
the middle were selected) for each experiment (n=9 per group).
Immunofluorescent staining
For immunofluorescent staining, H9c2 were rinsed in
Dulbecco's PBS (DPBS) for 5 min at room temperature, then fixed in
4% paraformaldehyde in DPBS at 25°C for 10 min. After being washed
with DPBS, the cells were permeabilized in 1% Triton X-100 for 1 h
and eventually blocked in 10% goat serum (blocking solution, cat.
no. 16210072; Gibco; Thermo Fisher Scientific, Inc.) for 1 h at
room temperature to inhibit nonspecific binding. Primary antibodies
included CD47 (cat. no. sc-53050; Santa Cruz Biotechnology, Inc.),
Beclin-1 (cat. no. sc-48341; Santa Cruz Biotechnology, Inc.),
SQSTM1/p62 (p62; cat. no. 23214; Cell Signal Technology, Inc., USA)
and Vimentin (cat. no. 5741; Cell Signal Technology, Inc.). All the
cells were treated with primary antibodies in the blocking solution
for 24 h at 4°C. Subsequently, the cells were rinsed in 1X DPBS and
incubated with secondary antibodies in the blocking solution for 1
h at 4°C. Alexa Fluor 546 goat anti-mouse lgG (H+L) (1:200; cat.
no. A11030; Invitrogen; Thermo Fisher Scientific, Inc.) and donkey
anti-rabbit-CY3 (1:200; cat. no. A21206; Molecular Probes; Thermo
Fisher Scientific, Inc.) were used as secondary antibodies. The
cells were then washed and stained with DAPI (Fluoroshield with
DAPI, F6057; Gibco; Thermo Fisher Scientific, Inc.). The stained
cells were examined using confocal images system (710; Zeiss AG,
Oberkochen Germany). All the sections were incubated with the
antibodies at the same concentration and under the same condition.
The tissue section images were captures at 490–560 nm with an
Olympus microscope (1–71; Olympus Corporation) and semi-quantified
with Image Pro Plus 6.0 software (Media Cybernetics, Inc.). The
integrated optical density (IOD) was collected for each photograph.
A total of five fields in each slice (five slides per animal) were
randomly selected for blinded measurements (n=6 per group). The
images were quantified by the immunoreactive area (IA) in
µm2 and the IOD. The staining intensity (SI) for each
image was calculated as SI=IOD/IA and the mean with standard
deviation was obtained for each series.
RT-qPCR
RT-qPCR was performed according to previously
described methods (27). Following
48 h of transfection with Si-CD47 or SiNC, total RNA from H9C2
cells was extracted using TRIzol reagent (B5704-1; Takara
Biotechnology, Co., Ltd., Dalian, China). Then, 1 µg of total RNA
was reverse transcribed to cDNA at 37°C for 15 min and 85°C for 5
sec using PrimeScript™ RT reagent kit (cat. no. RR037A; Takara
Biotechnology, Co., Ltd.). Subsequently, PCR was performed using a
Light Cycler PCR QC kit (Roche Applied Science) and 7300 Real-Time
PCR System (LC96; Roche Applied Science). In the 25 µl reaction
system, 300 nmol/l primers were added. The thermo cycling
conditions were: 2 min at 50°C and 10 min at 95°C, followed by 40
cycles of 15 sec at 95°C and 1 min at 60°C. The primers were as
follows: CD47 (NM_019195.2) forward:
5′-AGAGAATCATTCTGCTGCTGGTTGC-3′, reverse:
5′-TGGTGAAAGAGGTCATTCCAAAAGC-3′; GAPDH (NM_017008.4), forward:
5′-CTGGAGAAACCTGCCAAGTATG-3′, reverse: 5′-GGTGGAAGAATGGGAGTTGCT-3′.
The housekeeping gene GAPDH was used as an internal reference.
GraphPad Prism 5 software (GraphPad Software, Inc.) was used for
chart production.
Western blot analysis
The cells were harvested in RIPA lysis buffer
(Bioteke Corporation, Beijing, China) containing 1 mM
phenylmethylsulfonyl fluoride. Protein concentration was measured
using the Bio-Rad method. Samples (20 mg protein) were separated by
10% SDS-PAGE and transferred to a nitrocellulose membrane. The
membrane was blocked with 5% non-fat milk in TBST buffer (100 mM
NaCl, 10 mM Tris-HCl, pH 7.4, 0.1% Tween-20) at 25°C for 1 h,
incubated with the primary antibodies against LC3 (1:1,000, cat.
no. 4108; Cell Signal Technology, Inc.), Beclin-1 (1:1,000, cat.
no. sc-48341; Santa Cruz Biotechnology, Inc.), p62 (1:1,000, 23214;
Cell Signal Technology, Inc.), LAMP2 (1:1,000, cat. no. sc-71492;
Santa Cruz Biotechnology, Inc.), cleaved caspase-3 (1:1,000, cat.
no. 9664; Cell Signal Technology, Inc.), cleaved caspase-9
(1:1,000, cat. no. 7237; Cell Signal Technology, Inc.) and GAPDH
(1:1,000, cat. no. sc-166574, Santa Cruz Biotechnology, Inc.) at
4°C overnight, and re-incubated with goat anti-rabbit IgG
HRP-conjugated secondary antibodies (1:5,000, cat. no. sc-2004;
Santa Cruz Biotechnology, Inc.) or goat anti-mouse IgG
HRP-conjugated secondary antibodies (1:5,000, cat. no. sc-2005;
Santa Cruz Biotechnology, Inc.). Then, the membranes were washed
three times in TBST. The blots were imaged using the ChemiDoc XRS+
Molecular Imager (Bio-Rad Laboratories, Inc.) with the Pierce ECL
Western Blotting Substrate (cat. no. 32209; Thermo Fisher
Scientific, Inc.) and analyzed using image analysis software
(ImageJ 1.42; National Institutes of Health, Bethesda, MD, USA).
The housekeeping protein GAPDH was used as the internal control.
The western blotting quantification was corrected to GAPDH
expression prior to normalization.
Statistical analysis
All data were presented as the mean ± standard error
and analyzed using SPSS 13.0 (SPSS, Inc.). One-way analysis of
variance was used to determine statistical significance. Bonferroni
post hoc test was introduced as needed. P<0.05 was considered to
indicate a statistically significant difference. All experiments
were repeated three times.
Results
CD47 protects cardiomyocytes against
H/R-induced oxidative stress and apoptosis in cardiomyocytes
The effects of the CD47 antibody on cardiomyocyte
function and oxidative stress levels in H9c2 cells are presented in
Fig. 1A-D. There was no
significant difference in LDH, CK-MB, MDA and SOD levels between
the control and CD47 groups. Compared with the control group, H/R
treatment significantly increased LDH, CK-MB and MDA levels and
significantly decreased SOD activity in the culture media in the
H/R group (P<0.01). Compared with the H/R group, CD47 antibody
treatment significantly decreased LDH, CK-MB and MDA levels and
enhanced SOD activity in H/R+CD47 group (P<0.01). Compared with
the control group, the autophagy inhibitor CQ significantly
increased LDH, CK-MB and MDA levels and weakened SOD activity in
H/R+CD47+CQ group (P<0.01).
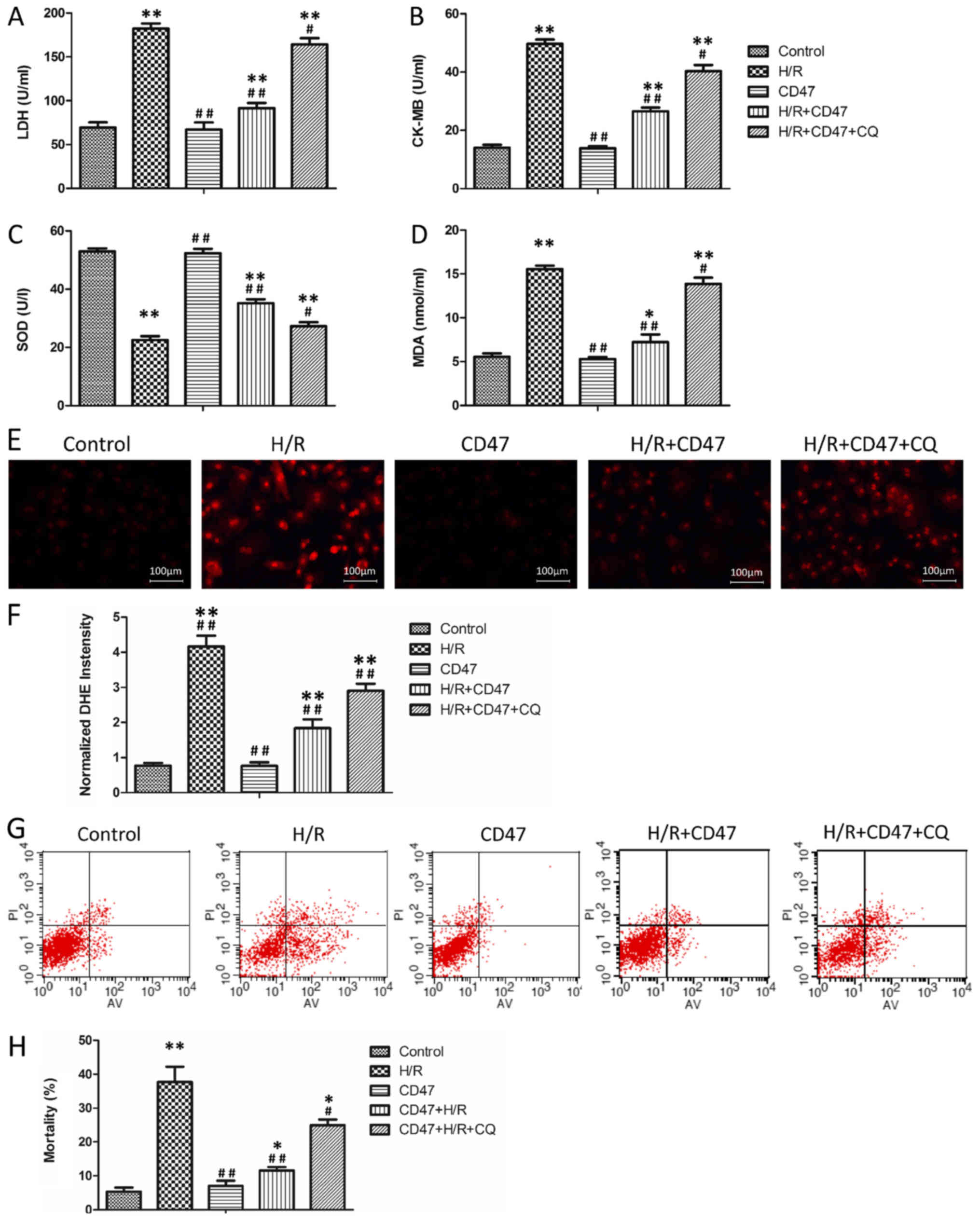 | Figure 1.CD47 protects cardiomyocytes against
H/R-induced function oxidative stress and apoptosis. The
biochemical parameters of the medium supernatant, including (A)
LDH, (B) CK-MB and (C) SOD activities and (D) MDA level, were
measured (n=9). (E) The ROS levels in the cells from all groups
were revealed by DHE staining. (F) The fluorescence intensity of
DHE staining was analyzed using Image-Pro Plus (n=9). (G)
Quantitative assessment of cell mortality by Annexin V-FITC/PI
staining. Intact cells are V−/PI−, early
apoptotic cells are V+/PI−, late apoptotic
cells are Annexin V+/PI+ and necrotic cells
are Annexin V−/PI+. The figures are
representative images of three different experiments. (H) Flow
cytometry results are displayed as quantitative bar graphs.
Mortality is defined as the percentage of Annexin V+
cells (n=9). The data were collected from three different
experiments. Data are presented as the mean ± standard deviation.
Statistical significance: *P<0.05 and **P<0.01 vs. the
control group, #P<0.05 and ##P<0.01 vs.
the H/R group, respectively. SOD, superoxide dismutase; MDA,
malondialdehyde; LDH, lactate dehydrogenase; CD, cluster of
differentiation; CK-MB, creatine kinase; PI, propidium iodide;
FITC, fluorescein isothiocyanate; CQ, chloroquine; H/R,
hypoxia/reoxygenation. |
To assess the effects of CD47 deficiency on ROS
production, intracellular generation of the ROS moiety
O2− was visualized with the fluoroprobe DHE
(Fig. 1E). The superoxide anion
oxidizes DHE to a novel product that binds to DNA, leading to
enhanced fluorescence. In this assay, confocal microscopy
demonstrated that cell slides from the H/R group exhibited
widespread and significant increases in DHE fluorescence compared
with those from the control group (P<0.01; Fig. 1F) and CD47 antibody treatment in
the H/R+CD47 group significantly decreased ROS fluorescence
intensity compared with H/R alone (P<0.01). However, the
autophagy inhibitor CQ significantly increased the ROS fluorescence
intensity in the H/R+CD47+CQ group (P<0.01).
Annexin V/PI double staining was used to assess the
apoptotic rate of the H9c2 cells (Fig.
1G). There was no significant difference in apoptotic rates
between the control and CD47 groups (Fig. 1H). Compared with the control group,
H/R treatment significantly increased the apoptotic rate in the H/R
group (P<0.01). Compared with the H/R group, CD47 antibody
treatment significantly decreased the apoptotic rate in the
H/R+CD47 group (P<0.01). Compared with the control group, the
autophagy inhibitor CQ significantly increased the apoptotic rate
in the H/R+CD47+CQ group (P<0.05).
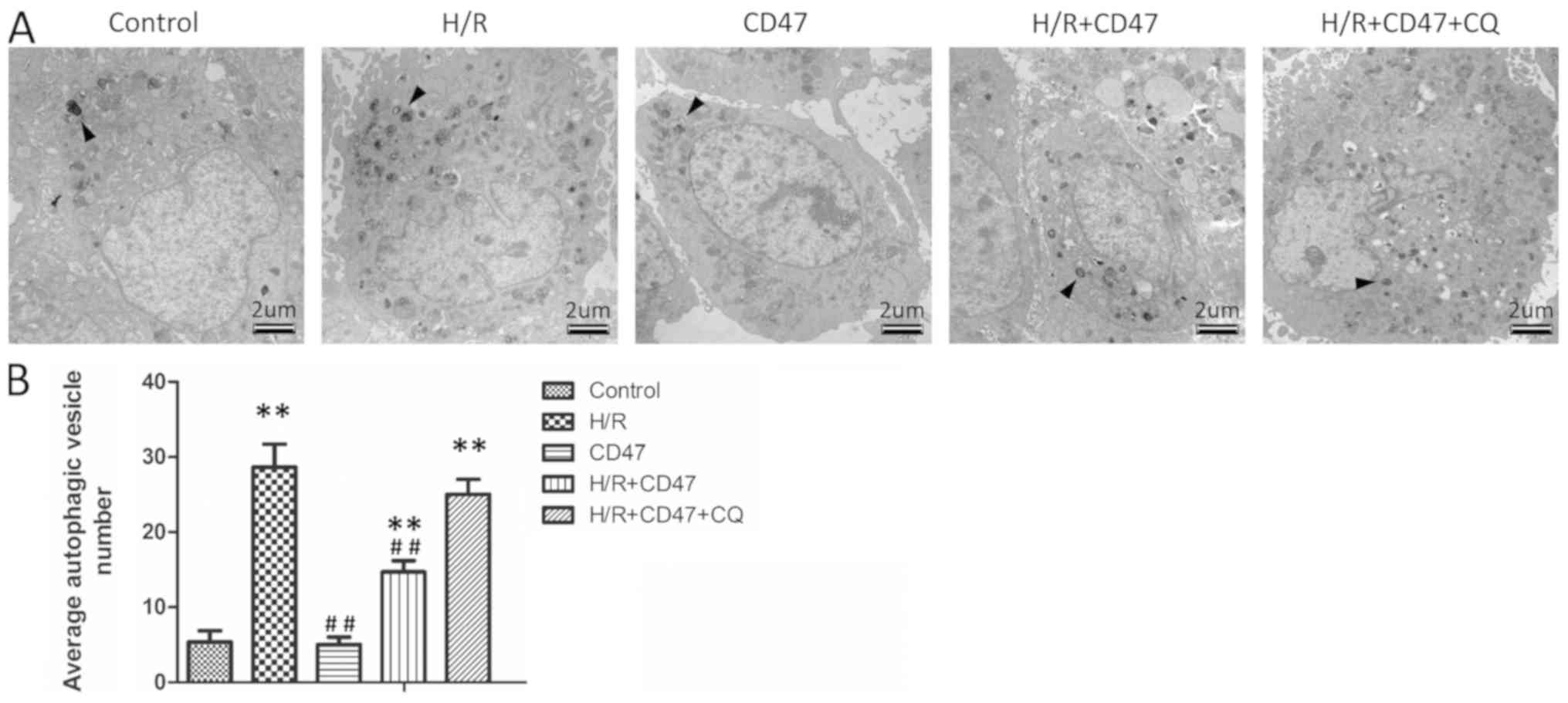
CD47 suppresses the accumulation of
autophagosomes in H/R-treated cardiomyocytes
TEM results demonstrated that there was no
significant difference in apoptotic rates between the control and
CD47 groups (Fig. 2A and B).
Compared with the control group, H/R treatment significantly
increased autophagic vesicle number in the H/R group (P<0.01).
Compared with the H/R group, CD47 treatment significantly decreased
the autophagic vesicle number in the H/R+CD47 group (P<0.01).
Compared with the control group, the autophagy inhibitor CQ
significantly increased the autophagic vesicle number in the
H/R+CD47+CQ group (P<0.01).
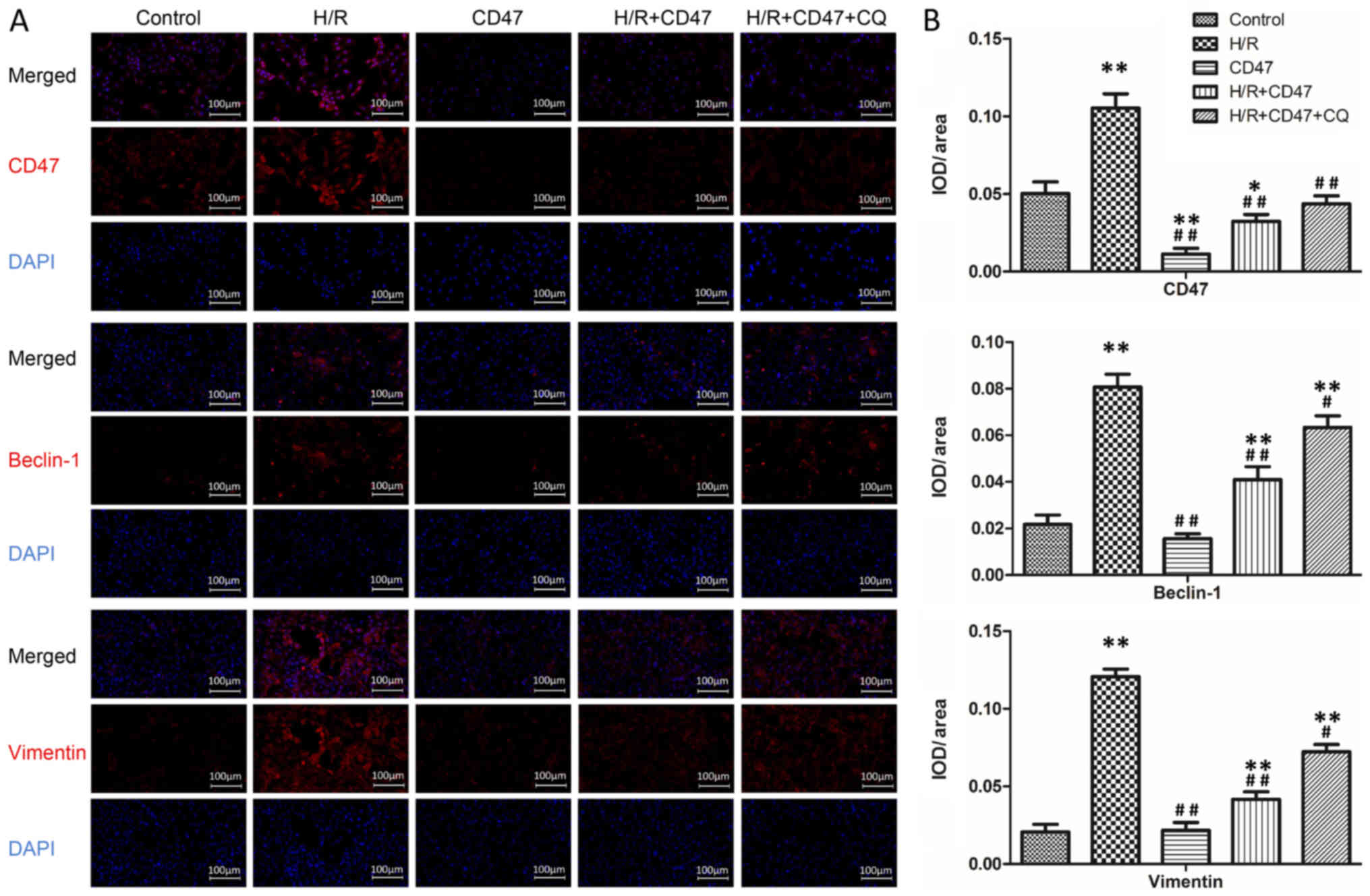
CD47 inhibits the accumulation of
protein aggregates in H/R-treated cardiomyocytes
As presented in Fig.
3, there was no significant difference in Beclin-1 and Vimentin
expression between the control and CD47 groups. Compared with the
control group, CD47 treatment significantly decreased CD47
fluorescence intensity in the CD47 group (P<0.01). Compared with
the control group, H/R treatment significantly increased CD47,
Beclin-1 and Vimentin expression in the H/R group (P<0.01).
Compared with the H/R group, CD47 antibody treatment in the
H/R+CD47 group significantly decreased CD47, Beclin-1 and Vimentin
expression (P<0.05 and P<0.01, respectively). Compared with
the control group, the autophagy inhibitor CQ significantly
increased Beclin-1 and Vimentin expression in the H/R+CD47+CQ group
(P<0.01).
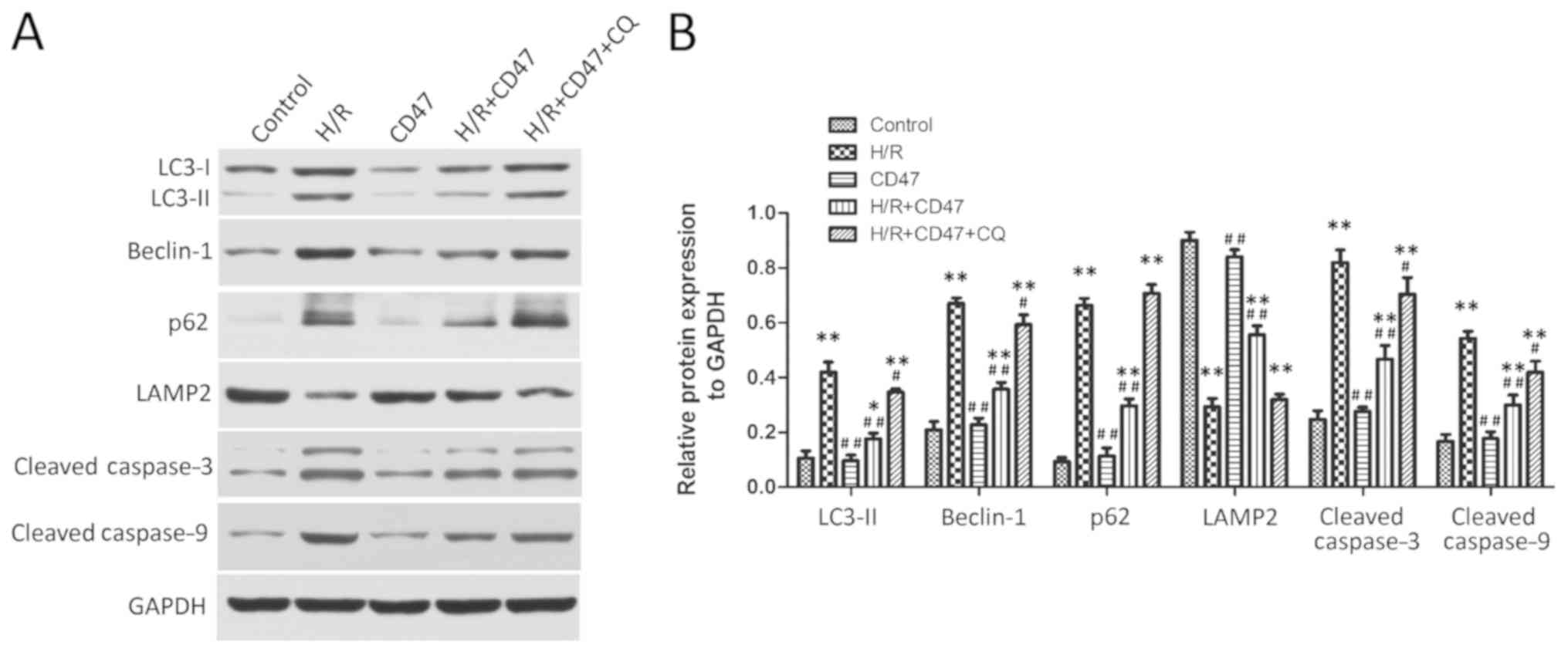
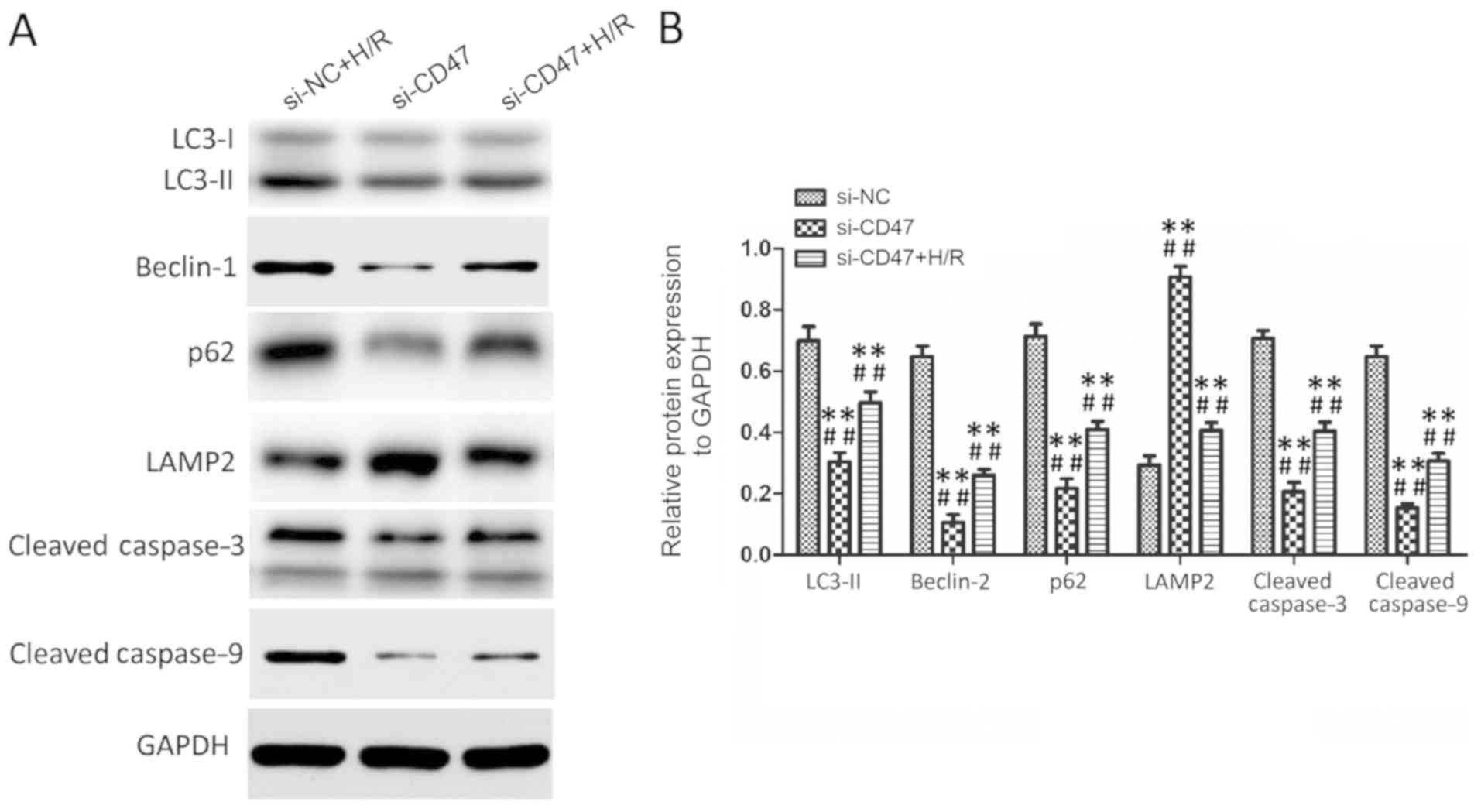
CD47 protects cardiomyocytes against
H/R injury through rescuing impaired autophagy flux
As presented in Fig.
4, the expression levels of LC3-II, Beclin-1, p62, LAMP2,
Cleaved caspase-3 and cleaved caspase-9 exhibited no significant
difference between the control and CD47 groups. Compared with the
control group, H/R treatment significantly increased LC3-II,
Beclin-1, p62, cleaved caspase-3 and cleaved caspase-9 protein
levels and significantly decreased LAMP2 protein level in the H/R
group (P<0.01). Compared with the H/R group, CD47 treatment
significantly decreased LC3-II, Beclin-1, p62 protein, cleaved
caspase-3 and cleaved caspase-9 levels and significantly increased
LAMP2 protein level in the H/R+CD47 group (P<0.01). However,
compared with the control group, the autophagy inhibitor CQ
significantly increased LC3-II, Beclin-1, p62 protein, cleaved
caspase-3 and cleaved caspase-9 levels and decreased LAMP2 protein
level in the H/R+CD47+CQ group (P<0.01 and P<0.05,
respectively).
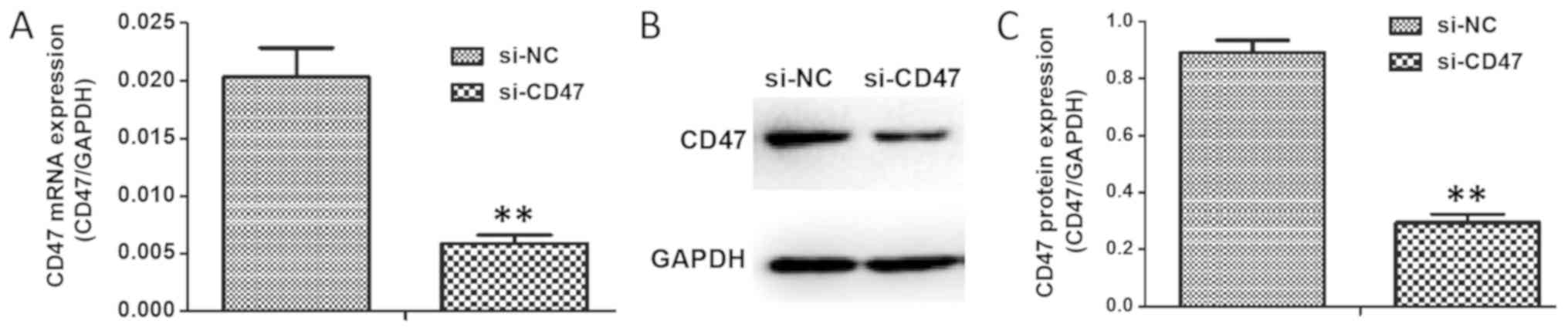
Downregulating CD47 expression
inhibits H/R-induced oxidative stress and apoptosis in
cardiomyocytes
To investigate the action of CD47 in H9C2 cells,
CD47 expression was knocked-down using RNA interference technology
(Si-CD47), along with a non-targeting Si-NC that was constructed to
use as a negative control in all assays. The interference
efficiency of Si-CD47 on H9c2 expression is demonstrated in
Fig. 5A. Compared with the Si-NC,
Si-CD47 exerted significantly increased efficiency in knocking down
CD47 mRNA expression (P<0.01; an inhibition rate of 62%).
Western blotting results (Fig. 5B and
C) indicated that the protein expression of CD47 in the Si-CD47
group was decreased compared with in the Si-NC group
(P<0.01).
The effects of si-CD47 on cardiomyocytic function
and oxidative stress levels in H9c2 are presented in Fig. 6A-D. Compared with the Si-CD47
group, H/R treatment significantly increased LDH, CK-MB and MDA
levels and significantly decreased SOD activity in H9C2 cells in
the Si-NC+H/R group (P<0.01). Compared with the Si-NC+H/R group,
CD47 downregulation decreased LDH, CK-MB and MDA levels and
increased SOD activity in the Si-CD47+H/R group (P<0.01).
Annexin V/PI double staining was used to assess the apoptotic rate
in the H9c2 cells (Fig. 6E).
Compared with the Si-CD47 group, H/R treatment significantly
increased the apoptotic rate in the Si-NC+H/R group (P<0.01;
Fig. 6F). Compared with the H/R
group, CD47 downregulation significantly decreased the apoptotic
rate in the Si-CD47+H/R group (P<0.01).
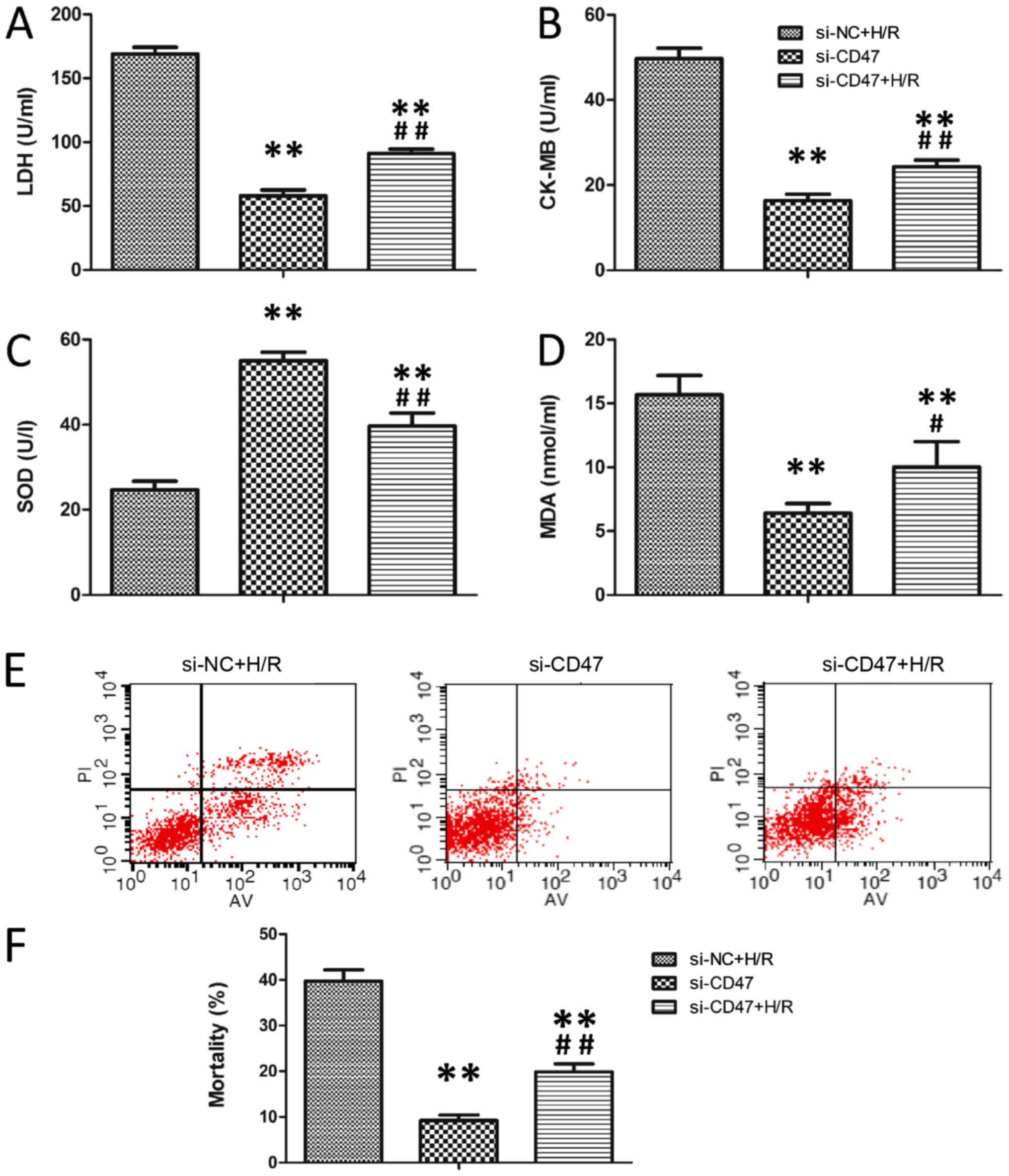 | Figure 6.CD47 downregulation protects
cardiomyocytes against H/R-induced oxidative stress. The
biochemical parameters of the medium supernatant, including (A)
LDH, (B) CK-MB and (C) SOD activities and (D) MDA level, were
measured (n=6). (E) Quantitative assessment of mortality by Annexin
V-FITC/PI staining. Intact cells are V−/PI−,
early apoptotic cells are V+/PI−, late
apoptotic cells are Annexin V+/PI+ and
necrotic cells are Annexin V−/PI+. The
figures are representative images of three different experiments.
(F) Flow cytometry results are displayed as quantitative bar
graphs. Cell mortality is defined as the percentage of Annexin
V+ cells (n=6). The data were collected from three
different experiments. All data are presented as the mean ±
standard deviation. Statistical significance: **P<0.01 vs. the
Si-NC group, #P<0.05 and ##P<0.01 vs.
the Si-CD47+H/R group, respectively. SOD, superoxide dismutase; PI,
propidium iodide; FITC, fluorescein isothiocyanate; LDH, lactate
dehydrogenase; MDA, malondialdehyde; H/R, hypoxia/reoxygenation;
CK-MB, creatinine kinase-MB; CD, cluster of differentiation. |
CD47 protects cardiomyocytes against
H/R injury through rescuing impaired autophagy flux
As presented in Fig.
7, H/R treatment significantly increased LC3-II, Beclin-1, p62,
cleaved caspase-3 and cleaved caspase-9 protein levels and
significantly decreased LAMP2 protein level in the Si-NC+H/R group
compared with the Si-CD47 group (P<0.01). Compared with the
Si-NC+H/R group, CD47 downregulation group significantly decreased
LC3-II, Beclin-1, p62 protein, cleaved caspase-3 and cleaved
caspase-9 levels and significantly increased LAMP2 protein level in
Si-CD47+H/R (P<0.01).
Discussion
CD47 deficiency has been reported to protect normal
cells and tissues from I/R injury-induced apoptosis (11,12,28–40).
However, the molecular mechanism behind this protection remains
obscure. In the present study, CD47 downregulation protected
cardiomyocytes against H/R injury in H9c2 cells, an effect largely
attributed to the inhibition of autophagy. It was demonstrated that
more obvious cardiomyocyte function damage, oxidative stress,
protein aggregation, cardiomyocyte apoptosis and autophagy
inhibition appeared in untreated H/R H9c2 cells, and that all these
changes were decreased by CD47 downregulation through autophagy
promotion; moreover, the autophagosome-lysosome fusion inhibitor CQ
reversed the effect of CD47 treatment.
Oxidative stress and apoptosis are two major
mechanisms involved in cardiomyocyte injury following hypoxia and
I/R (31–33). Autophagy regulates intracellular
homeostasis and cardiomyocytic survival. Mounting evidence supports
the idea that ROS and autophagosome accumulation induce autophagy
(34–36). Autophagosome accumulation hampers
the clearance of damaged intracellular organelles and proteins,
resulting in overproduction of ROS (37). Oxidative stress serves a central
role in myocardial I/R injury (38,39).
During injury, the blood supply is re-established in the ischemic
myocardium and superabundant oxygen free radicals are generated to
trigger oxidative stress and aggravate myocardium I/R injury
(40). Through autophagy protein
aggregates and damaged organelles are removed from the cytoplasm
(3,41). Once autophagy is disrupted, the
resultant autophagosome accumulation increases ROS production and
mitochondrial permeability, a process ending in cell death
(6). As demonstrated in the
present study, this process can be attenuated by CD47 antibody or
Si-CD47 pretreatment. The results of the present study indicate
that CD47 downregulation can inhibit apoptosis by the oxidative
stress pathway.
I/R injury in H9c2 cells is a complex biochemical
cascade that is initiated after ischemia and is exacerbated after
the return of blood flow. I/R causes both cardiomyocytic death and
autophagy in H9c2 cells (41).
Autophagy has a dual role in cell development during heart ischemia
and reperfusion (42). Autophagy
maintains the homeostasis of cellular ATP and ensures cell survival
in the ischemic heart (43).
However, excessive autophagy can cause cell death during
reperfusion (44). In the present
study, both H/R-induced apoptosis and autophagy in H9c2 cells were
attenuated by CD47 downregulation through increasing autophagosome
procession. However, the lysosomal acidification inhibitor CQ
prevents autophagosome-lysosome fusion (44) and can neutralize the effect of CD47
antibody treatment.
Autophagy is regulated by several proteins,
including LC3-II, Beclin-1, p62 and LAMP2 (45–48).
Therefore, the present study examined the levels of these
autophagy-associated proteins in myocytes during H/R. In mammalian
cells, LC3-II becomes membrane-bounding and easy to localize on
autophagosomes (49), which in
turn makes LC3-II an ideal autophagosomal marker (50). In the present study, the increase
of LC3-II expression was associated with reduced cardiomyocytic
viability during H/R, suggesting that autophagy during the
reperfusion phase may be detrimental for myocytes. Notably, CD47
antibody or Si-CD47 treatment downregulated the expression of
LC3-II and increased the survival rate of H9c2 cells, indicating
that CD47 downregulation can promote cell survival through
inhibiting the excessive autophagy induced by H/R.
To further explore the role of CD47 in autophagic
flux, the expression of p62 and Beclin-1, which link ubiquitinated
aggregates for destruction within autophagosomes, get degraded upon
autophagosome processing and increased in I/R-treated hearts, were
assessed (49,51). The p62 protein was degraded,
indicating that CD47 signaling normally limits cell survival by
preventing autophagic flux. Otherwise, Beclin-1, an
early-autophagy-related gene (45), activates autophagy during
reperfusion and promotes cardiomyocytic apoptosis (3). Downregulating Beclin-1 expression in
myocytes can inhibit I/R-induced autophagy and increase cell
survival rate (52). Bcl-2 can
bind to Beclin-1 to form a Bcl-2-Beclin-1 complex that prevents
Beclin-1 from assembling the pre-autophagosomal structure, a
process that inhibits autophagy and apoptosis (53,54).
Bcl-2 is a potent anti-apoptotic protein (55) and inducing its partner Beclin-1 by
loss of CD47 may be a master regulator of the fate decision between
apoptotic cell death and protective autophagy. The present study
identified Beclin-1 as another target of CD47 signaling
pathway.
Another finding of this study was the rapid decline
in LAMP2 abundance in the H/R group and the improvement of
autophagosome processing by upregulating LAMP2 in the CD47
treatment groups. However, CQ can disrupt the effect of CD47 by
inhibiting autophagosome processing. Both ischemia and reperfusion
induce the decline of LAMP2, a lysosome membrane protein that
participates in autophagosome-lysosome fusion (47,48).
This decline can be accelerated by ROS generation (49). LAMP2 knockdown impairs autophagy in
ventricular myocytes of adult rats and causes cell apoptosis at a
level comparable to that achieved by 3-Methyladenine, another
autophagy inhibitor (56).
Ablation of LAMP2 (57) or loss of
the LAMP2 (58) protein in Danon
disease (59), which is
characterized by autophagosome accumulation in myocardium and
cardiomyopathy (58), can cause
extensive myocardial fibrosis (60), suggesting that autophagosome
accumulation induced by inhibition of autophagic flux is a
pathogenic mechanism responsible for H/R injury in
cardiomyocytes.
In conclusion, the CD47 antibody or Si-CD47 can
protect cardiomyocytes against H/R injury through increasing
autophagic flux and autophagic clearance. CD47 deficiency may offer
a potential therapeutic approach for preventing myocardial I/R
injury, which should be tested with in vivo experiments.
Acknowledgements
The authors would like to thank Dr Wei Sun and Dr
Peng Li (Department of Cardiology, The First Affiliated Hospital of
Nanjing Medical University) for their technical assistance.
Funding
The present study was supported by the Natural
Science Foundation of China (grant no. 81627802).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DH designed the study and drafted the manuscript. YL
and KZ undertook cell culture and characterization. PZ, HF and YZ
helped perform the experiments regarding molecular biological
technique. DB and QC helped to perform TEM and immunofluorescence
staining. YY, LL and YD assisted with the biochemical index
detection. XK provided constructive suggestions on experimental
design, performed the data analysis and revised the manuscript. All
authors reviewed the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All experiments with animals were approved by the
Institutional Animal Care and Use Committee (IACUC) of the Nanjing
Medical University and the methods were carried out in accordance
with the approved guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Keeley EC, Boura JA and Grines CL: Primary
angioplasty versus intravenous thrombolytic therapy for acute
myocardial infarction: A quantitative review of 23 randomised
trials. Lancet. 361:13–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Przyklenk K, Dong Y, Undyala VV and
Whittaker P: Autophagy as a therapeutic target for
ischaemia/reperfusion injury? Concepts, controversies, and
challenges. Cardiovasc Res. 94:197–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jennings RB: Historical perspective on the
pathology of myocardial ischemia/reperfusion injury. Circ Res.
113:428–438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Verma S, Fedak PW, Weisel RD, Butany J,
Rao V, Maitland A, Li RK, Dhillon B and Yau TM: Fundamentals of
reperfusion injury for the clinical cardiologist. Circulation.
105:2332–2336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y and Ren J: Targeting autophagy for
the therapeutic application of histone deacetylase inhibitors in
ischemia/reperfusion heart injury. Circulation. 129:1088–1091.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ong SB and Gustafsson AB: New roles for
mitochondria in cell death in the reperfused myocardium. Cardiovasc
Res. 94:190–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y,
Chen J, Wang P and Fei Y: NF-κB-mediated induction of autophagy in
cardiac ischemia/reperfusion injury. Biochem Biophys Res Commun.
436:180–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wagner C, Tillack D, Simonis G, Strasser
RH and Weinbrenner C: Ischemic post-conditioning reduces infarct
size of the in vivo rat heart: Role of PI3-K, mTOR, GSK-3beta, and
apoptosis. Mol Cell Biochem. 339:135–147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ and Diwan A: Autophagy is impaired in cardiac
ischemia-reperfusion injury. Autophagy. 8:1394–1396. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Isenberg JS, Maxhimer JB, Powers P, Tsokos
M, Frazier WA and Roberts DD: Treatment of liver
ischemia/reperfusion injury by limiting thrombospondin-1/CD47
signaling. Surgery. 144:752–761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maxhimer JB, Shih HB, Isenberg JS, Miller
TW and Roberts DD: Thrombospondin-1/CD47 blockade following
ischemia-reperfusion injury is tissue protective. Plast Reconstr
Surg. 124:1880–1889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharifi-Sanjani M, Shoushtari AH, Quiroz
M, Baust J, Sestito SF, Mosher M, Ross M, McTiernan CF, St Croix
CM, Bilonick RA, et al: Cardiac CD47 drives left ventricular heart
failure through Ca2+-CaMKII-regulated induction of
HDAC3. J Am Heart Assoc. 3:e0006702014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sezaki S, Hirohata S, Iwabu A, Nakamura K,
Toeda K, Miyoshi T, Yamawaki H, Demircan K, Kusachi S, Shiratori Y
and Ninomiya Y: Thrombospondin-1 is induced in rat myocardial
infarction and its induction is accelerated by
ischemia/reperfusion. Exp Biol Med. 230:621–630. 2005. View Article : Google Scholar
|
|
15
|
Rogers NM, Thomson AW and Isenberg JS:
Activation of parenchymal CD47 promotes renal ischemia-reperfusion
injury. J Am Soc Nephrol. 23:1538–1550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao ZY, Banan B, Jia J, Manning PT,
Hiebsch RR, Gunasekaran M, Upadhya GA, Frazier WA, Mohanakumar T,
Lin Y and Chapman WC: CD47 blockade reduces ischemia/reperfusion
injury and improves survival in a rat liver transplantation model.
Liver Transpl. 21:468–477. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin Y, Manning PT, Jia J, Gaut JP, Xiao Z,
Capoccia BJ, Chen CC, Hiebsch RR, Upadhya G, Mohanakumar T, et al:
CD47 blockade reduces ischemia-reperfusion injury and improves
outcomes in a rat kidney transplant model. Transplantation.
98:394–401. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Isenberg JS, Pappan LK, Romeo MJ, Abu-Asab
M, Tsokos M, Wink DA, Frazier WA and Roberts DD: Blockade of
thrombospondin-1-CD47 interactions prevents necrosis of full
thickness skin grafts. Ann Surg. 247:180–190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rogers NM, Zhang ZJ, Wang JJ, Thomson AW
and Isenberg JS: CD47 regulates renal tubular epithelial cell
self-renewal and proliferation following renal ischemia
reperfusion. Kidney Int. 90:334–347. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maxhimer JB, Soto-Pantoja DR, Ridnour LA,
Shih HB, Degraff WG, Tsokos M, Wink DA, Isenberg JS and Roberts DD:
Radioprotection in normal tissue and delayed tumor growth by
blockade of CD47 signaling. Sci Transl Med. 1:3ra72009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Isenberg JS, Maxhimer JB, Hyodo F, Pendrak
ML, Ridnour LA, DeGraff WG, Tsokos M, Wink DA and Roberts DD:
Thrombospondin-1 and CD47 limit cell and tissue survival of
radiation injury. Am J Pathol. 173:1100–1112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soto-Pantoja DR, Miller TW, Pendrak ML,
DeGraff WG, Sullivan C, Ridnour LA, Abu-Asab M, Wink DA, Tsokos M
and Roberts DD: CD47 deficiency confers cell and tissue
radioprotection by activation of autophagy. Autophagy. 8:1628–1642.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao X, Chen A, Yang P, Song X, Liu Y, Li
Z, Wang X, Wang L and Li Y: Alpha-lipoic acid protects
cardiomyocytes against hypoxia/reoxygenation injury by inhibiting
autophagy. Biochem Biophys Res Commun. 441:935–940. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang B, Zhou M, Li C, Zhou J, Li H, Zhu
D, Wang Z, Chen A and Zhao Q: MicroRNA-92a inhibition attenuates
Hypoxia/Reoxygenation-induced myocardiocyte apoptosis by targeting
Smad7. PLoS One. 9:e1002982014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Yin C, Feng L, Wang C and Sheng G:
Ara-C and anti-CD47 antibody combination therapy eliminates acute
monocytic leukemia THP-1 cells in vivo and in vitro. Genet Mol Res.
14:5630–5641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma X, Godar RJ, Liu H and Diwan A:
Enhancing lysosome biogenesis attenuates BNIP3-induced
cardiomyocyte death. Autophagy. 8:297–309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang HB and Yang J, Ding JW, Chen LH, Li
S, Liu XW, Yang CJ, Fan ZX and Yang J: RNAi-mediated
down-regulation of CD47 protects against
ischemia/reperfusion-induced myocardial damage via activation of
eNOS in a rat model. Cell Physiol Biochem. 40:1163–1174. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Isenberg JS, Hyodo F, Pappan LK, Abu-Asab
M, Tsokos M, Krishna MC, Frazier WA and Roberts DD: Blocking
thrombospondin-1/CD47 signaling alleviates deleterious effects of
aging on tissue responses to ischemia. Arterioscler Thromb Vasc
Biol. 27:2582–2588. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thakar CV, Zahedi K, Revelo MP, Wang Z,
Burnham CE, Barone S, Bevans S, Lentsch AB, Rabb H and Soleimani M:
Identification of thrombospondin 1 (TSP-1) as a novel mediator of
cell injury in kidney ischemia. J Clin Invest. 115:3451–3459. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen HW, Chien CT, Yu SL, Lee YT and Chen
WJ: Cyclosporine A regulate oxidative stress-induced apoptosis in
cardiomyocytes: Mechanisms via ROS generation, iNOS and Hsp70. Br J
Pharmacol. 137:771–781. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Webster KA, Discher DJ, Kaiser S,
Hernandez O, Sato B and Bishopric NH: Hypoxia activated apoptosis
of cardiac myocytes requires reoxygenation or a pH shift and is
independent of p53. J Clin Invest. 104:239–252. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee Y and Gustafsson AB: Role of apoptosis
in cardiovascular disease. Apoptosis. 14:536–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jaber N, Dou Z, Chen JS, Catanzaro J,
Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J and Zong
WX: Class III PI3K Vps34 plays an essential role in autophagy and
in heart and liver function. Proc Natl Acad Sci USA. 109:2003–2008.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saiki S, Sasazawa Y, Imamichi Y, Kawajiri
S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K,
et al: Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tannous P, Zhu H, Nemchenko A, Berry JM,
Johnstone JL, Shelton JM, Miller FJ Jr, Rothermel BA and Hill JA:
Intracellular protein aggregation is a proximal trigger of
cardiomyocyte autophagy. Circulation. 117:3070–3078. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Classification of cell death:
Recommendations of the Nomenclature Committee on Cell Death. Cell
Death Differ. 16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016:16564502016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang W, Han Y, Meng G, Bai W, Xie L, Lu
H, Shao Y, Wei L, Pan S, Zhou S, et al: Direct renin inhibition
with aliskiren protects against myocardial ischemia/reperfusion
injury by activating nitric oxide synthase signaling in
spontaneously hypertensive rats. J Am Heart Assoc. 3:e0006062014.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen C, Chen W, Nong Z, Ma Y, Qiu S and Wu
G: Cardioprotective effects of combined therapy with hyperbaric
oxygen and diltiazem pretreatment on myocardial
ischemia-reperfusion injury in rats. Cell Physiol Biochem.
38:2015–2019. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin-1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takagi H, Matsui Y and Sadoshima J: The
role of autophagy in mediating cell survival and death during
ischemia and reperfusion in the heart. Antioxid Redox Signal.
9:1373–1381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takagi H, Matsui Y, Hirotani S, Sakoda H,
Asano T and Sadoshima J: AMPK mediates autophagy during myocardial
ischemia in vivo. Autophagy. 3:405–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
He C and Levine B: The Beclin 1
interactome. Curr Opin Cell Biol. 22:140–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Levine B and Ranganathan R: Autophagy:
Snapshot of the network. Nature. 466:38–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Eskelinen EL, Illert AL, Tanaka Y,
Schwarzmann G, Blanz J, Von Figura K and Saftig P: Role of LAMP-2
in lysosome biogenesis and autophagy. Mol Biol Cell. 13:3355–3368.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huynh KK, Eskelinen EL, Scott CC,
Malevanets A, Saftig P and Grinstein S: LAMP proteins are required
for fusion of lysosomes with phagosomes. EMBO J. 26:313–324. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
52
|
Valentim L, Laurence KM, Townsend PA,
Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS and
Stephanou A: Urocortin inhibits Beclin1.mediated autophagic cell
death in cardiac myocytes exposed to ischaemia/reperfusion injury.
J Mol Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang XH, Kleeman LK, Jiang HH, Gordon G,
Goldman JE, Berry G, Herman B and Levine B: Protection against
fatal Sindbis virus encephalitis by beclin, a novel
Bcl-2-interacting protein. J Virol. 72:8586–8596. 1998.PubMed/NCBI
|
|
54
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Akar U, Chaves-Reyez A, Barria M, Tari A,
Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G and Ozpolat
B: Silencing of Bcl-2 expression by small interfering RNA induces
autophagic cell death in MCF-7 breast cancer cells. Autophagy.
4:669–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Maruyama R, Goto K, Takemura G, Ono K,
Nagao K, Horie T, Tsujimoto A, Kanamori H, Miyata S, Ushikoshi H,
et al: Morphological and biochemical characterization of basal and
starvation-induced autophagy in isolated adult rat cardiomyocytes.
Am J Physiol Heart Circ Physiol. 295:H1599–H1607. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tanaka Y, Guhde G, Suter A, Eskelinen EL,
Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K and
Saftig P: Accumulation of autophagic vacuoles and cardiomyopathy in
LAMP-2-deficient mice. Nature. 406:902–906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nishino I, Fu J, Tanji K, Yamada T,
Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al: Primary
LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and
myopathy (Danon disease). Nature. 406:906–910. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Maron BJ, Roberts WC, Arad M, Haas TS,
Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP, Ho CY, et al:
Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy.
JAMA. 301:1253–1259. 2009. View Article : Google Scholar : PubMed/NCBI
|